Attenuating the Biologic Drive for Weight Regain Following Weight Loss: Must What Goes Down Always Go Back Up?


{kind=link}
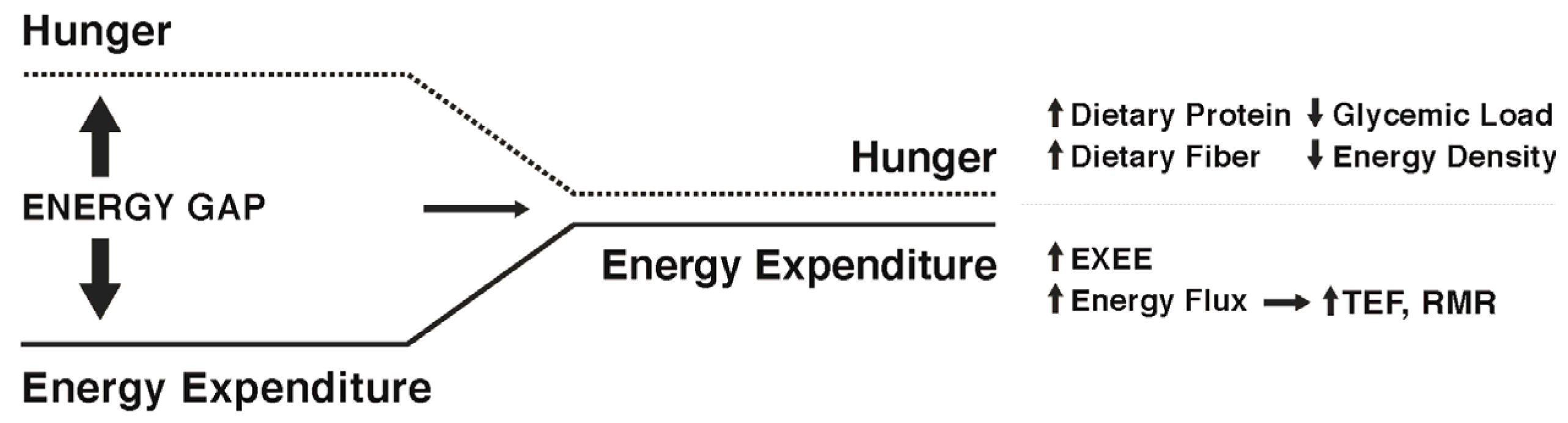{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Dynamic Energy Balance
3. Why Is Weight Loss So Difficult to Maintain?
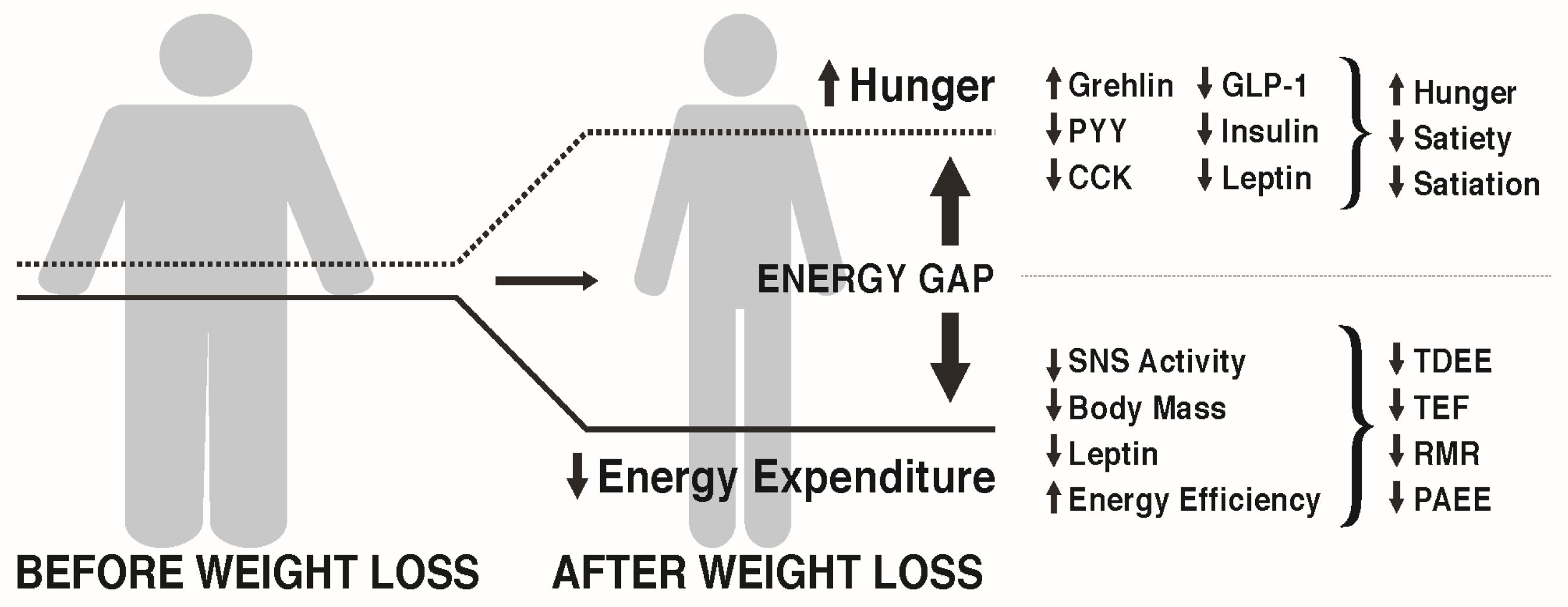
3.1. The Energy Gap Concept
3.2. How Does Weight Loss Affect Hunger and Satiety?
3.3. How Does Weight Loss Affect Energy Expenditure?
4. Can the Weight Loss-Induced Energy Gap Be Attenuated by Lifestyle Factors to Enhance Weight Maintenance?
5. Approaches to Attenuate the Increased Hunger Following Weight Loss
Diet Composition
6. Approaches to Attenuate the Decline in Energy Expenditure Following Weight Loss
6.1. Resting Metabolic Rate
6.2. Thermic Effect of Food
6.3. Non-Exercise Activity Thermogenesis
6.4. Exercise Energy Expenditure
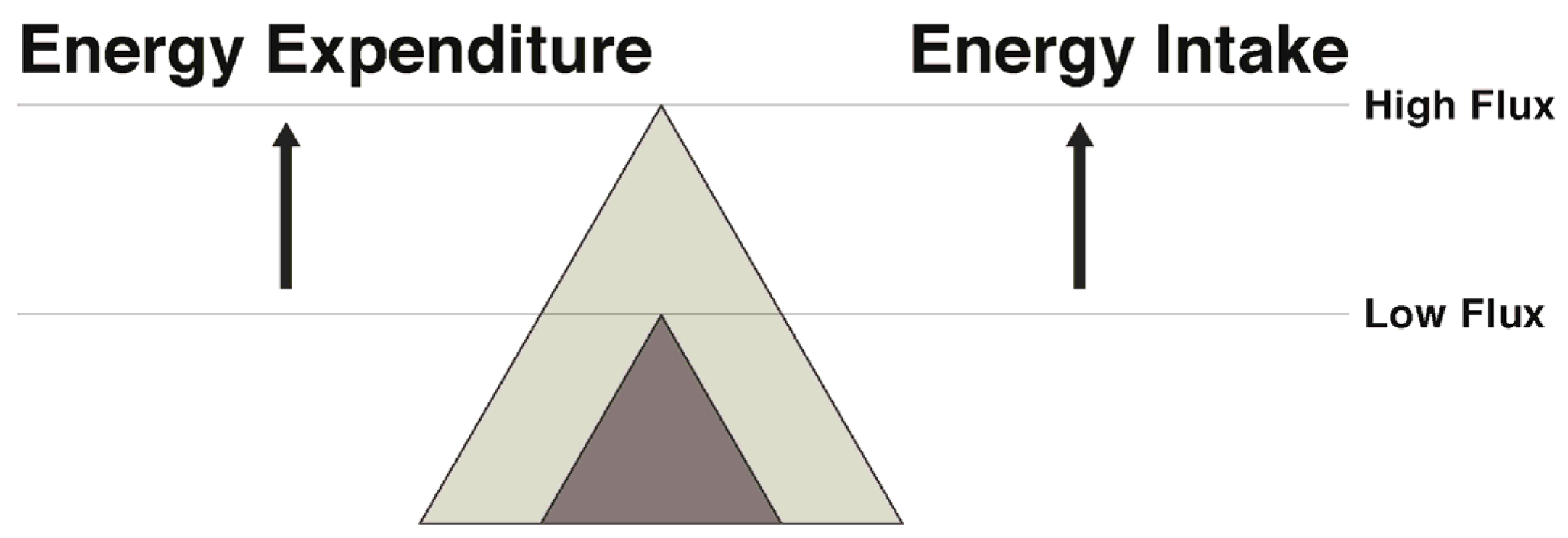
7. Narrowing the Energy Gap by Increased Energy Flux: Effects on Both Appetite and Energy Expenditure
8. Summary and Conclusions
Author Contributions
Conflicts of Interest
References
- American Medical Association House of Delegates. Obesity as a Disease; American Medical Association: Chicago, IL, USA, 2013. [Google Scholar]
- Abumrad, N.A.; Klein, S. Update on the pathophysiology of obesity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Barnett, A.C.; Bruce, C.R.; Schenk, S.; Horowitz, J.F.; Hoy, A.J. Regulation of plasma ceramide levels with fatty acid oversupply: Evidence that the liver detects and secretes de novo synthesised ceramide. Diabetologia 2012, 55, 2741–2746. [Google Scholar] [CrossRef] [PubMed]
- Petelin, A.; Bizjak, M.; Cernelic-Bizjak, M.; Jurdana, M.; Jakus, T.; Jenko-Praznikar, Z. Low-grade inflammation in overweight and obese adults is affected by weight loss program. J. Endocrinol. Investig. 2014, 37, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Wing, R.R.; Phelan, S. Long-term weight loss maintenance. Am. J. Clin. Nutr. 2005, 82, 222S–225S. [Google Scholar] [PubMed]
- Hall, K.D. Predicting metabolic adaptation, body weight change, and energy intake in humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E449–E466. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.D. Modeling metabolic adaptations and energy regulation in humans. Annu. Rev. Nutr. 2012, 32, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.J.; Enderle, J.; Bosy-Westphal, A. Changes in Energy Expenditure with Weight Gain and Weight Loss in Humans. Curr. Obes. Rep. 2016, 5, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Tremblay, A.; Despres, J.P.; Nadeau, A.; Lupien, P.J.; Theriault, G.; Dussault, J.; Moorjani, S.; Pinault, S.; Fournier, G. The response to long-term overfeeding in identical twins. N. Engl. J. Med. 1990, 322, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Tremblay, A.; Despres, J.P.; Theriault, G.; Nadeau, A.; Lupien, P.J.; Moorjani, S.; Prudhomme, D.; Fournier, G. The response to exercise with constant energy intake in identical twins. Obes. Res. 1994, 2, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.A.; Eberhardt, N.L.; Jensen, M.D. Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science 1999, 283, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Leibel, R.L. Adaptive thermogenesis in humans. Int. J. Obes. 2010, 34, S47–S55. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.; Perusse, L.; Bouchard, C. Energy balance and body-weight stability: Impact of gene-environment interactions. Br. J. Nutr. 2004, 92, S63–S66. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, S.A.; Nabben, M.; Bijland, S.; Voshol, P.J.; van Klinken, J.B.; Havekes, L.M.; Romijn, J.A.; Hoeks, J.; Hesselink, M.K.; Schrauwen, P.; et al. High levels of whole-body energy expenditure are associated with a lower coupling of skeletal muscle mitochondria in C57Bl/6 mice. Metab. Clin. Exp. 2010, 59, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, S.A.; van Marken Lichtenbelt, W.; van Dijk Willems, K.; Schrauwen, P. Skeletal muscle mitochondrial uncoupling, adaptive thermogenesis and energy expenditure. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Speliotes, E.K.; Loos, R.J.; Li, S.; Lindgren, C.M.; Heid, I.M.; Berndt, S.I.; Elliott, A.L.; Jackson, A.U.; Lamina, C.; et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009, 41, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Lango, A.H.; Lindgren, C.M.; Luan, J.; Magi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, J.H.; Luan, J.; Luben, R.N.; Rodwell, S.A.; Khaw, K.T.; Ong, K.K.; Wareham, N.J.; Loos, R.J. Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. Am. J. Clin. Nutr. 2010, 91, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Maccani, J.Z.; Hawley, N.L.; Wing, R.R.; Kelsey, K.T.; McCaffery, J.M. Epigenetic patterns in successful weight loss maintainers: A pilot study. Int. J. Obes. 2015, 39, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C. Genetics and genomics of obesity: Current status. Prog. Mol. Biol. Transl. Sci. 2010, 94, 1–8. [Google Scholar] [PubMed]
- Bray, M.S.; Loos, R.J.; McCaffery, J.M.; Ling, C.; Franks, P.W.; Weinstock, G.M.; Snyder, M.P.; Vassy, J.L.; Agurs-Collins, T.; Conference Working Group. NIH working group report-using genomic information to guide weight management: From universal to precision treatment. Obesity 2016, 24, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.J.; Janssens, A.C. Predicting Polygenic Obesity Using Genetic Information. Cell Metab. 2017, 25, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, E.; Guo, J.; Howard, L.; Kerns, J.C.; Knuth, N.D.; Brychta, R.; Chen, K.Y.; Skarulis, M.C.; Walter, M.; Walter, P.J.; et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity 2016, 24, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Keesey, R.E.; Powley, T.L. Body energy homeostasis. Appetite 2008, 51, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Maclean, P.S.; Bergouignan, A.; Cornier, M.A.; Jackman, M.R. Biology’s response to dieting: The impetus for weight regain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R581–R600. [Google Scholar] [CrossRef] [PubMed]
- MacLean, P.S.; Higgins, J.A.; Giles, E.D.; Sherk, V.D.; Jackman, M.R. The role for adipose tissue in weight regain after weight loss. Obes. Rev. 2015, 16, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sumithran, P.; Proietto, J. The defence of body weight: A physiological basis for weight regain after weight loss. Clin. Sci. 2013, 124, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Kissileff, H.R.; Mayer, L.E.; Hirsch, J.; Leibel, R.L. Energy intake in weight-reduced humans. Brain Res. 2010, 1350, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Niswender, K.D.; Baskin, D.G.; Schwartz, M.W. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol. Metab. TEM 2004, 15, 362–369. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Niswender, K.D. Adiposity signaling and biological defense against weight gain: Absence of protection or central hormone resistance? J. Clin. Endocrinol. Metab. 2004, 89, 5889–5897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J. Leptin and the Regulation of Food Intake and Body Weight. J. Nutr. Sci. Vitaminol. 2015, 61, S202. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. Thrifty genes for obesity, an attractive but flawed idea, and an alternative perspective: The ‘drifty gene’ hypothesis. Int. J. Obes. 2008, 32, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. Evolutionary perspectives on the obesity epidemic: Adaptive, maladaptive, and neutral viewpoints. Annu. Rev. Nutr. 2013, 33, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. If body fatness is under physiological regulation, then how come we have an obesity epidemic? Physiology 2014, 29, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Kissileff, H.R.; Thornton, J.C.; Torres, M.I.; Pavlovich, K.; Mayer, L.S.; Kalari, V.; Leibel, R.L.; Rosenbaum, M. Leptin reverses declines in satiation in weight-reduced obese humans. Am. J. Clin. Nutr. 2012, 95, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Sumithran, P.; Prendergast, L.A.; Delbridge, E.; Purcell, K.; Shulkes, A.; Kriketos, A.; Proietto, J. Long-term persistence of hormonal adaptations to weight loss. N. Engl. J. Med. 2011, 365, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Naslund, E.; Andersson, I.; Degerblad, M.; Kogner, P.; Kral, J.G.; Rossner, S.; Hellstrom, P.M. Associations of leptin, insulin resistance and thyroid function with long-term weight loss in dieting obese men. J. Inter. Med. 2000, 248, 299–308. [Google Scholar] [CrossRef]
- Moran, L.J.; Noakes, M.; Clifton, P.M.; Wittert, G.A.; Le Roux, C.W.; Ghatei, M.A.; Bloom, S.R.; Norman, R.J. Postprandial ghrelin, cholecystokinin, peptide YY, and appetite before and after weight loss in overweight women with and without polycystic ovary syndrome. Am. J. Clin. Nutr. 2007, 86, 1603–1610. [Google Scholar] [PubMed]
- Bloom, S. Hormonal regulation of appetite. Obes. Rev. 2007, 8, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S. Control of appetite by gut hormones. Regul. Pept. 2010, 164, 22. [Google Scholar] [CrossRef]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Camina, J.P. Cell biology of the ghrelin receptor. J. Neuroendocrinol. 2006, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, C.W.; Patterson, M.; Vincent, R.P.; Hunt, C.; Ghatei, M.A.; Bloom, S.R. Postprandial plasma ghrelin is suppressed proportional to meal calorie content in normal-weight but not obese subjects. J. Clin. Endocrinol. Metab. 2005, 90, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- English, P.J.; Ghatei, M.A.; Malik, I.A.; Bloom, S.R.; Wilding, J.P. Food fails to suppress ghrelin levels in obese humans. J. Clin. Endocrinol. Metab. 2002, 87, 2984. [Google Scholar] [CrossRef] [PubMed]
- Huda, M.S.; Dovey, T.; Wong, S.P.; English, P.J.; Halford, J.; McCulloch, P.; Cleator, J.; Martin, B.; Cashen, J.; Hayden, K.; et al. Ghrelin restores ‘lean-type’ hunger and energy expenditure profiles in morbidly obese subjects but has no effect on postgastrectomy subjects. Int. J. Obes. 2009, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gueugnon, C.; Mougin, F.; Nguyen, N.U.; Bouhaddi, M.; Nicolet-Guenat, M.; Dumoulin, G. Ghrelin and PYY levels in adolescents with severe obesity: Effects of weight loss induced by long-term exercise training and modified food habits. Eur. J. Appl. Physiol. 2012, 112, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E.; Weigle, D.S.; Frayo, R.S.; Breen, P.A.; Ma, M.K.; Dellinger, E.P.; Purnell, J.Q. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N. Engl. J. Med. 2002, 346, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Cornier, M.A.; Grunwald, G.K.; Johnson, S.L.; Bessesen, D.H. Effects of short-term overfeeding on hunger, satiety, and energy intake in thin and reduced-obese individuals. Appetite 2004, 43, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.E.; Finlayson, G.; Gibbons, C.; Caudwell, P.; Hopkins, M. The biology of appetite control: Do resting metabolic rate and fat-free mass drive energy intake? Physiol. Behav. 2015, 152, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.; Finlayson, G.; Duarte, C.; Whybrow, S.; Ritz, P.; Horgan, G.W.; Blundell, J.E.; Stubbs, R.J. Modelling the associations between fat-free mass, resting metabolic rate and energy intake in the context of total energy balance. Int. J. Obes. 2016, 40, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Dulloo, A.G. Collateral fattening: When a deficit in lean body mass drives overeating. Obesity 2017, 25, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Dulloo, A.G.; Jacquet, J.; Miles-Chan, J.L.; Schutz, Y. Passive and active roles of fat-free mass in the control of energy intake and body composition regulation. Eur. J. Clin. Nutr. 2017, 71, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Chaput, J.P.; Drapeau, V.; Hetherington, M.; Lemieux, S.; Provencher, V.; Tremblay, A. Psychobiological effects observed in obese men experiencing body weight loss plateau. Depression Anxiety 2007, 24, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Gilhooly, C.H.; Das, S.K.; Golden, J.K.; McCrory, M.A.; Dallal, G.E.; Saltzman, E.; Kramer, F.M.; Roberts, S.B. Food cravings and energy regulation: The characteristics of craved foods and their relationship with eating behaviors and weight change during 6 months of dietary energy restriction. Int. J. Obes. 2007, 31, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Leibel, R.L.; Hirsch, J. Diminished energy requirements in reduced-obese patients. Metab. Clin. Exp. 1984, 33, 164–170. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Hirsch, J.; Gallagher, D.A.; Leibel, R.L. Long-term persistence of adaptive thermogenesis in subjects who have maintained a reduced body weight. Am. J. Clin. Nutr. 2008, 88, 906–912. [Google Scholar] [PubMed]
- Van Baak, M.A. Meal-induced activation of the sympathetic nervous system and its cardiovascular and thermogenic effects in man. Physiol. Behav. 2008, 94, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Melby, C.L.; Schmidt, W.D.; Corrigan, D. Resting metabolic rate in weight-cycling collegiate wrestlers compared with physically active, noncycling control subjects. Am. J. Clin. Nutr. 1990, 52, 409–414. [Google Scholar] [PubMed]
- Knuth, N.D.; Johannsen, D.L.; Tamboli, R.A.; Marks-Shulman, P.A.; Huizenga, R.; Chen, K.Y.; Abumrad, N.N.; Ravussin, E.; Hall, K.D. Metabolic adaptation following massive weight loss is related to the degree of energy imbalance and changes in circulating leptin. Obesity 2014, 22, 2563–2569. [Google Scholar] [CrossRef] [PubMed]
- Camps, S.G.; Verhoef, S.P.; Westerterp, K.R. Weight loss, weight maintenance, and adaptive thermogenesis. Am. J. Clin. Nutr. 2013, 97, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Melby, C.L.; Sylliaasen, S.; Rhodes, T. Diet-Induced Weight-Loss and Metabolic Changes in Obese Women with High versus Low Prior Weight-Loss Regain. Nutr. Res. 1991, 11, 971–978. [Google Scholar] [CrossRef]
- Leibel, R.L.; Rosenbaum, M.; Hirsch, J. Changes in energy expenditure resulting from altered body weight. N. Engl. J. Med. 1995, 332, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.D.; Corrigan, D.; Melby, C.L. Two seasons of weight cycling does not lower resting metabolic rate in college wrestlers. Med. Sci. Sports Exerc. 1993, 25, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Vandenborne, K.; Goldsmith, R.; Simoneau, J.A.; Heymsfield, S.; Joanisse, D.R.; Hirsch, J.; Murphy, E.; Matthews, D.; Segal, K.R.; et al. Effects of experimental weight perturbation on skeletal muscle work efficiency in human subjects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R183–R192. [Google Scholar] [CrossRef] [PubMed]
- Camps, S.G.; Verhoef, S.P.; Westerterp, K.R. Leptin and energy restriction induced adaptation in energy expenditure. Metab. Clin. Exp. 2015, 64, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Hames, K.C.; Coen, P.M.; King, W.C.; Anthony, S.J.; Stefanovic-Racic, M.; Toledo, F.G.; Lowery, J.B.; Helbling, N.L.; Dube, J.J.; DeLany, J.P.; et al. Resting and exercise energy metabolism in weight-reduced adults with severe obesity. Obesity 2016, 24, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Nicolson, M.; Hirsch, J.; Murphy, E.; Chu, F.; Leibel, R.L. Effects of weight change on plasma leptin concentrations and energy expenditure. J. Clin. Endocrinol. Metab. 1997, 82, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Strohacker, K.; McCaffery, J.M.; MacLean, P.S.; Wing, R.R. Adaptations of leptin, ghrelin or insulin during weight loss as predictors of weight regain: A review of current literature. Int. J. Obes. 2014, 38, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Papandonatos, G.D.; Pan, Q.; Pajewski, N.M.; Delahanty, L.M.; Peter, I.; Erar, B.; Ahmad, S.; Harden, M.; Chen, L.; Fontanillas, P.; et al. Genetic Predisposition to Weight Loss and Regain With Lifestyle Intervention: Analyses From the Diabetes Prevention Program and the Look AHEAD Randomized Controlled Trials. Diabetes 2015, 64, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Gorin, A.A.; Phelan, S.; Wing, R.R.; Hill, J.O. Promoting long-term weight control: Does dieting consistency matter? Int. J. Obes. Relat. Metab. Disord. 2004, 28, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Phelan, S.; Liu, T.; Gorin, A.; Lowe, M.; Hogan, J.; Fava, J.; Wing, R.R. What distinguishes weight-loss maintainers from the treatment-seeking obese? Analysis of environmental, behavioral, and psychosocial variables in diverse populations. Ann. Behav. Med. 2009, 38, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Phelan, S.; Wing, R.R.; Raynor, H.A.; Dibello, J.; Nedeau, K.; Peng, W. Holiday weight management by successful weight losers and normal weight individuals. J. Consul. Clin. Psychol. 2008, 76, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Phelan, S.; Wyatt, H.; Nassery, S.; Dibello, J.; Fava, J.L.; Hill, J.O.; Wing, R.R. Three-year weight change in successful weight losers who lost weight on a low-carbohydrate diet. Obesity 2007, 15, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Phelan, S.; Wyatt, H.R.; Hill, J.O.; Wing, R.R. Are the eating and exercise habits of successful weight losers changing? Obesity 2006, 14, 710–716. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.T.; Wing, R.R.; Klem, M.L.; Lang, W.; Hill, J.O. What predicts weight regain in a group of successful weight losers? J. Consul. Clin. Psychol. 1999, 67, 177–185. [Google Scholar] [CrossRef]
- Thomas, J.G.; Bond, D.S.; Phelan, S.; Hill, J.O.; Wing, R.R. Weight-loss maintenance for 10 years in the National Weight Control Registry. Am. J. Prev. Med. 2014, 46, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, C.A.; Fappa, E.; Karfopoulou, E.; Gkza, A.; Yannakoulia, M. Weight loss maintenance in relation to locus of control: The MedWeight study. Behav. Res. Ther. 2015, 71, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Brikou, D.; Zannidi, D.; Karfopoulou, E.; Anastasiou, C.A.; Yannakoulia, M. Breakfast consumption and weight-loss maintenance: Results from the MedWeight study. Br. J. Nutr. 2016, 115, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Karfopoulou, E.; Anastasiou, C.A.; Avgeraki, E.; Kosmidis, M.H.; Yannakoulia, M. The role of social support in weight loss maintenance: Results from the MedWeight study. J. Behav. Med. 2016, 39, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Karfopoulou, E.; Brikou, D.; Mamalaki, E.; Bersimis, F.; Anastasiou, C.A.; Hill, J.O.; Yannakoulia, M. Dietary patterns in weight loss maintenance: Results from the MedWeight study. Eur. J. Nutr. 2016, 56, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Karfopoulou, E.; Mouliou, K.; Koutras, Y.; Yannakoulia, M. Behaviours associated with weight loss maintenance and regaining in a Mediterranean population sample. A qualitative study. Clin. Obes. 2013, 3, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.J.; Kroff, J.; Clamp, L.D.; Lambert, E.V. Compensations for Weight Loss in Successful and Unsuccessful Dieters. Am. J. Health Behav. 2015, 39, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Chaix, A.; Panda, S. Daily Eating Patterns and Their Impact on Health and Disease. Trends Endocrinol. Metab. TEM 2016, 27, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Chaix, A.; Yooseph, S.; Panda, S. Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 2014, 20, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Davy, B.M.; Dennis, E.A.; Dengo, A.L.; Wilson, K.L.; Davy, K.P. Water consumption reduces energy intake at a breakfast meal in obese older adults. J. Am. Diet. Assoc. 2008, 108, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Flack, K.D.; Davy, B.M. Beverage consumption and adult weight management: A review. Eat. Behav. 2009, 10, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Flood, J.E.; Rolls, B.J. Soup preloads in a variety of forms reduce meal energy intake. Appetite 2007, 49, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Leidy, H.J.; Bossingham, M.J.; Mattes, R.D.; Campbell, W.W. Increased dietary protein consumed at breakfast leads to an initial and sustained feeling of fullness during energy restriction compared to other meal times. Br. J. Nutr. 2009, 101, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Leidy, H.J.; Mattes, R.D.; Campbell, W.W. Effects of acute and chronic protein intake on metabolism, appetite, and ghrelin during weight loss. Obesity 2007, 15, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Leidy, H.J.; Tang, M.; Armstrong, C.L.; Martin, C.B.; Campbell, W.W. The effects of consuming frequent, higher protein meals on appetite and satiety during weight loss in overweight/obese men. Obesity 2011, 19, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Westerterp-Plantenga, M.S.; Lejeune, M.P.; Nijs, I.; van Ooijen, M.; Kovacs, E.M. High protein intake sustains weight maintenance after body weight loss in humans. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, M.P.; Kovacs, E.M.; Westerterp-Plantenga, M.S. Additional protein intake limits weight regain after weight loss in humans. Br. J. Nutr. 2005, 93, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Aller, E.E.; Larsen, T.M.; Claus, H.; Lindroos, A.K.; Kafatos, A.; Pfeiffer, A.; Martinez, J.A.; Handjieva-Darlenska, T.; Kunesova, M.; Stender, S.; et al. Weight loss maintenance in overweight subjects on ad libitum diets with high or low protein content and glycemic index: the DIOGENES trial 12-month results. Int. J. Obes. 2014, 38, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Leidy, H.J.; Clifton, P.M.; Astrup, A.; Wycherley, T.P.; Westerterp-Plantenga, M.S.; Luscombe-Marsh, N.D.; Woods, S.C.; Mattes, R.D. The role of protein in weight loss and maintenance. Am. J. Clini. Nutr. 2015, 101, 1320S–1329S. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, M.P.; Westerterp, K.R.; Adam, T.C.; Luscombe-Marsh, N.D.; Westerterp-Plantenga, M.S. Ghrelin and glucagon-like peptide 1 concentrations, 24-h satiety, and energy and substrate metabolism during a high-protein diet and measured in a respiration chamber. Am. J. Clin. Nutr. 2006, 83, 89–94. [Google Scholar] [PubMed]
- Batterham, R.L.; Heffron, H.; Kapoor, S.; Chivers, J.E.; Chandarana, K.; Herzog, H.; Le Roux, C.W.; Thomas, E.L.; Bell, J.D.; Withers, D.J. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. 2006, 4, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Blatt, A.D.; Williams, R.A.; Roe, L.S.; Rolls, B.J. Effects of energy content and energy density of pre-portioned entrees on energy intake. Obesity 2012, 20, 2010–2018. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.D.; Wyatt, H.R.; Hill, J.O.; McGuckin, B.G.; Brill, C.; Mohammed, B.S.; Szapary, P.O.; Rader, D.J.; Edman, J.S.; Klein, S. A randomized trial of a low-carbohydrate diet for obesity. N. Engl. J. Med. 2003, 348, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Samaha, F.F.; Iqbal, N.; Seshadri, P.; Chicano, K.L.; Daily, D.A.; McGrory, J.; Williams, T.; Williams, M.; Gracely, E.J.; Stern, L. A low-carbohydrate as compared with a low-fat diet in severe obesity. N. Engl. J. Med. 2003, 348, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.K.; Rosenbaum, D.; Han, H.; Geiselman, P.J.; Wyatt, H.R.; Hill, J.O.; Brill, C.; Bailer, B.; Miller, B.V., 3rd; Stein, R.; et al. Change in food cravings, food preferences, and appetite during a low-carbohydrate and low-fat diet. Obesity 2011, 19, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Swain, J.F.; Feldman, H.A.; Wong, W.W.; Hachey, D.L.; Garcia-Lago, E.; Ludwig, D.S. Effects of dietary composition on energy expenditure during weight-loss maintenance. JAMA 2012, 307, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.N.; Hodges, V.A.; Gillham, M.B. Normal-weight adults consume more fiber and fruit than their age- and height-matched overweight/obese counterparts. J. Am. Diet. Assoc. 2006, 106, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Willett, W.C.; Manson, J.E.; Hu, F.B.; Rosner, B.; Colditz, G. Relation between changes in intakes of dietary fiber and grain products and changes in weight and development of obesity among middle-aged women. Am. J. Clin. Nutr. 2003, 78, 920–927. [Google Scholar] [PubMed]
- Tucker, L.A.; Thomas, K.S. Increasing total fiber intake reduces risk of weight and fat gains in women. J. Nutr. 2009, 139, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.H.; Bacon, J.A.; Weinsier, R.L. The effects of high and low energy density diets on satiety, energy intake, and eating time of obese and nonobese subjects. Am. J. Clin. Nutr. 1983, 37, 763–767. [Google Scholar] [PubMed]
- Geliebter, A.; Grillot, C.L.; Aviram-Friedman, R.; Haq, S.; Yahav, E.; Hashim, S.A. Effects of oatmeal and corn flakes cereal breakfasts on satiety, gastric emptying, glucose, and appetite-related hormones. Ann. Nutr. Metab. 2015, 66, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Spetter, M.S.; de Graaf, C.; Mars, M.; Viergever, M.A.; Smeets, P.A. The sum of its parts—Effects of gastric distention, nutrient content and sensory stimulation on brain activation. PLoS ONE 2014, 9, e90872. [Google Scholar] [CrossRef] [PubMed]
- Lafond, D.W.; Greaves, K.A.; Maki, K.C.; Leidy, H.J.; Romsos, D.R. Effects of two dietary fibers as part of ready-to-eat cereal (RTEC) breakfasts on perceived appetite and gut hormones in overweight women. Nutrients 2015, 7, 1245–1266. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.J.; Slavin, J.L. The effect of fiber on satiety and food intake: A systematic review. J. Am. Coll. Nutr. 2013, 32, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Poehlman, E.T.; Melby, C.L.; Badylak, S.F. Resting metabolic rate and postprandial thermogenesis in highly trained and untrained males. Am. J. Clin. Nutr. 1988, 47, 793–798. [Google Scholar]
- Burke, C.M.; Bullough, R.C.; Melby, C.L. Resting metabolic rate and postprandial thermogenesis by level of aerobic fitness in young women. Eur. J. Clin. Nutr. 1993, 47, 575–585. [Google Scholar] [PubMed]
- Bell, C.; Day, D.S.; Jones, P.P.; Christou, D.D.; Petitt, D.S.; Osterberg, K.; Melby, C.L.; Seals, D.R. High energy flux mediates the tonically augmented beta-adrenergic support of resting metabolic rate in habitually exercising older adults. J. Clin. Endocrinol. Metab. 2004, 89, 3573–3578. [Google Scholar] [CrossRef] [PubMed]
- Bullough, R.C.; Gillette, C.A.; Harris, M.A.; Melby, C.L. Interaction of Acute Changes in Exercise Energy-Expenditure and Energy-Intake on Resting Metabolic-Rate. Am. J. Clin. Nutr. 1995, 61, 473–481. [Google Scholar] [PubMed]
- Elia, M.; Livesey, G. Energy expenditure and fuel selection in biological systems: The theory and practice of calculations based on indirect calorimetry and tracer methods. World Rev. Nutr. Diet. 1992, 70, 68–131. [Google Scholar] [PubMed]
- Javed, F.; He, Q.; Davidson, L.E.; Thornton, J.C.; Albu, J.; Boxt, L.; Krasnow, N.; Elia, M.; Kang, P.; Heshka, S.; et al. Brain and high metabolic rate organ mass: Contributions to resting energy expenditure beyond fat-free mass. Am. J. Clin. Nutr. 2010, 91, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Geliebter, A.; Maher, M.M.; Gerace, L.; Gutin, B.; Heymsfield, S.B.; Hashim, S.A. Effects of strength or aerobic training on body composition, resting metabolic rate, and peak oxygen consumption in obese dieting subjects. Am. J. Clin. Nutr. 1997, 66, 557–563. [Google Scholar] [PubMed]
- Melby, C.; Scholl, C.; Edwards, G.; Bullough, R. Effect of acute resistance exercise on postexercise energy expenditure and resting metabolic rate. J. Appl. Physiol. 1993, 75, 1847–1853. [Google Scholar] [PubMed]
- Osterberg, K.L.; Melby, C.L. Effect of acute resistance exercise on postexercise oxygen consumption and resting metabolic rate in young women. Int. J. Sport Nutr. Exerc. Metab. 2000, 10, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Wing, R.R.; Hill, J.O. Successful weight loss maintenance. Ann. Rev. Nutr. 2001, 21, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, K.R.; Wilson, S.A.; Rolland, V. Diet induced thermogenesis measured over 24h in a respiration chamber: Effect of diet composition. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.L.; Peng, C.; Carreira, M.C.; Halder, L.; Hattersley, J.; Piya, M.K.; Tripathi, G.; Randeva, H.S.; Casanueva, F.F.; McTernan, P.G.; et al. Enhanced thermic effect of food, postprandial NEFA suppression and raised adiponectin in obese women who eat slowly. Clin. Endocrinol. 2015, 82, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, N.D.; Clifton, P.M.; Noakes, M.; Farnsworth, E.; Wittert, G. Effect of a high-protein, energy-restricted diet on weight loss and energy expenditure after weight stabilization in hyperinsulinemic subjects. Int. J. Obes. Relat. Metab. 2003, 27, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, N.D.; Clifton, P.M.; Noakes, M.; Parker, B.; Wittert, G. Effects of energy-restricted diets containing increased protein on weight loss, resting energy expenditure, and the thermic effect of feeding in type 2 diabetes. Diabetes Care 2002, 25, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.A.; Vander Weg, M.W.; Hill, J.O.; Klesges, R.C. Non-exercise activity thermogenesis: The crouching tiger hidden dragon of societal weight gain. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Goran, M.I.; Poehlman, E.T. Endurance training does not enhance total energy expenditure in healthy elderly persons. Am. J. Physiol. 1992, 263, E950–E957. [Google Scholar] [PubMed]
- Luke, A.; Cooper, R.S. Physical activity does not influence obesity risk: Time to clarify the public health message. Int. J. Epidemiol. 2013, 42, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Jakicic, J.M.; Marcus, B.H.; Lang, W.; Janney, C. Effect of exercise on 24-month weight loss maintenance in overweight women. Arch. Int. Med. 2008, 168, 1550–1559. [Google Scholar] [CrossRef] [PubMed]
- Jakicic, J.M.; Otto, A.D.; Lang, W.; Semler, L.; Winters, C.; Polzien, K.; Mohr, K.I. The effect of physical activity on 18-month weight change in overweight adults. Obesity 2011, 19, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Sevits, K.J.; Melanson, E.L.; Swibas, T.; Binns, S.E.; Klochak, A.L.; Lonac, M.C.; Peltonen, G.L.; Scalzo, R.L.; Schweder, M.M.; Smith, A.M.; et al. Total daily energy expenditure is increased following a single bout of sprint interval training. Physiol. Rep. 2013, 1, e00131. [Google Scholar] [CrossRef] [PubMed]
- Kessler, H.S.; Sisson, S.B.; Short, K.R. The potential for high-intensity interval training to reduce cardiometabolic disease risk. Sports Med. 2012, 42, 489–509. [Google Scholar] [CrossRef] [PubMed]
- Weston, K.S.; Wisloff, U.; Coombes, J.S. High-intensity interval training in patients with lifestyle-induced cardiometabolic disease: A systematic review and meta-analysis. Br. J. Sports Med. 2014, 48, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Boutcher, S.H. High-intensity intermittent exercise and fat loss. J. Obes. 2011, 2011, 868305. [Google Scholar] [CrossRef] [PubMed]
- Jackman, M.R.; Steig, A.; Higgins, J.A.; Johnson, G.C.; Fleming-Elder, B.K.; Bessesen, D.H.; MacLean, P.S. Weight regain after sustained weight reduction is accompanied by suppressed oxidation of dietary fat and adipocyte hyperplasia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1117–R1129. [Google Scholar] [CrossRef] [PubMed]
- Steig, A.J.; Jackman, M.R.; Giles, E.D.; Higgins, J.A.; Johnson, G.C.; Mahan, C.; Melanson, E.L.; Wyatt, H.R.; Eckel, R.H.; Hill, J.O.; et al. Exercise reduces appetite and traffics excess nutrients away from energetically efficient pathways of lipid deposition during the early stages of weight regain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R656–R667. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Roy, P.; Mitra, K.P. Relation between caloric intake, body weight, and physical work: Studies in an industrial male population in West Bengal. Am. J. Clin. Nutr. 1956, 4, 169–175. [Google Scholar] [PubMed]
- Stubbs, R.J.; Hughes, D.A.; Johnstone, A.M.; Horgan, G.W.; King, N.; Blundell, J.E. A decrease in physical activity affects appetite, energy, and nutrient balance in lean men feeding ad libitum. Am. J. Clin. Nutr. 2004, 79, 62–69. [Google Scholar] [PubMed]
- MacLean, P.S.; Higgins, J.A.; Wyatt, H.R.; Melanson, E.L.; Johnson, G.C.; Jackman, M.R.; Giles, E.D.; Brown, I.E.; Hill, J.O. Regular exercise attenuates the metabolic drive to regain weight after long-term weight loss. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R793–R802. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.E.; Stubbs, R.J.; Hughes, D.A.; Whybrow, S.; King, N.A. Cross talk between physical activity and appetite control: Does physical activity stimulate appetite? Proc. Nutr. Soc. 2003, 62, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Hazell, T.J.; Islam, H.; Townsend, L.K.; Schmale, M.S.; Copeland, J.L. Effects of exercise intensity on plasma concentrations of appetite-regulating hormones: Potential mechanisms. Appetite 2016, 98, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.E.; Hill, J.O.; Jacobsen, D.J.; Potteiger, J.; Sullivan, D.K.; Johnson, S.L.; Heelan, K.; Hise, M.; Fennessey, P.V.; Sonko, B.; et al. Effects of a 16-month randomized controlled exercise trial on body weight and composition in young, overweight men and women: The Midwest Exercise Trial. Arch. Intern. Med. 2003, 163, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.E.; Gibbons, C.; Caudwell, P.; Finlayson, G.; Hopkins, M. Appetite control and energy balance: Impact of exercise. Obes. Rev. 2015, 16, 67–76. [Google Scholar] [CrossRef] [PubMed]
- King, N.A.; Hopkins, M.; Caudwell, P.; Stubbs, R.J.; Blundell, J.E. Individual variability following 12 weeks of supervised exercise: Identification and characterization of compensation for exercise-induced weight loss. Int. J. Obes. 2008, 32, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Paris, H.L.; Foright, R.M.; Werth, K.A.; Larson, L.C.; Beals, J.W.; Cox-York, K.; Bell, C.; Melby, C.L. Increasing Energy Flux to Decrese the Biological Drive toward Weight Regain after Weight Loss-a Proof-of-Concept Pilot Study. Clin. Nutr. ESPEN 2016, 11, e12–e20. [Google Scholar] [CrossRef]
- Wadden, T.A.; Neiberg, R.H.; Wing, R.R.; Clark, J.M.; Delahanty, L.M.; Hill, J.O.; Krakoff, J.; Otto, A.; Ryan, D.H.; Vitolins, M.Z.; et al. Four-year weight losses in the Look AHEAD study: Factors associated with long-term success. Obesity 2011, 19, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.J.; Yokum, S.; Stice, E. Low energy intake plus low energy expenditure (low energy flux), not energy surfeit, predicts future body fat gain. Am. J. Clin. Nutr. 2016, 103, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
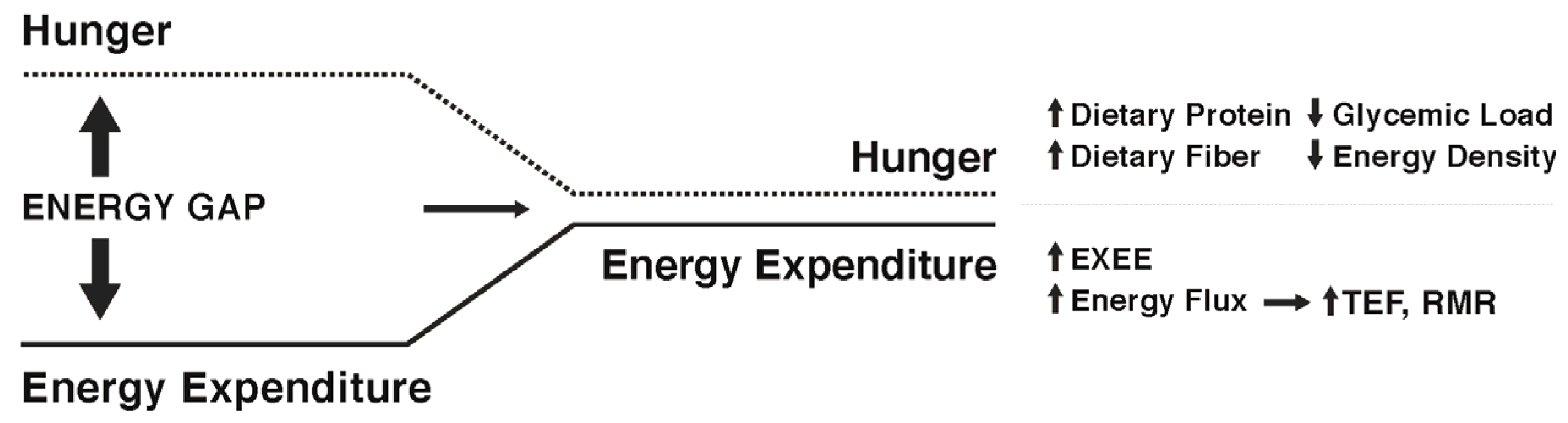

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melby, C.L.; Paris, H.L.; Foright, R.M.; Peth, J. Attenuating the Biologic Drive for Weight Regain Following Weight Loss: Must What Goes Down Always Go Back Up? Nutrients 2017, 9, 468. https://doi.org/10.3390/nu9050468
Melby CL, Paris HL, Foright RM, Peth J. Attenuating the Biologic Drive for Weight Regain Following Weight Loss: Must What Goes Down Always Go Back Up? Nutrients. 2017; 9(5):468. https://doi.org/10.3390/nu9050468
Chicago/Turabian StyleMelby, Christopher L., Hunter L. Paris, Rebecca M. Foright, and James Peth. 2017. "Attenuating the Biologic Drive for Weight Regain Following Weight Loss: Must What Goes Down Always Go Back Up?" Nutrients 9, no. 5: 468. https://doi.org/10.3390/nu9050468
APA StyleMelby, C. L., Paris, H. L., Foright, R. M., & Peth, J. (2017). Attenuating the Biologic Drive for Weight Regain Following Weight Loss: Must What Goes Down Always Go Back Up? Nutrients, 9(5), 468. https://doi.org/10.3390/nu9050468