Development and Validation of a Liquid Chromatography High-Resolution Mass Spectrometry Method for the Simultaneous Determination of Mycotoxins and Phytoestrogens in Plant-Based Fish Feed and Exposed Fish



 and
and 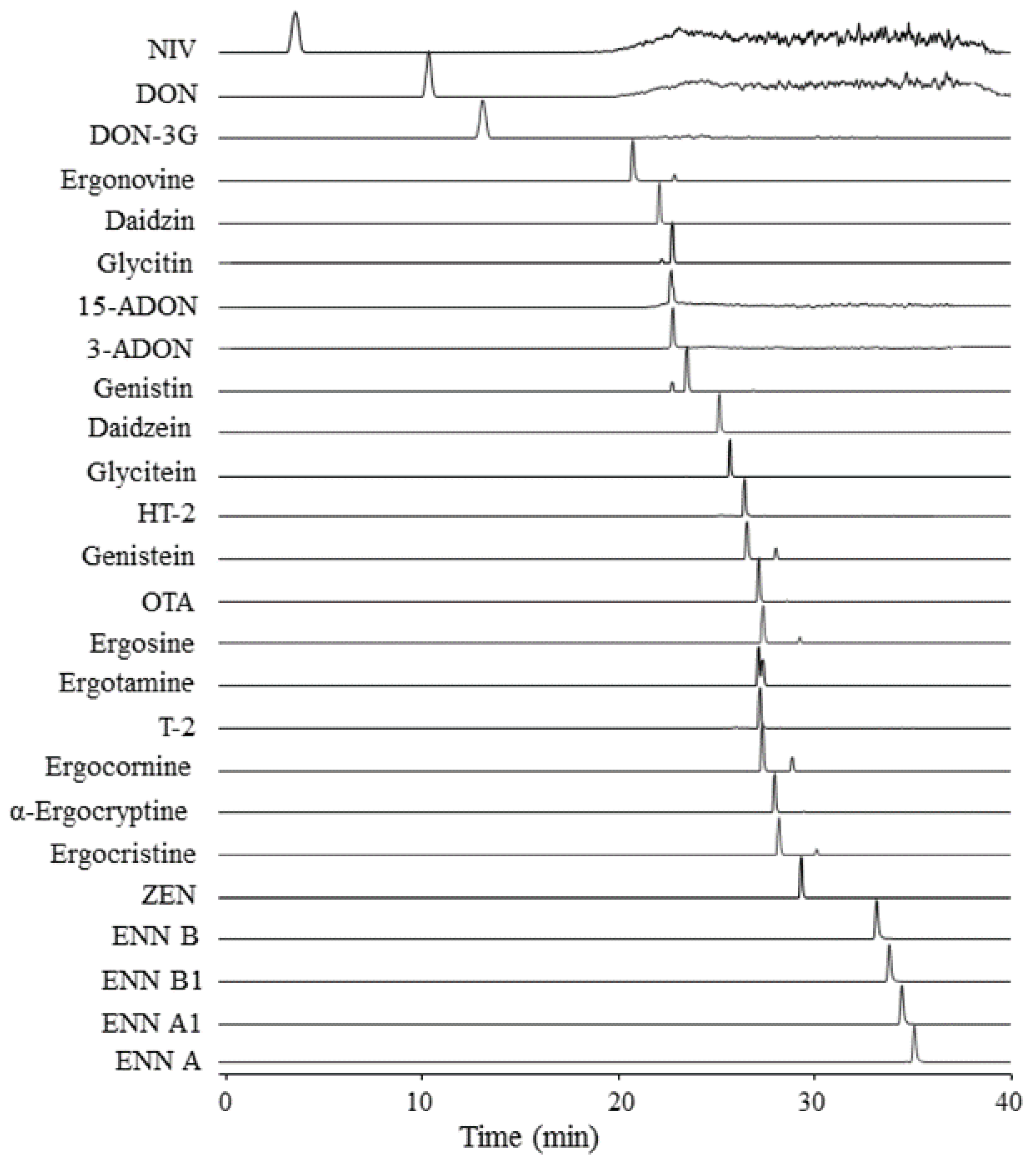{kind=link}
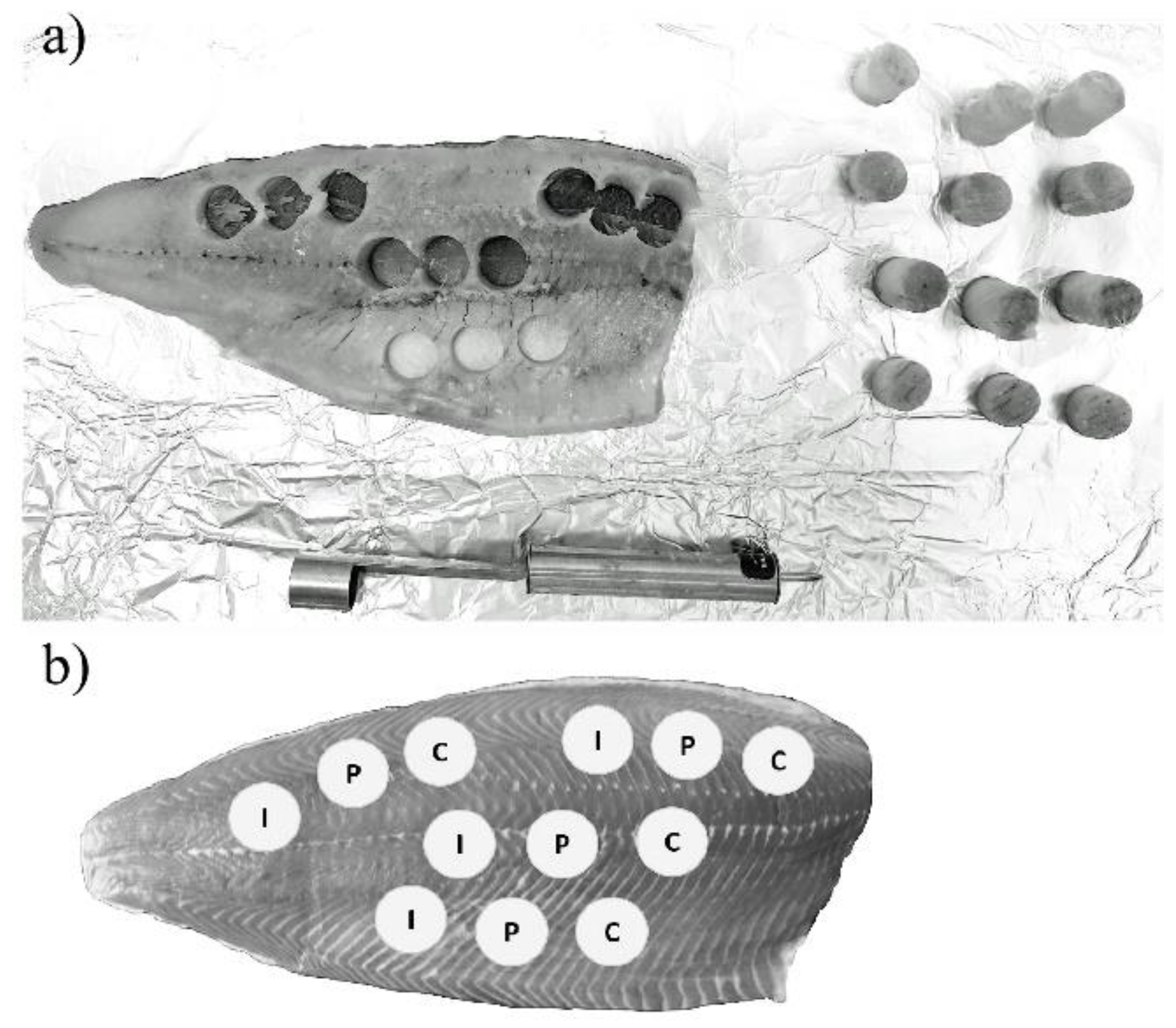{kind=link}
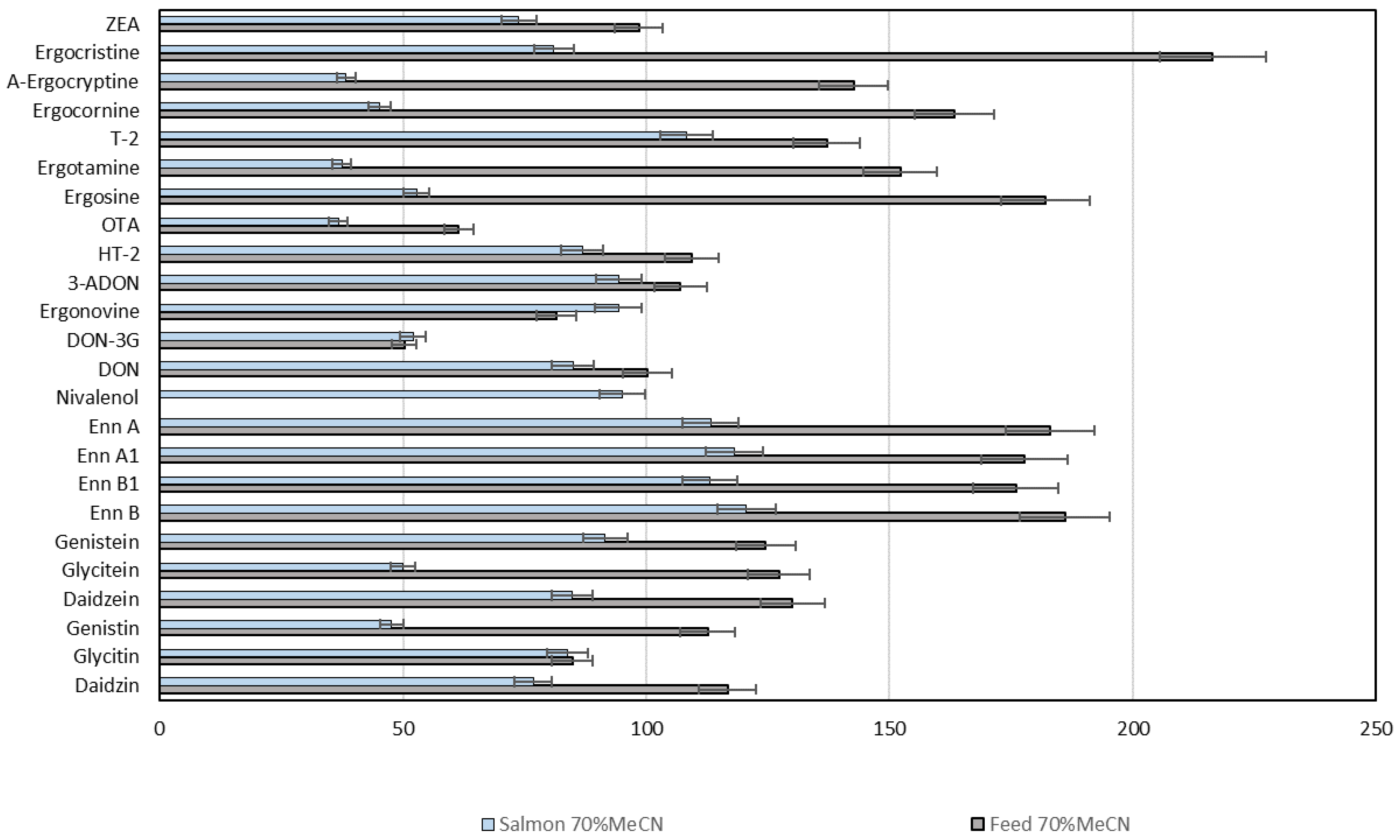{kind=link}
Abstract
Share and Cite
Johny, A.; Fæste, C.K.; Bogevik, A.S.; Berge, G.M.; Fernandes, J.M.O.; Ivanova, L. Development and Validation of a Liquid Chromatography High-Resolution Mass Spectrometry Method for the Simultaneous Determination of Mycotoxins and Phytoestrogens in Plant-Based Fish Feed and Exposed Fish. Toxins 2019, 11, 222. https://doi.org/10.3390/toxins11040222
Johny A, Fæste CK, Bogevik AS, Berge GM, Fernandes JMO, Ivanova L. Development and Validation of a Liquid Chromatography High-Resolution Mass Spectrometry Method for the Simultaneous Determination of Mycotoxins and Phytoestrogens in Plant-Based Fish Feed and Exposed Fish. Toxins. 2019; 11(4):222. https://doi.org/10.3390/toxins11040222
Chicago/Turabian StyleJohny, Amritha, Christiane Kruse Fæste, André S. Bogevik, Gerd Marit Berge, Jorge M.O. Fernandes, and Lada Ivanova. 2019. "Development and Validation of a Liquid Chromatography High-Resolution Mass Spectrometry Method for the Simultaneous Determination of Mycotoxins and Phytoestrogens in Plant-Based Fish Feed and Exposed Fish" Toxins 11, no. 4: 222. https://doi.org/10.3390/toxins11040222
APA StyleJohny, A., Fæste, C. K., Bogevik, A. S., Berge, G. M., Fernandes, J. M. O., & Ivanova, L. (2019). Development and Validation of a Liquid Chromatography High-Resolution Mass Spectrometry Method for the Simultaneous Determination of Mycotoxins and Phytoestrogens in Plant-Based Fish Feed and Exposed Fish. Toxins, 11(4), 222. https://doi.org/10.3390/toxins11040222