Toxin ζ Reduces the ATP and Modulates the Uridine Diphosphate-N-acetylglucosamine Pool

 and
and 
Abstract
1. Introduction
2. Results
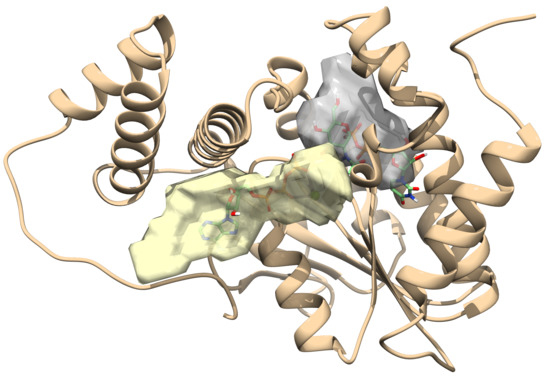
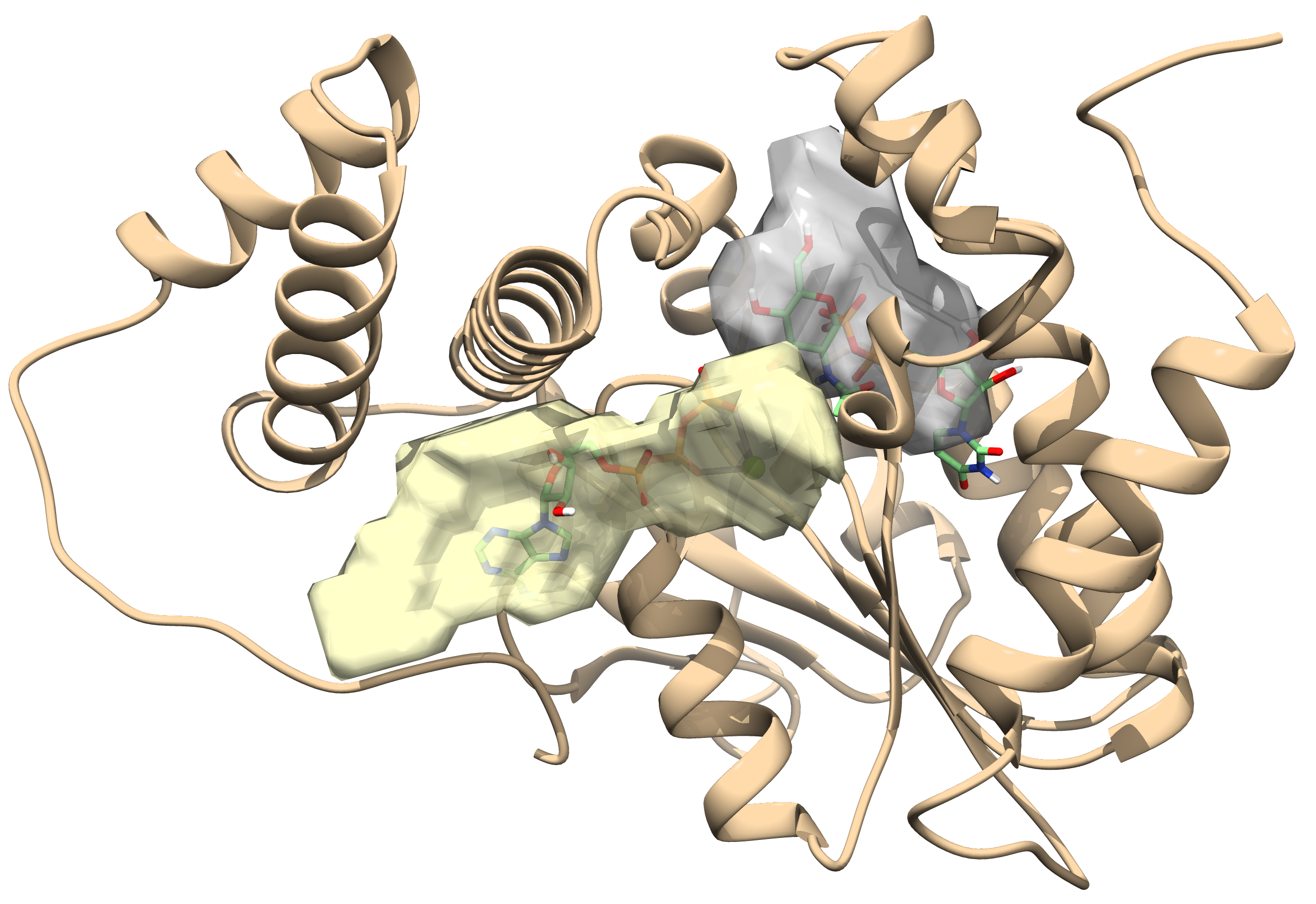
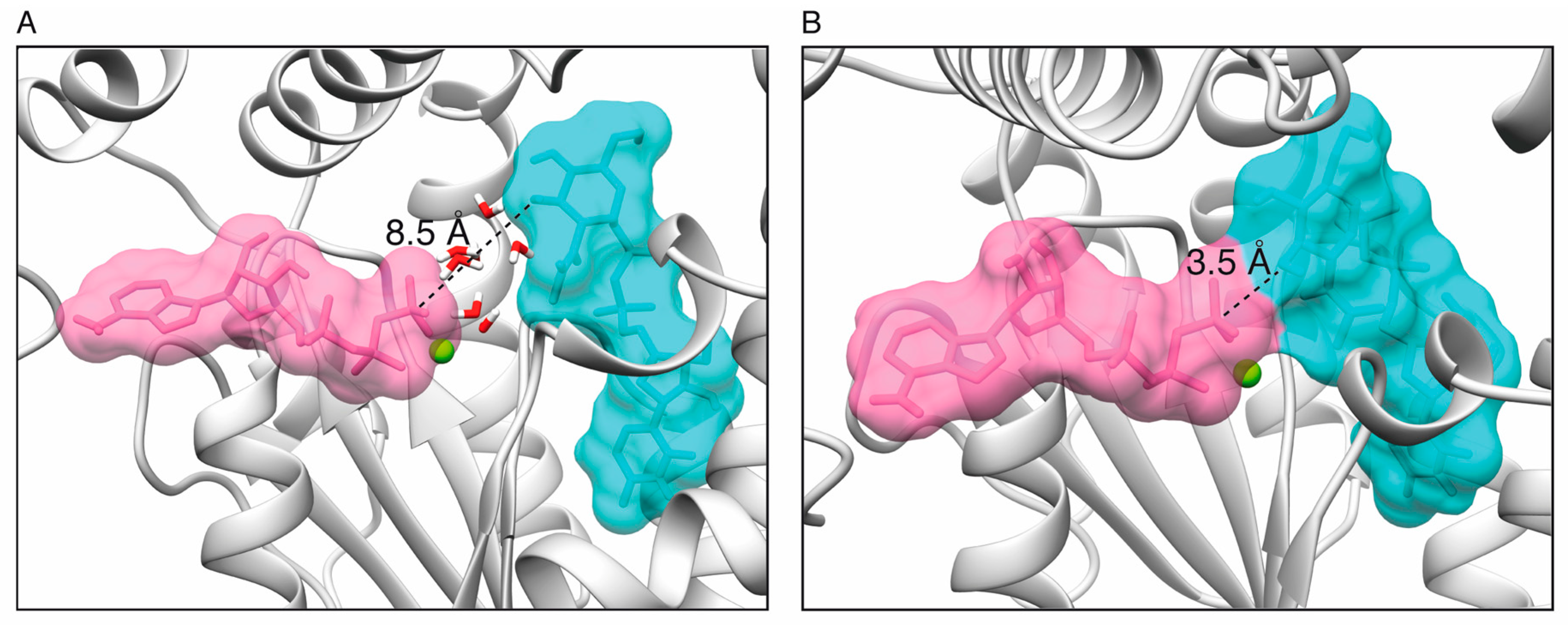
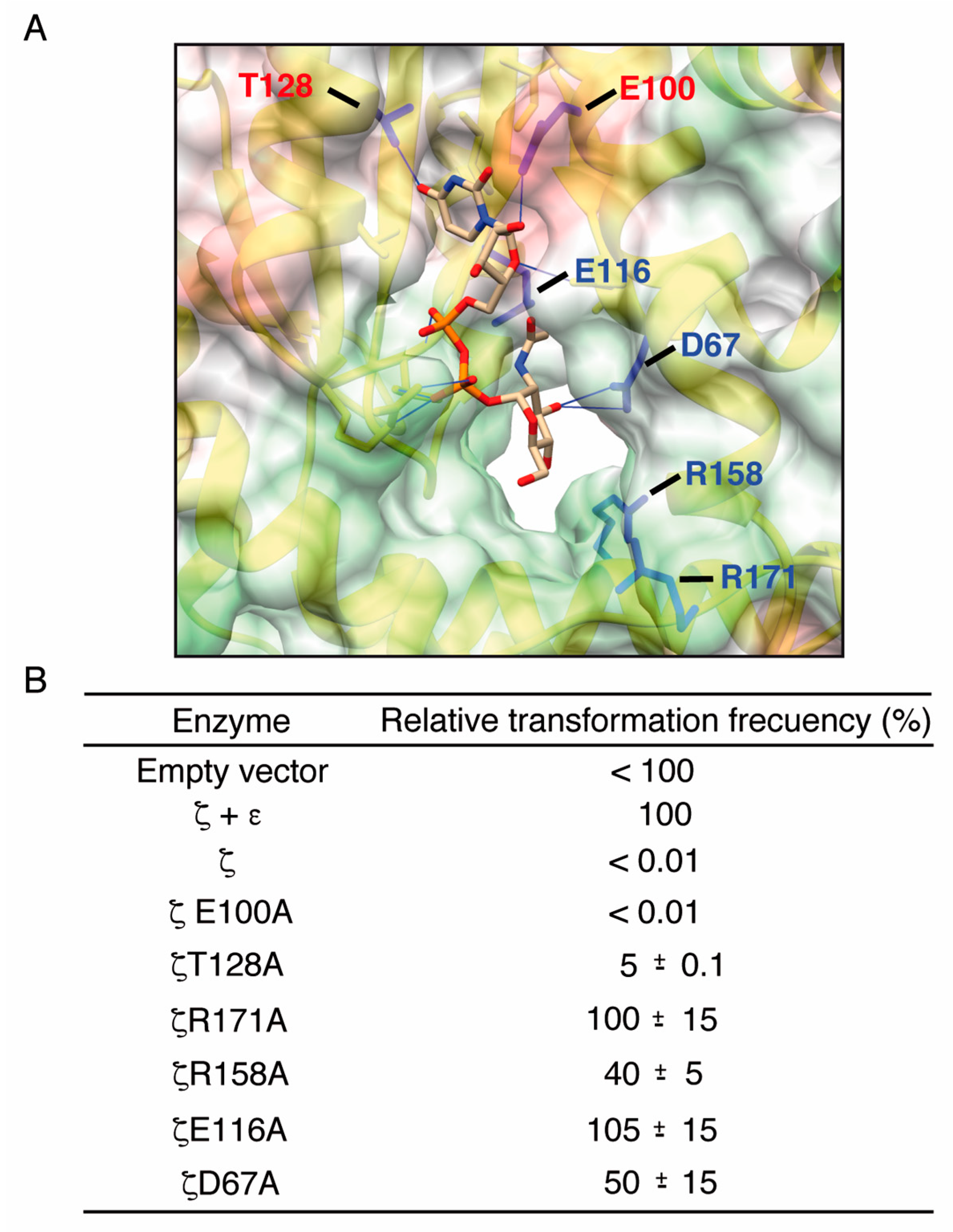
2.1. Toxin ζ Has a Large ATP Site Pocket
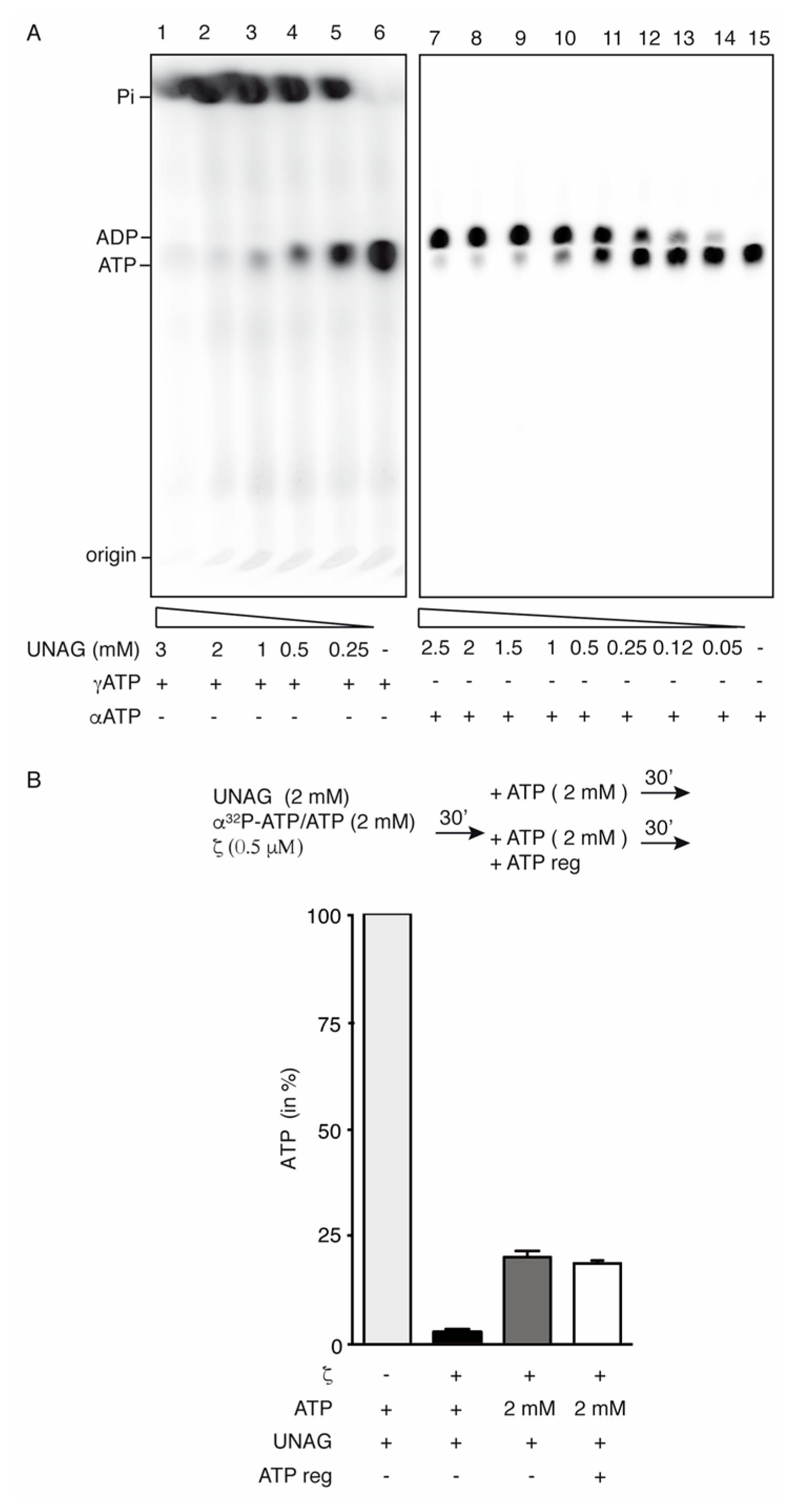
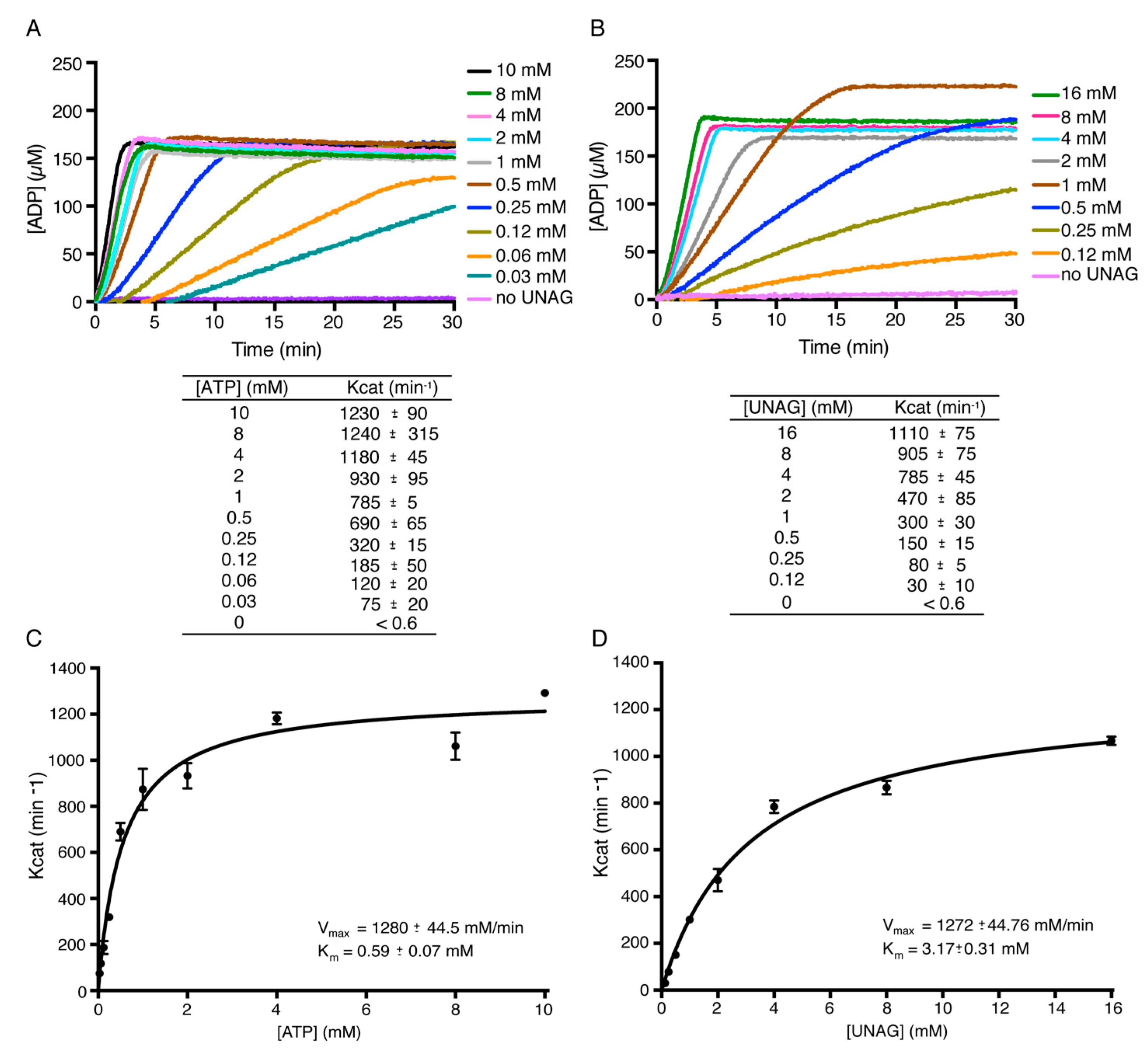
2.2. Toxin ζ Is a UNAG-Dependent ATPase
2.3. Alanine Mutagenesis of Relevant Residues
2.4. Toxin ζ Has a Low Affinity for UNAG
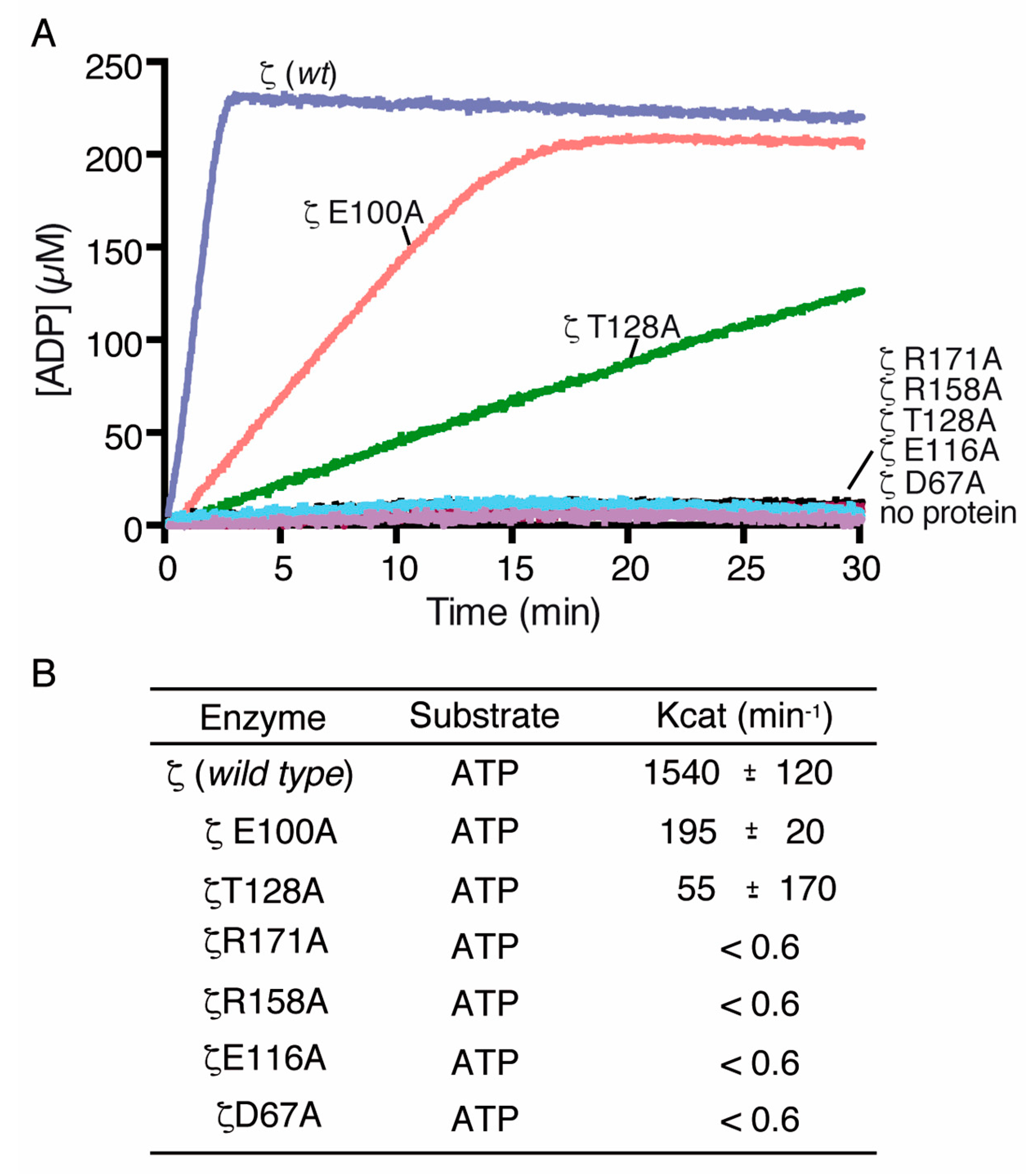
2.5. Toxin ζ Variants Have a Low In Vitro Activity
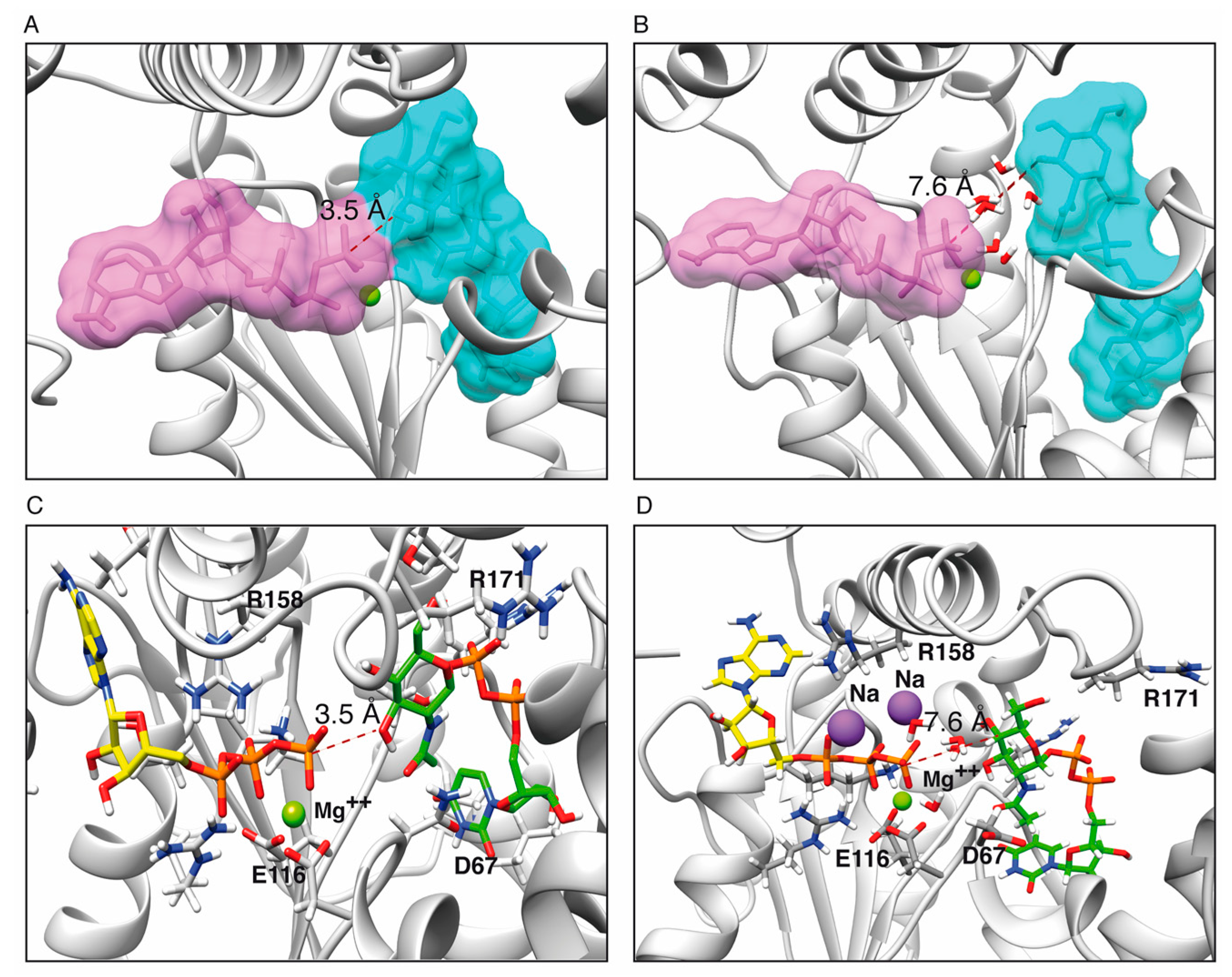
2.6. Reaction Mechanism
2.6.1. One Step Phosphotransference Reaction
2.6.2. Two Step Water-Mediated Reaction
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Plasmids
4.2. Biochemical Assays
4.3. In Silico Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.Y. Tadb 2.0: An updated database of bacterial type ii toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Saavedra De Bast, M. Bacterial toxin-antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 2011, 9, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Amato, S.M.; Orman, M.A.; Brynildsen, M.P. Metabolic control of persister formation in Escherichia coli. Mol. Cell 2013, 50, 475–487. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Gerdes, K.; Lewis, K.; McKinney, J.D. A problem of persistence: Still more questions than answers? Nat. Rev. Microbiol. 2013, 11, 587–591. [Google Scholar] [CrossRef]
- Goeders, N.; Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins 2014, 6, 304–324. [Google Scholar] [CrossRef]
- Michiels, J.E.; Van den Bergh, B.; Verstraeten, N.; Michiels, J. Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat. 2016, 29, 76–89. [Google Scholar] [CrossRef]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, targets, and triggers: An overview of toxin-antitoxin biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, F.; Fraikin, N.; Putrins, M.; Hallaert, T.; Hauryliuk, V.; Garcia-Pino, A.; Sjodin, A.; Kasvandik, S.; Udekwu, K.; Tenson, T.; et al. Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli type II persister cells. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Lioy, V.S.; Machon, C.; Tabone, M.; Gonzalez-Pastor, J.E.; Daugelavicius, R.; Ayora, S.; Alonso, J.C. The ζ toxin induces a set of protective responses and dormancy. PLoS ONE 2012, 7, e30282. [Google Scholar] [CrossRef] [PubMed]
- Tabone, M.; Lioy, V.S.; Ayora, S.; Machon, C.; Alonso, J.C. Role of toxin ζ and starvation responses in the sensitivity to antimicrobials. PLoS ONE 2014, 9, e86615. [Google Scholar] [CrossRef]
- Kim, J.S.; Wood, T.K. Persistent persister misperceptions. Front. Microbiol. 2016, 7, 2134. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.T.; Espinosa, M.; Yeo, C.C. Keeping the wolves at bay: Antitoxins of prokaryotic type II toxin-antitoxin systems. Front. Mol. Biosci. 2016, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Orejas, R.; Espinosa, M.; Yeo, C.C. The importance of the expendable: Toxin-antitoxin genes in plasmids and chromosomes. Front. Microbiol. 2017, 8, 1479. [Google Scholar] [CrossRef]
- Mutschler, H.; Meinhart, A. ε/ζ systems: Their role in resistance, virulence, and their potential for antibiotic development. J. Mol. Med. 2011, 89, 1183–1194. [Google Scholar] [CrossRef]
- Khoo, S.K.; Loll, B.; Chan, W.T.; Shoeman, R.L.; Ngoo, L.; Yeo, C.C.; Meinhart, A. Molecular and structural characterization of the PezAT chromosomal toxin-antitoxin system of the human pathogen Streptococcus pneumoniae. J. Biol. Chem. 2007, 282, 19606–19618. [Google Scholar] [CrossRef]
- Yao, X.; Chen, T.; Shen, X.; Zhao, Y.; Wang, M.; Rao, X.; Yin, S.; Wang, J.; Gong, Y.; Lu, S.; et al. The chromosomal SezAT toxin-antitoxin system promotes the maintenance of the SSPI-1 pathogenicity island in epidemic Streptococcus suis. Mol. Microbiol. 2015, 98, 243–257. [Google Scholar] [CrossRef]
- Meinhart, A.; Alings, C.; Strater, N.; Camacho, A.G.; Alonso, J.C.; Saenger, W. Crystallization and preliminary X-ray diffraction studies of the εζ addiction system encoded by Streptococcus pyogenes plasmid pSM19035. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Meinhart, A.; Alonso, J.C.; Strater, N.; Saenger, W. Crystal structure of the plasmid maintenance system ε/ζ: Functional mechanism of toxin ζ and inactivation by ε2ζ2 complex formation. Proc. Natl. Acad. Sci. USA 2003, 100, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Gebhardt, M.; Shoeman, R.L.; Meinhart, A. A novel mechanism of programmed cell death in bacteria by toxin-antitoxin systems corrupts peptidoglycan synthesis. PLoS Biol. 2011, 9, e1001033. [Google Scholar] [CrossRef] [PubMed]
- Tabone, M.; Ayora, S.; Alonso, J.C. Toxin ζ reversible induces dormancy and reduces the UDP-N-acetylglucosamine pool as one of the protective responses to cope with stress. Toxins 2014, 6, 2787–2803. [Google Scholar] [CrossRef] [PubMed]
- Moreno-del Alamo, M.; Tabone, M.; Lioy, V.S.; Alonso, J.C. Toxin ζ triggers a survival response to cope with stress and persistence. Front. Microbiol. 2017, 8, 1130. [Google Scholar] [CrossRef] [PubMed]
- Eschenburg, S.; Priestman, M.A.; Abdul-Latif, F.A.; Delachaume, C.; Fassy, F.; Schonbrunn, E. A novel inhibitor that suspends the induced fit mechanism of UDP-N-acetylglucosamine enolpyruvyl transferase (MurA). J. Biol. Chem. 2005, 280, 14070–14075. [Google Scholar] [CrossRef] [PubMed]
- Lioy, V.S.; Martin, M.T.; Camacho, A.G.; Lurz, R.; Antelmann, H.; Hecker, M.; Hitchin, E.; Ridge, Y.; Wells, J.M.; Alonso, J.C. pSM19035-encoded ζ toxin induces stasis followed by death in a subpopulation of cells. Microbiology 2006, 152, 2365–2379. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Voss, N.R.; Gerstein, M. 3v: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555–W562. [Google Scholar] [CrossRef]
- Izard, T.; Ellis, J. The crystal structures of Chloramphenicol Phosphotransferase reveal a novel inactivation mechanism. EMBO J. 2000, 19, 2690–2700. [Google Scholar] [CrossRef]
- Sherrer, R.L.; Araiso, Y.; Aldag, C.; Ishitani, R.; Ho, J.M.; Soll, D.; Nureki, O. C-terminal domain of archaeal O-phosphoseryl-tRNA kinase displays large-scale motion to bind the 7-bp D-stem of archaeal tRNA(sec). Nucleic Acids Res. 2011, 39, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Das, U.; Smith, P.; Shuman, S. Structure and mechanism of the polynucleotide kinase component of the bacterial PnkP-Hen1 RNA repair system. RNA 2012, 18, 2277–2286. [Google Scholar] [CrossRef]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera- a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Allouche, A.R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. Application of the PM6 method to modeling proteins. J. Mol. Model. 2009, 15, 765–805. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. Dock 6: Combining techniques to model rna-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. Comput. Aided Mol. Des. 2009, 16, 11–26. [Google Scholar] [CrossRef]
- Neudert, G.; Klebe, G. Dsx: A knowledge-based scoring function for the assessment of protein-ligand complexes. J. Chem. Inf. Model. 2011, 51, 2731–2745. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Zhou, C.; Wu, S.; Liu, Y.; Triplett, L.; Miao, J.; Tokuhisa, J.; Deblais, L.; Robinson, H.; Leach, J.E.; et al. Crystal structure of Xanthomonas AvrRxo1-Orf1, a type iii effector with a polynucleotide kinase domain, and its interactor AvrRxo1-Orf2. Structure 2015, 23, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Prokop, M.; Adam, J.; Kriz, Z.; Wimmerova, M.; Koca, J. TRITON: A graphical tool for ligand-binding protein engineering. Bioinformatics 2008, 24, 1955–1956. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein structure modeling with modeller. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [PubMed]
- Bittner, A.N.; Kriel, A.; Wang, J.D. Lowering GTP level increases survival of amino acid starvation but slows growth rate for Bacillus subtilis cells lacking (p)ppGpp. J. Bacteriol. 2014, 196, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- López, J.M.; Marks, C.L.; Freese, E. The decrease of guanine nucleotides initiates sporulation of Bacillus subtilis. Biochim. Biophys. Acta 1979, 587, 238–252. [Google Scholar] [CrossRef]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; Van Dien, S.J.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef]
- Namboori, S.C.; Graham, D.E. Enzymatic analysis of uridine diphosphate N-acetyl-d-glucosamine. Anal. Biochem. 2008, 381, 94–100. [Google Scholar] [CrossRef]
- Rocker, A.; Peschke, M.; Kittila, T.; Sakson, R.; Brieke, C.; Meinhart, A. The ng_ζ1 toxin of the gonococcal ε/ζ toxin/antitoxin system drains precursors for cell wall synthesis. Nat. Commun. 2018, 9, 1686. [Google Scholar] [CrossRef]
- Bachega, J.F.; Timmers, L.F.; Assirati, L.; Bachega, L.R.; Field, M.J.; Wymore, T. GTKDynamo: A PyMOL plug-in for QC/MM hybrid potential simulations. J. Comput. Chem. 2013, 34, 2190–2196. [Google Scholar] [CrossRef]
- Field, M.J. The pDynamo program for molecular simulations using hybrid quantum chemical and molecular mechanical potentials. J. Chem. Theory Comput. 2008, 4, 1151–1161. [Google Scholar] [CrossRef]
- Wang, C.; Huang, W.; Liao, J.L. QM/MM investigation of ATP hydrolysis in aqueous solution. J. Phys. Chem. B 2015, 119, 3720–3726. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.P.; Kaila, V.R.I.; Sundholm, D. Ab initio, density functional theory, and semi-empirical calculations. In Biomocular Simulations; Luca Monticelli, L., Salonen, E., Eds.; Springer Science + Business Media: New York, NY, USA, 2003; Volume 924, pp. 3–27. [Google Scholar]
- Yilmazer, N.D.; Korth, M. Comparison of molecular mechanics, semi-empirical quantum mechanical, and density functional theory methods for scoring protein-ligand interactions. J. Phys. Chem. B 2013, 117, 8075–8084. [Google Scholar] [CrossRef] [PubMed]
- Rayne, S.; Forest, K. Comment on “comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCSs) and perfluorocarboxylates (PFCS) emitted from direct sources”. Environ. Sci. Technol. 2009, 43, 7155–7156. [Google Scholar] [CrossRef] [PubMed]
- Frison, G.; Ohanessian, G. A comparative study of semiempirical, ab initio, and DFT methods in evaluating metal-ligand bond strength, proton affinity, and interactions between first and second shell ligands in Zn-biomimetic complexes. J. Comput. Chem. 2008, 29, 416–433. [Google Scholar] [CrossRef]
- Warshel, A.; Parson, W.W. Computer simulations of electron-transfer reactions in solution and in photosynthetic reaction centers. Annu. Rev. Phys. Chem. 1991, 42, 279–309. [Google Scholar] [CrossRef]
- Allen, K.N.; Dunaway-Mariano, D. Phosphoryl group transfer: Evolution of a catalytic scaffold. Trends Biochem. Sci. 2004, 29, 495–503. [Google Scholar] [CrossRef]
- Harrison, C.B.; Schulten, K. Quantum and classical dynamics simulations of ATP hydrolysis in solution. J. Chem. Theory Comput. 2012, 8, 2328–2335. [Google Scholar] [CrossRef]
- Akola, J.; Jones, R.O. ATP hydrolysis in water—A density functional study. J. Phys. Chem. B 2003, 107, 11774–11783. [Google Scholar] [CrossRef]
- Volante, A.; Carrasco, B.; Tabone, M.; Alonso, J.C. The interaction of ω2 with the RNA polymerase β’ subunit functions as an activation to repression switch. Nucleic Acids Res. 2015, 43, 9249–9261. [Google Scholar] [CrossRef]
- De La Cruz, E.M.; Sweeney, H.L.; Ostap, E.M. ADP inhibition of myosin V ATPase activity. Biophys. J. 2000, 79, 1524–1529. [Google Scholar] [CrossRef]
- Yadav, T.; Carrasco, B.; Myers, A.R.; George, N.P.; Keck, J.L.; Alonso, J.C. Genetic recombination in Bacillus subtilis: A division of labor between two single-strand DNA-binding proteins. Nucleic Acids Res. 2012, 40, 5546–5559. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.; Nguyen, C.; Salomon-Ferrer, R.; Walker, R.C.; Gilson, M.K.; Kurtzman, T. Solvation thermodynamic mapping of molecular surfaces in ambertools: Gist. J. Comput. Chem. 2016, 37, 2029–2037. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE–antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 2002, 12, 14–20. [Google Scholar] [CrossRef]
- Herraez, A. Biomolecules in the computer: Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006, 34, 255–261. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | ATP | ADP | UNAG | UNAG-3P |
|---|---|---|---|---|
| H-bonds | 4 | 1 | 5 | 8 |
| Contacts | 12 | 7 | 14 | 16 |
| Atom contacts | 92 | 35 | 60 | 82 |
| DSX | −110.835 | −61.036 | −99.592 | −119.225 |
| Xscore | −6.73 | 4.57 | −7.36 | −7.81 |
| Condition | wt | K46A | D67A | E100A | E116A | T128A | R158A | R171A |
|---|---|---|---|---|---|---|---|---|
| H-bonds | 9 | 1 | 2 | 4 | 3 | 3 | 2 | 4 |
| Contacts | 13 | 10 | 11 | 13 | 9 | 8 | 8 | 12 |
| Atom | 39 | 41 | 50 | 46 | 39 | 32 | 46 | 53 |
| Xscore | −7.36 | −6.77 | −6.87 | −7.09 | −8.81 | −7.00 | −7.17 | −7.23 |
| DSX | −99.592 | −74.601 | −85.754 | −77.066 | −79.611 | −85.631 | −83.521 | −84.271 |
| ATP-Pγ Near to UNAG-O3’ | ΔG‡ (Kcal/mol) | ΔG° (Kcal/mol) |
| ATP + Mg2+ + UNAG (H2O) | 13.81 | 33.63 |
| ζ + ATP + Mg2+ + UNAG (H2O) | 18.28 | 10.76 |
| ATP + Mg2+ + UNAG (NaCl) | 7.28 | −17.84 |
| ζ + ATP + Mg2+ + UNAG (NaCl) | 2.98 | −4.62 |
| ATP-Pγ Far from UNAG-O3’ | ΔG‡ (Kcal/mol) | ΔG° (Kcal/mol) |
| ATP + Mg2+ + UNAG (water) | 5.45 | −0.61 |
| ATP + Mg2+ + UNAG + H2O (water) | 23.98 | 16.05 |
| ζ + ATP + Mg2+ + UNAG (water, tot) | 79.35 | 14.5 |
| ζ + ATP + Mg2+ + UNAG (water, react) | 16.56 | −48.29 |
| ATP + Mg2+ + UNAG (NaCl) | 161.57 | 95.3 |
| ATP + Mg2+ + UNAG + H2O (NaCl) | 23.16 | 9.77 |
| ζ + ATP + Mg2+ + UNAG (NaCl) | 38.64 | 8.57 |
| ATP-Pγ Far from UNAG-O3’ (two steps) | ΔG‡ (Kcal/mol) | ΔG° (Kcal/mol) |
| ζ + ATP + Mg2+ + H2O1059 + UNAG (water, step 1) | 9.61 | 0.68 |
| ζ + ADP + Mg2+ + Pi1059 + UNAG (water, step 2) | 25.2 | −9 |
| ζ + ATP + Mg2+ + H2O2259 + UNAG (water, step 1) | 36.14 | 2.29 |
| ζ + ADP + Mg2+ + Pi2259 + UNAG (water, step 2) | 24.32 | 49.23 |
| ζ + ATP + Mg2+ + H2O2273 + UNAG (water, step 1) | 8.81 | −11.05 |
| ζ + ADP + Mg2+ + Pi2273 + UNAG (water, step 2) | 33.03 | 45.31 |
| ζ + ATP + Mg2+ + H2O3646 + UNAG (water, step 1) | 7.35 | −14.44 |
| ζ + ADP + Mg2+ + Pi3646 + UNAG (water, step 2) | 44.52 | −22.06 |
| ζ + ATP + Mg2+ + H2O2204 + UNAG (NaCl, step 1) | 2 | −16.86 |
| ζ + ADP + Mg2+ + Pi2204 + UNAG (NaCl, step 2) | 54.53 | 52.21 |
| ζ + ATP + Mg2+ + H2O2637 + UNAG (NaCl, step 1) | 4 | −9.12 |
| ζ + ADP + Mg2+ + Pi2637 + UNAG (NaCl, step 2) | 46.12 | 47.54 |
| ζ + ATP + Mg2+ + H2O4589 + UNAG (NaCl, step 1) | 2.77 | 18.74 |
| ζ + ADP + Mg2+ + Pi4589 + UNAG (NaCl, step 2) | 47.22 | 39.47 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-del Álamo, M.; Tabone, M.; Muñoz-Martínez, J.; Valverde, J.R.; Alonso, J.C. Toxin ζ Reduces the ATP and Modulates the Uridine Diphosphate-N-acetylglucosamine Pool. Toxins 2019, 11, 29. https://doi.org/10.3390/toxins11010029
Moreno-del Álamo M, Tabone M, Muñoz-Martínez J, Valverde JR, Alonso JC. Toxin ζ Reduces the ATP and Modulates the Uridine Diphosphate-N-acetylglucosamine Pool. Toxins. 2019; 11(1):29. https://doi.org/10.3390/toxins11010029
Chicago/Turabian StyleMoreno-del Álamo, María, Mariangela Tabone, Juan Muñoz-Martínez, José R. Valverde, and Juan C. Alonso. 2019. "Toxin ζ Reduces the ATP and Modulates the Uridine Diphosphate-N-acetylglucosamine Pool" Toxins 11, no. 1: 29. https://doi.org/10.3390/toxins11010029
APA StyleMoreno-del Álamo, M., Tabone, M., Muñoz-Martínez, J., Valverde, J. R., & Alonso, J. C. (2019). Toxin ζ Reduces the ATP and Modulates the Uridine Diphosphate-N-acetylglucosamine Pool. Toxins, 11(1), 29. https://doi.org/10.3390/toxins11010029