Repeated Oral Administration of a KDEL-Tagged Recombinant Cholera Toxin B Subunit Effectively Mitigates DSS Colitis despite a Robust Immunogenic Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
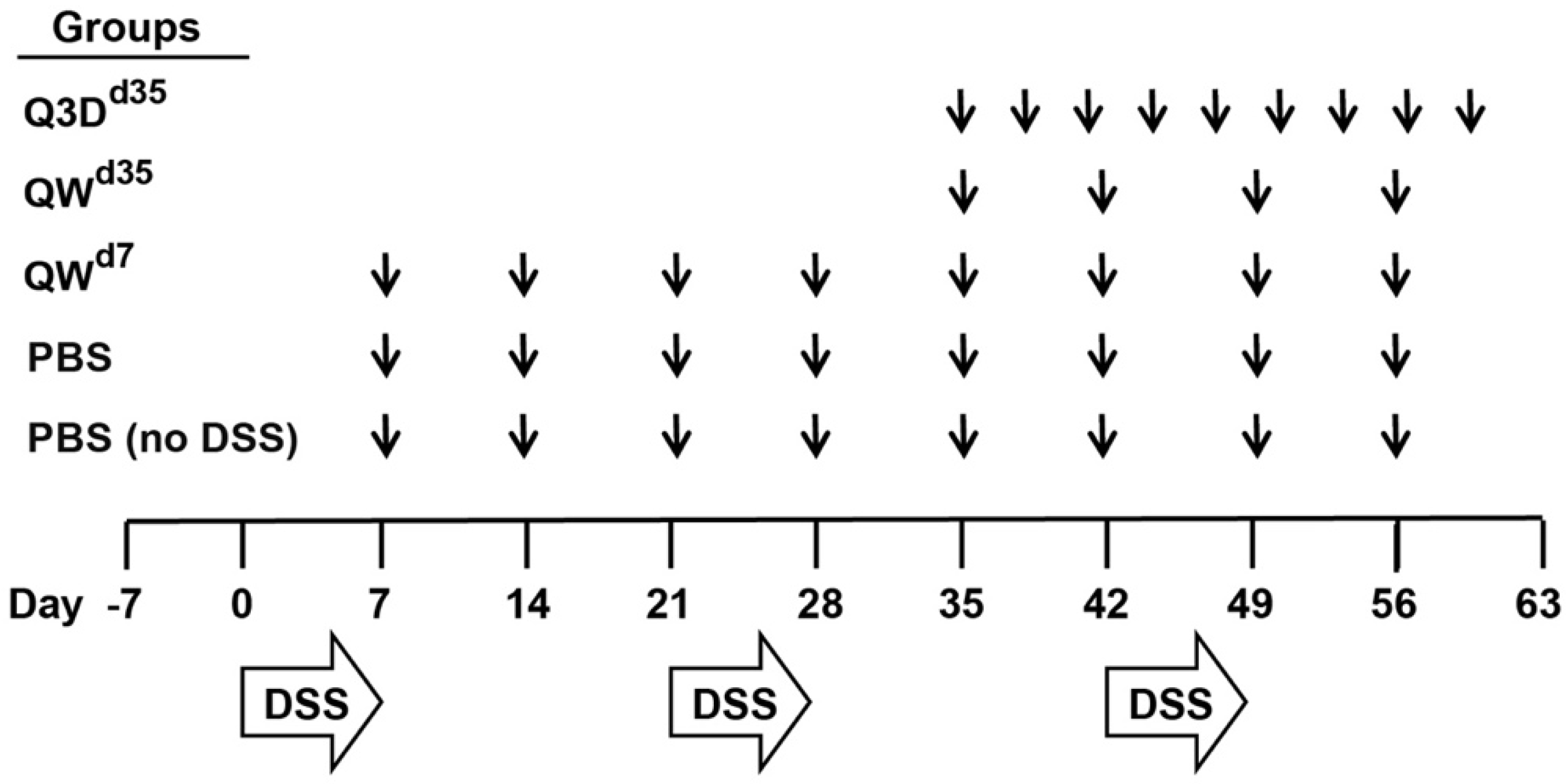
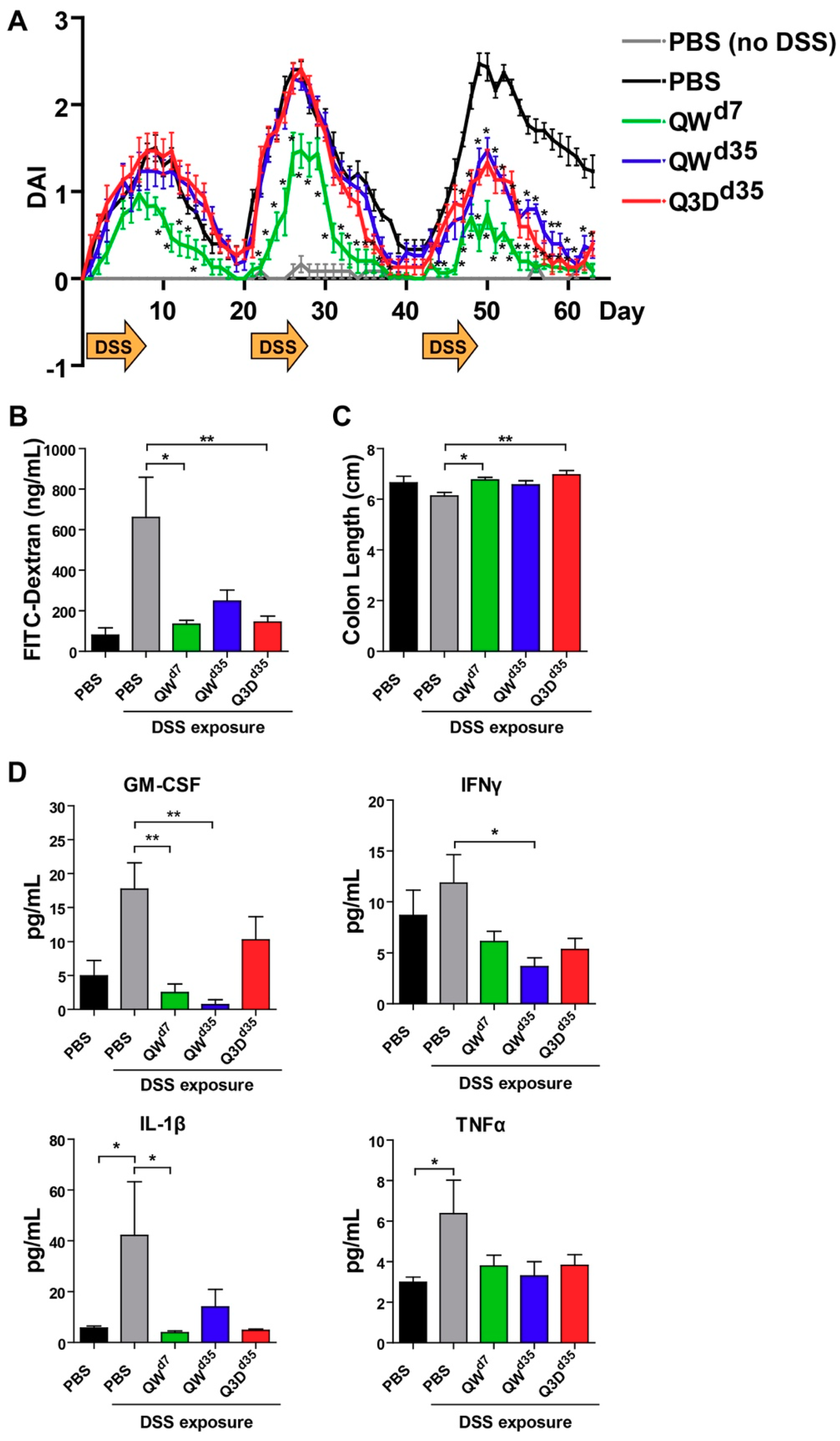
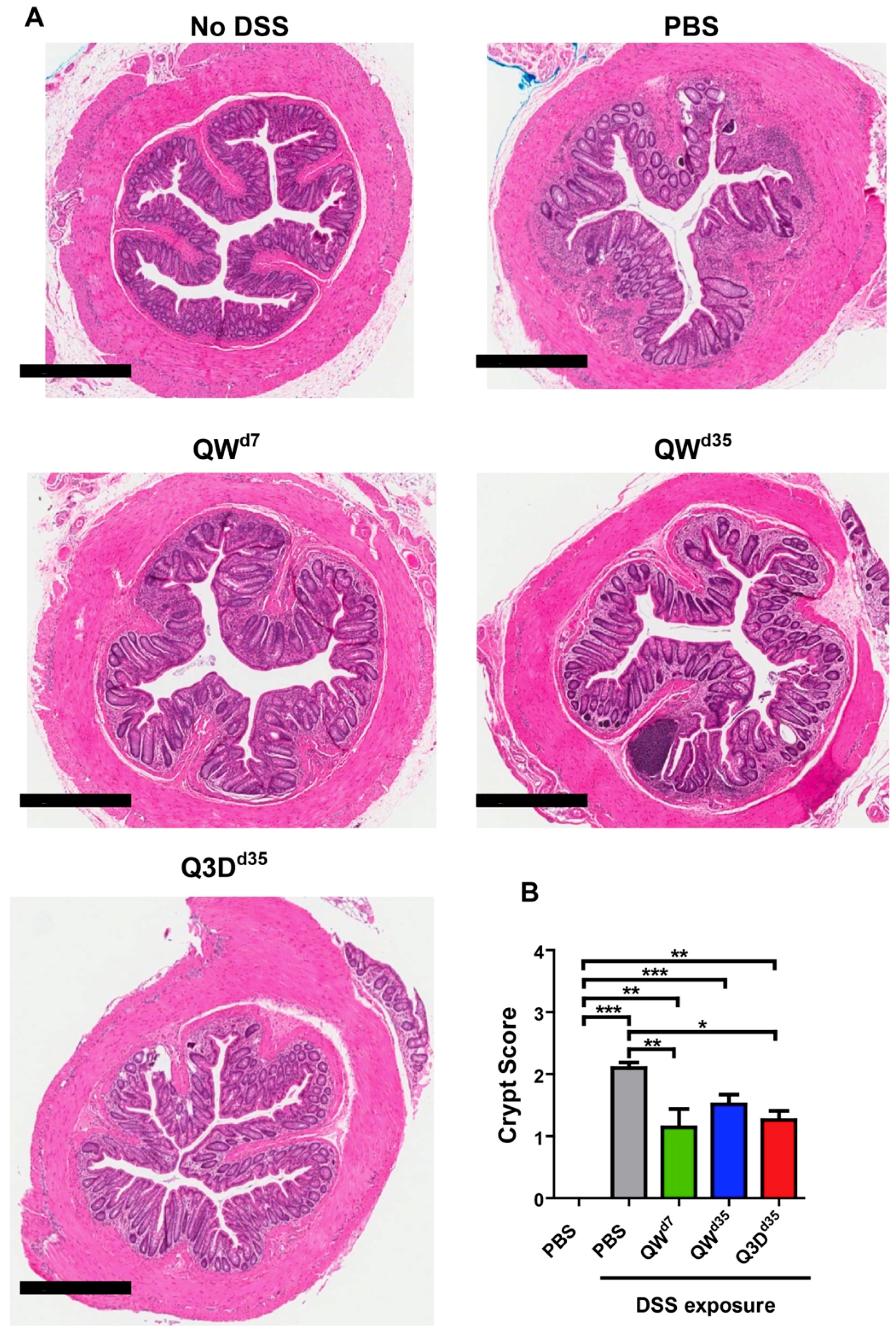
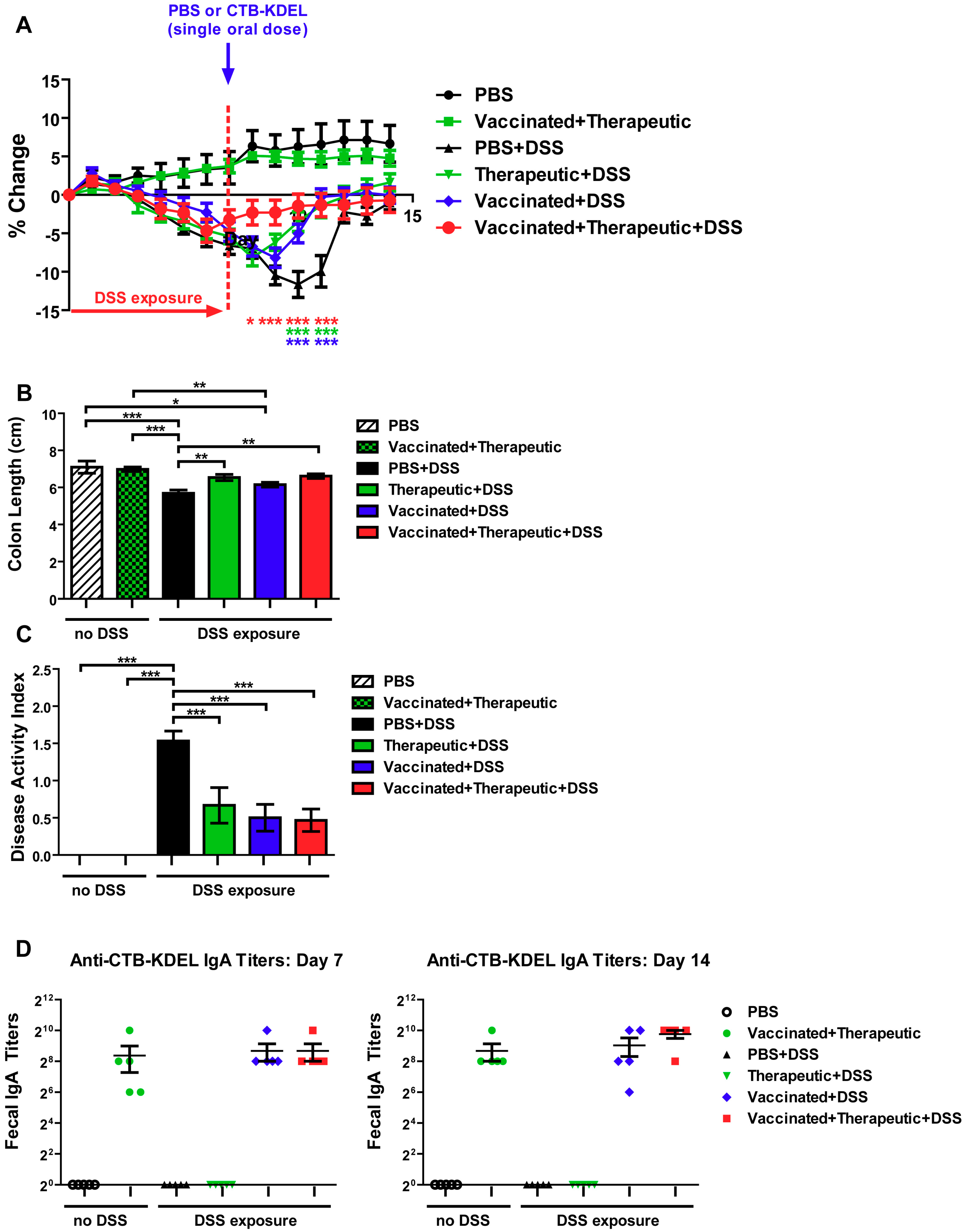
2.1. CTB-KDEL’s Therapeutic Effects in a Mouse Colitis Model Exposed to Repeated DSS Cycles
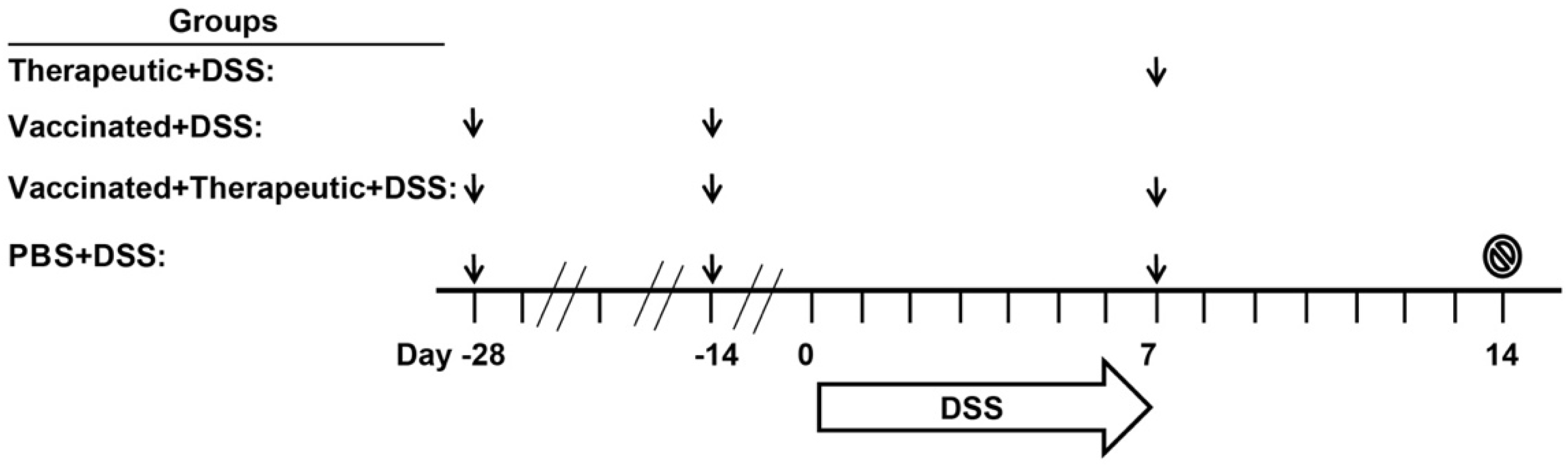
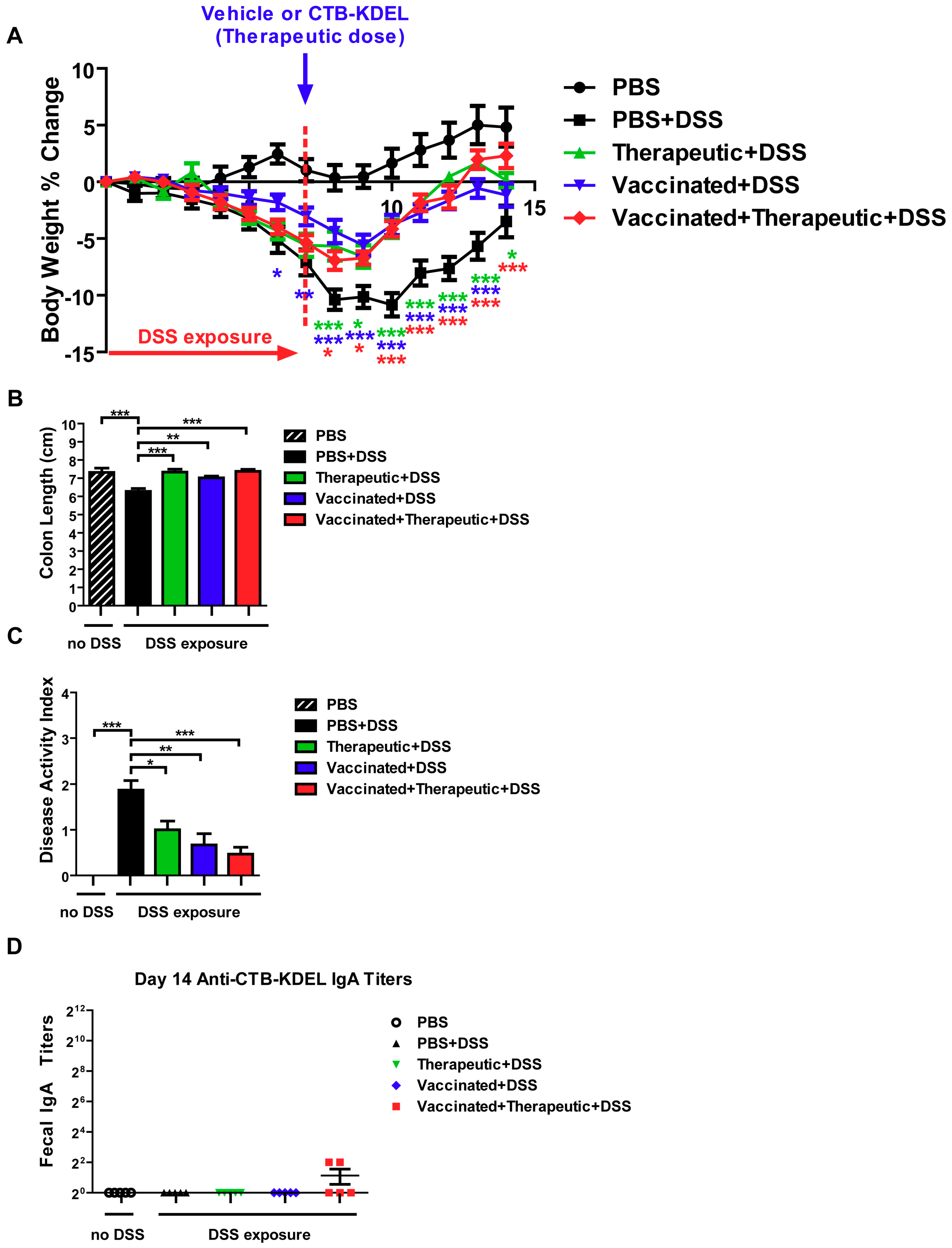
2.2. The Impact of CTB-KDEL’s Immunogenicity on Its Therapeutic Effect in Acute DSS Colitis Models
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. CTB-KDEL
5.3. Chronic DSS Colitis Model
5.4. Intestinal Permeability Analysis
5.5. Protein Isolation and Quantification
5.6. Histology
5.7. Acute DSS Colitis Models to Study the Impact of CTB-KDEL Immunogenicity
5.8. Detection of Fecal IgAs
5.9. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baldauf, K.J.; Royal, J.M.; Kouokam, J.C.; Haribabu, B.; Jala, V.R.; Yaddanapudi, K.; Hamorsky, K.T.; Dryden, G.W.; Matoba, N. Oral administration of a recombinant cholera toxin B subunit promotes mucosal healing in the colon. Mucosal Immunol. 2017, 10, 887–900. [Google Scholar] [CrossRef]
- Royal, J.M.; Oh, Y.J.; Grey, M.J.; Lencer, W.I.; Ronquillo, N.; Galandiuk, S.; Matoba, N. A modified cholera toxin B subunit containing an ER retention motif enhances colon epithelial repair via an unfolded protein response. FASEB J. 2019. [Google Scholar] [CrossRef]
- Dahlhamer, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of inflammatory bowel disease among adults aged >/=18 Years—United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1166–1169. [Google Scholar] [CrossRef]
- Neurath, M.F. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol. 2014, 7, 6–19. [Google Scholar] [CrossRef]
- Walsh, A.J.; Bryant, R.V.; Travis, S.P. Current best practice for disease activity assessment in IBD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 567–579. [Google Scholar] [CrossRef]
- Pagnini, C.; Menasci, F.; Festa, S.; Rizzatti, G.; Delle Fave, G. “Mucosal healing” in ulcerative colitis: Between clinical evidence and market suggestion. World J. Gastrointest. Pathophysiol. 2014, 5, 54–62. [Google Scholar] [CrossRef]
- Park, S.C.; Jeen, Y.T. Current and emerging biologics for ulcerative colitis. Gut Liver 2015, 9, 18–27. [Google Scholar] [CrossRef]
- Saleh, M.; Trinchieri, G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat. Rev. Immunol. 2011, 11, 9. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2005, 353, 2462–2476. [Google Scholar] [CrossRef]
- Perse, M.; Cerar, A. Dextran sodium sulphate colitis mouse model: Traps and tricks. J. Biomed. Biotechnol. 2012, 2012, 718617. [Google Scholar] [CrossRef]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Das, S.; Batra, S.K.K.; Rachagani, S. Mouse model of dextran sodium sulfate (DSS)-induced colitis. Bio Protoc. 2017, 7, e2515. [Google Scholar] [CrossRef]
- Bergquist, C.; Johansson, E.L.; Lagergard, T.; Holmgren, J.; Rudin, A. Intranasal vaccination of humans with recombinant cholera toxin B subunit induces systemic and local antibody responses in the upper respiratory tract and the vagina. Infect. Immun. 1997, 65, 2676–2684. [Google Scholar]
- Jertborn, M.; Nordstrom, I.; Kilander, A.; Czerkinsky, C.; Holmgren, J. Local and systemic immune responses to rectal administration of recombinant cholera toxin B subunit in humans. Infect. Immun. 2001, 69, 4125–4128. [Google Scholar] [CrossRef]
- Kozlowski, P.A.; Cu-Uvin, S.; Neutra, M.R.; Flanigan, T.P. Comparison of the oral, rectal, and vaginal immunization routes for induction of antibodies in rectal and genital tract secretions of women. Infect. Immun. 1997, 65, 1387–1394. [Google Scholar]
- Royal, J.M.; Matoba, N. Therapeutic potential of cholera toxin B subunit for the treatment of inflammatory diseases of the mucosa. Toxins 2017, 9, 379. [Google Scholar] [CrossRef]
- Pasetti, M.F.; Simon, J.K.; Sztein, M.B.; Levine, M.M. Immunology of gut mucosal vaccines. Immunol. Rev. 2011, 239, 125–148. [Google Scholar] [CrossRef]
- Cuatrecasas, P. Interaction of Vibrio cholerae enterotoxin with cell membranes. Biochemistry 1973, 12, 3547–3558. [Google Scholar] [CrossRef]
- Kuziemko, G.M.; Stroh, M.; Stevens, R.C. Cholera toxin binding affinity and specificity for gangliosides determined by surface plasmon resonance. Biochemistry 1996, 35, 6375–6384. [Google Scholar] [CrossRef]
- MacKenzie, C.R.; Hirama, T.; Lee, K.K.; Altman, E.; Young, N.M. Quantitative analysis of bacterial toxin affinity and specificity for glycolipid receptors by surface plasmon resonance. J. Biol. Chem. 1997, 272, 5533–5538. [Google Scholar] [CrossRef]
- Dawson, R.M. Characterization of the binding of cholera toxin to ganglioside GM1 immobilized onto microtitre plates. J. Appl. Toxicol. JAT 2005, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Hamorsky, K.T.; Kouokam, J.C.; Bennett, L.J.; Baldauf, K.J.; Kajiura, H.; Fujiyama, K.; Matoba, N. Rapid and scalable plant-based production of a cholera toxin B subunit variant to aid in mass vaccination against cholera outbreaks. PLoS Negl. Trop. Dis. 2013, 7, e2046. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Chen, X.; Vicini, P.; Rup, B.; Hickling, T.P. Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products. Cell. Immunol. 2015, 295, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Swanson, S.J.; Bussiere, J. Immunogenicity assessment in non-clinical studies. Curr. Opin. Microbiol. 2012, 15, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993, 69, 238–249. [Google Scholar] [PubMed]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Griseri, T.; McKenzie, B.S.; Schiering, C.; Powrie, F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity 2012, 37, 1116–1129. [Google Scholar] [CrossRef]
- Cong, Y.; Bowdon, H.R.; Elson, C.O. Identification of an immunodominant T cell epitope on cholera toxin. Eur. J. Immunol. 1996, 26, 2587–2594. [Google Scholar] [CrossRef]
- Takahashi, I.; Marinaro, M.; Kiyono, H.; Jackson, R.J.; Nakagawa, I.; Fujihashi, K.; Hamada, S.; Clements, J.D.; Bost, K.L.; McGhee, J.R. Mechanisms for mucosal immunogenicity and adjuvancy of Escherichia coli labile enterotoxin. J. Infect. Dis. 1996, 173, 627–635. [Google Scholar] [CrossRef][Green Version]
- DeVoss, J.; Diehl, L. Murine models of inflammatory bowel disease (IBD): Challenges of modeling human disease. Toxicol. Pathol. 2014, 42, 99–110. [Google Scholar] [CrossRef]
- Soderholm, J.D.; Streutker, C.; Yang, P.C.; Paterson, C.; Singh, P.K.; McKay, D.M.; Sherman, P.M.; Croitoru, K.; Perdue, M.H. Increased epithelial uptake of protein antigens in the ileum of Crohn’s disease mediated by tumour necrosis factor alpha. Gut 2004, 53, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Graham, W.V.; Wang, Y.; Witkowski, E.D.; Schwarz, B.T.; Turner, J.R. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 2005, 166, 409–419. [Google Scholar] [CrossRef]
- Marini, M.; Bamias, G.; Rivera-Nieves, J.; Moskaluk, C.A.; Hoang, S.B.; Ross, W.G.; Pizarro, T.T.; Cominelli, F. TNF-alpha neutralization ameliorates the severity of murine Crohn’s-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8366–8371. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Watanabe, M. Cellular and molecular mechanisms of the epithelial repair in IBD. Dig. Dis. Sci. 2005, 50, S34–S38. [Google Scholar] [CrossRef] [PubMed]
- Gulwani, H. Ulcerative Colitis. PathologyOutlines.com Website. Available online: http://www.pathologyoutlines.com/topic/colonuc.html (accessed on 6 October 2019).
- Tursi, A. Histologic healing in inflammatory bowel disease clinical practice: A reliable target? Clin. Gastroenterol. Hepatol. 2015, 13, 1211–1212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peyrin-Biroulet, L.; Bressenot, A.; Kampman, W. Histologic remission: The ultimate therapeutic goal in ulcerative colitis? Clin. Gastroenterol. Hepatol. 2014, 12, 929–934.e2. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.R.; Rodriguez, J.R. Clinical presentation of Crohn’s, ulcerative colitis, and indeterminate colitis: Symptoms, extraintestinal manifestations, and disease phenotypes. Semin. Pediatric Surg. 2017, 26, 349–355. [Google Scholar] [CrossRef]
- Schellekens, H. The immunogenicity of therapeutic proteins. Discov. Med. 2010, 9, 560–564. [Google Scholar]
- Sauna, Z.E.; Lagasse, D.; Pedras-Vasconcelos, J.; Golding, B.; Rosenberg, A.S. Evaluating and mitigating the immunogenicity of therapeutic proteins. Trends Biotechnol. 2018, 36, 1068–1084. [Google Scholar] [CrossRef]
- Brinks, V.; Jiskoot, W.; Schellekens, H. Immunogenicity of therapeutic proteins: The use of animal models. Pharm. Res. 2011, 28, 2379–2385. [Google Scholar] [CrossRef]
- Reinholdt, J.; Husby, S. IgA and Mucosal Homeostasis. In Madame Curie Bioscience Database; Landes Bioscience: Auatin, TX, USA, 2013. [Google Scholar]
- Corthesy, B. Role of secretory IgA in infection and maintenance of homeostasis. Autoimmun Rev. 2013, 12, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Gutzeit, C.; Magri, G.; Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 2014, 260, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.; Pais, B.; Favre, L.; Benyacoub, J.; Corthesy, B. Role of secretory IgA in the mucosal sensing of commensal bacteria. Gut Microbes 2014, 5, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Mkaddem, S.B.; Christou, I.; Rossato, E.; Berthelot, L.; Lehuen, A.; Monteiro, R.C. IgA, IgA receptors, and their anti-inflammatory properties. Curr. Top. Microbiol. Immunol. 2014, 382, 221–235. [Google Scholar]
- Smits, H.H.; Gloudemans, A.K.; van Nimwegen, M.; Willart, M.A.; Soullie, T.; Muskens, F.; de Jong, E.C.; Boon, L.; Pilette, C.; Johansen, F.E.; et al. Cholera toxin B suppresses allergic inflammation through induction of secretory IgA. Mucosal Immunol. 2009, 2, 331–339. [Google Scholar] [CrossRef]
- Rouquette-Jazdanian, A.K.; Foussat, A.; Lamy, L.; Pelassy, C.; Lagadec, P.; Breittmayer, J.P.; Aussel, C. Cholera toxin B-subunit prevents activation and proliferation of human CD4+ T cells by activation of a neutral sphingomyelinase in lipid rafts. J. Immunol. (Baltimore, MD, USA, 1950) 2005, 175, 5637–5648. [Google Scholar] [CrossRef]
- Coccia, E.M.; Remoli, M.E.; Di Giacinto, C.; Del Zotto, B.; Giacomini, E.; Monteleone, G.; Boirivant, M. Cholera toxin subunit B inhibits IL-12 and IFN production and signaling in experimental colitis and Crohn’s disease. Gut 2005, 54, 1558–1564. [Google Scholar] [CrossRef]
- D’Ambrosio, A.; Colucci, M.; Pugliese, O.; Quintieri, F.; Boirivant, M. Cholera toxin B subunit promotes the induction of regulatory T cells by preventing human dendritic cell maturation. J. Leukoc. Biol. 2008, 84, 661–668. [Google Scholar] [CrossRef]
- Moore, L.; Hamorsky, K.; Matoba, N. Production of recombinant cholera toxin B subunit in Nicotiana benthamiana using GENEWARE(R) tobacco mosaic virus vector. Methods Mol. Biol. 2016, 1385, 129–137. [Google Scholar]
- Gupta, J.; Nebreda, A.R. Analysis of intestinal permeability in mice. Bio Protoc. 2014, 4, e1289. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Royal, J.M.; Reeves, M.A.; Matoba, N. Repeated Oral Administration of a KDEL-Tagged Recombinant Cholera Toxin B Subunit Effectively Mitigates DSS Colitis despite a Robust Immunogenic Response. Toxins 2019, 11, 678. https://doi.org/10.3390/toxins11120678
Royal JM, Reeves MA, Matoba N. Repeated Oral Administration of a KDEL-Tagged Recombinant Cholera Toxin B Subunit Effectively Mitigates DSS Colitis despite a Robust Immunogenic Response. Toxins. 2019; 11(12):678. https://doi.org/10.3390/toxins11120678
Chicago/Turabian StyleRoyal, Joshua M., Micaela A. Reeves, and Nobuyuki Matoba. 2019. "Repeated Oral Administration of a KDEL-Tagged Recombinant Cholera Toxin B Subunit Effectively Mitigates DSS Colitis despite a Robust Immunogenic Response" Toxins 11, no. 12: 678. https://doi.org/10.3390/toxins11120678
APA StyleRoyal, J. M., Reeves, M. A., & Matoba, N. (2019). Repeated Oral Administration of a KDEL-Tagged Recombinant Cholera Toxin B Subunit Effectively Mitigates DSS Colitis despite a Robust Immunogenic Response. Toxins, 11(12), 678. https://doi.org/10.3390/toxins11120678