Fructose Consumption in the Development of Obesity and the Effects of Different Protocols of Physical Exercise on the Hepatic Metabolism


 ,
, {kind=link}
{kind=link}
Abstract
:1. New Story/Old Enemy
2. Methodology
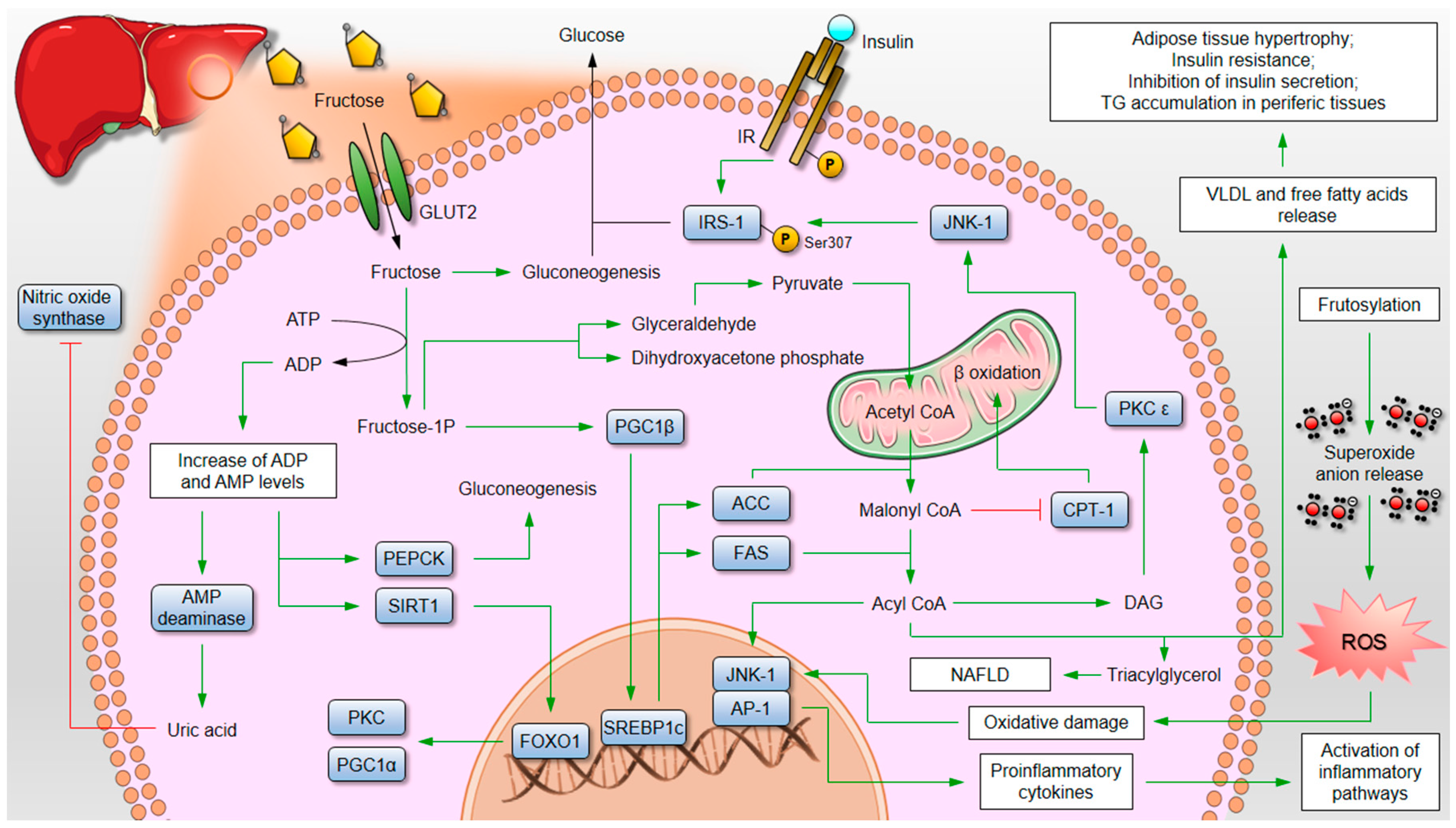
3. High Fructose Intake and Its Consequences on Metabolic Health
3.1. Animal Evidence
3.2. Human Evidence
4. The History of Fructose Consumption
5. Sweet Poison
6. How to Deal with the Enemy
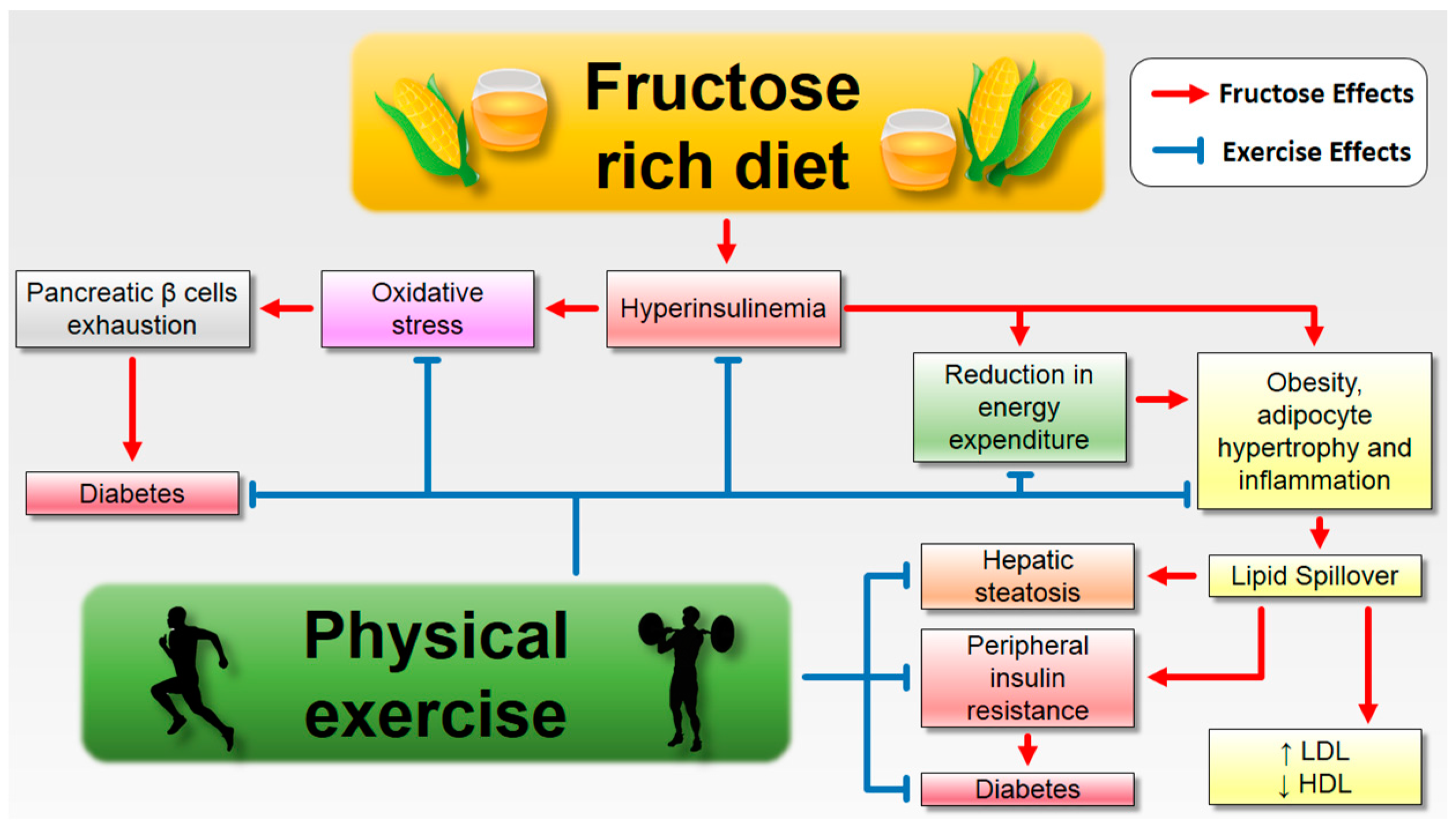
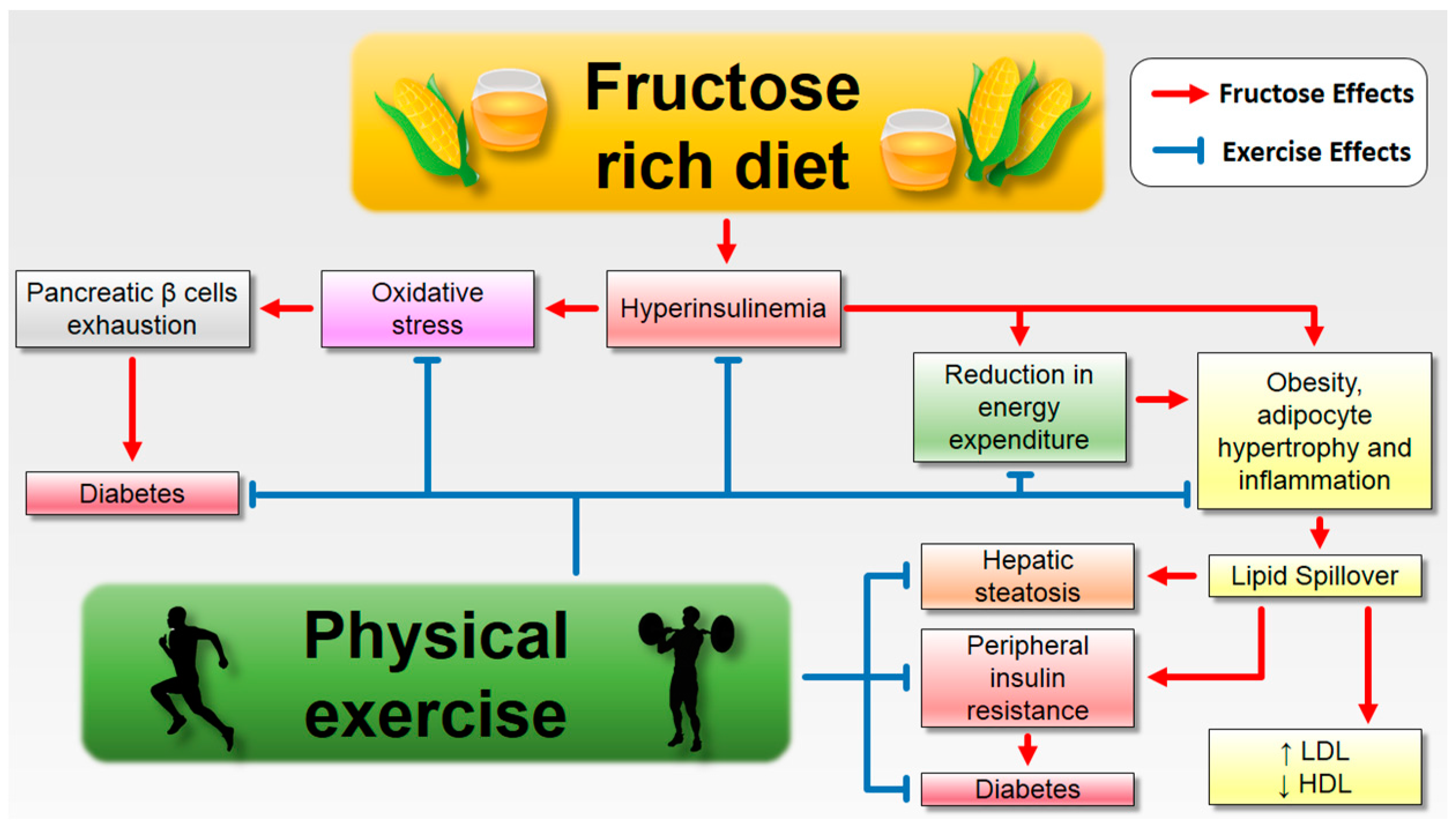
6.1. Fructose Consumption and Its Complications: The Role of Aerobic Exercise
6.1.1. Animal Evidence
6.1.2. Human Evidence
6.2. Fructose Consumption and Its Complicatons: The Role of Strength Exercise
6.2.1. Animal Evidence
6.2.2. Human Evidence
6.3. Fructose Consumption and Its Complicatons: The Role of Combined Exercise
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA Carboxylase |
| ADP | Adenosine Diphosphate |
| Akt | Protein kinase B |
| AMP | Adenosine Monophosphate |
| AMPK | AMP-activated protein kinase |
| AP-1 | Activator Protein-1 |
| AQP7 | Aquaporin 7 |
| ATP | Adenosine Triphosphate |
| BMI | Body mass index |
| CB1 | Cannabinoid 1 |
| CPT-1 | Carnitine Palmitoyl Transferase 1 |
| DAG | Diacylglycerol |
| DNA | Deoxyribonucleic Acid |
| eIF2α | Eukaryotic initiation factor 2-α |
| FAS | Fatty Acyl-CoA Synthase |
| FoxO1 | Forkhead box protein 01 |
| Fructose 1-P | Fructose 1-Phosphate |
| G6Pase | Glucose-6-phosphatase |
| GLUT2 | Glucose Transporter 2 |
| GLUT4 | Glucose Transporter 4 |
| GLUT5 | Glucose Transporter 5 |
| HDL | High-Density Lipoprotein |
| HFCS-42 | High-Fructose Corn Syrup with 42% of Fructose |
| HFCS-55 | High-Fructose Corn Syrup with 55% of Fructose |
| HFCS | High-Fructose Corn Syrup |
| IκBα | I-kappa-B-alpha |
| IR | Insulin Receptor |
| IRS-1 | Insulin Receptor Substrate 1 |
| JNK 1 | C-Jun-N terminal kinase-1 |
| LDL | Low-Density Lipoprotein |
| mTOR | Mechanistic target of rapamycin |
| NAD+/NADH | Nicotinamide Adenine Dinucleotide |
| NAFLD | Non-Alcoholic Fat Liver Disease |
| NF-κB | Nuclear factor-kappa B |
| NPY | Neuropeptide-Y |
| PEPCK | Phosphoenolpyruvate Carboxykinase |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PGC-1α | Peroxisome Proliferator-Activated Receptor-Gama Coactivator 1 Alpha |
| PGC-1β | Peroxisome Proliferator-Activated Receptor-Gama Coactivator 1 Beta |
| PKC | Protein Kinase C |
| POMC | Proopiomelanocortin |
| PTP-1B | Protein-tyrosine phosphatase 1B |
| ROS | Reactive Oxygen Species |
| S6K1 | Ribosomal protein S6 kinase beta-1 |
| SCD-1 | Stearoyl-CoA desaturase-1 |
| SIRT-1 | Sirtuin-1 |
| SREBP1c | Sterol Regulatory Element-Binding Protein 1c |
| TG | Triglycerides |
| TNF-α | Tumor necrosis factor alpha |
| TRB3 | Tribbles homolog 3 |
| VLDL | Very Low Density Lipoprotein |
| WAT | White Adipose Tissue |
References
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. Lond. 2005, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tuttle, K.R.; Short, R.A.; Johnson, R.J. Hypothesis: Fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat. Clin. Pract. Nephrol. 2005, 1, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Sullivan, L.; Jacques, P.F.; Wang, T.J.; Fox, C.S.; Meigs, J.B.; D’Agostino, R.B.; Gaziano, J.M.; Vasan, R.S. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation 2007, 116, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.; Gersch, M.S.; Benner, S.; Sánchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [PubMed]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Cao, H. Adipocytokines in obesity and metabolic disease. J. Endocrinol. 2014, 220, T47–T59. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar]
- Toop, C.R.; Gentili, S. Fructose Beverage Consumption Induces a Metabolic Syndrome Phenotype in the Rat: A Systematic Review and Meta-Analysis. Nutrients 2016, 8, 577. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Welsh, J.A.; Le, N.-A.; Holzberg, J.; Sharma, P.; Martin, D.R.; Vos, M.B. Dietary fructose reduction improves markers of cardiovascular disease risk in Hispanic-American adolescents with NAFLD. Nutrients 2014, 6, 3187–3201. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Feng, Q.; Zhang, Q.; Wang, T.; Xiao, X. Early Life Fructose Exposure and Its Implications for Long-Term Cardiometabolic Health in Offspring. Nutrients 2016, 8, 685. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B.; Malik, V.S. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: Epidemiologic evidence. Physiol. Behav. 2010, 100, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Peterson, K.E.; Gortmaker, S.L. Relation between consumption of sugar-sweetened drinks and childhood obesity: A prospective, observational analysis. Lancet 2001, 357, 505–508. [Google Scholar] [CrossRef]
- Green, B.N.; Johnson, C.D.; Adams, A. Writing narrative literature reviews for peer-reviewed journals: Secrets of the trade. J. Chiropr. Med. 2006, 5, 101–117. [Google Scholar] [CrossRef]
- Bocarsly, M.E.; Powell, E.S.; Avena, N.M.; Hoebel, B.G. High-fructose corn syrup causes characteristics of obesity in rats: Increased body weight, body fat and triglyceride levels. Pharmacol. Biochem. Behav. 2010, 97, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.-W. Liquiritigenin attenuates cardiac injury induced by high fructose-feeding through fibrosis and inflammation suppression. Biomed. Pharmacother. 2017, 86, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Senthilkumar, G.P.; Sivaraman, K.; Bobby, Z.; Paneerselvam, S.; Harichandrakumar, K.T. Effect of s-methyl-l-cysteine on oxidative stress, inflammation and insulin resistance in male wistar rats fed with high fructose diet. Iran. J. Med. Sci. 2015, 40, 45–50. [Google Scholar] [PubMed]
- Rodrigues, D.F.; Henriques, M.C.; Oliveira, M.C.; Menezes-Garcia, Z.; Marques, P.E.; Souza, D.G.; Menezes, G.B.; Teixeira, M.M.; Ferreira, A.V.M. Acute intake of a high-fructose diet alters the balance of adipokine concentrations and induces neutrophil influx in the liver. J. Nutr. Biochem. 2014, 25, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, R.-Q.; Zeng, X.-Y.; Zhou, X.; Li, S.; Jo, E.; Molero, J.C.; Ye, J.-M. Restoration of autophagy alleviates hepatic ER stress and impaired insulin signalling transduction in high fructose-fed male mice. Endocrinology 2015, 156, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Baelemans, A.; Erlanson-Albertsson, C. Effects of sucrose, glucose and fructose on peripheral and central appetite signals. Regul. Pept. 2008, 150, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.-W.; Chiang, M.-T.; Yao, H.-T.; Chiang, W. The effect of high-fat and high-fructose diets on glucose tolerance and plasma lipid and leptin levels in rats. Diabetes Obes. Metab. 2004, 6, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, M.; Raji, L.; Prabhu, D.; Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. High-fructose diet is as detrimental as high-fat diet in the induction of insulin resistance and diabetes mediated by hepatic/pancreatic endoplasmic reticulum (ER) stress. Mol. Cell. Biochem. 2016, 423, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Jarukamjorn, K.; Jearapong, N.; Pimson, C.; Chatuphonprasert, W. A High-Fat, High-Fructose Diet Induces Antioxidant Imbalance and Increases the Risk and Progression of Nonalcoholic Fatty Liver Disease in Mice. Sci. Cairo 2016, 2016, 5029414. [Google Scholar] [CrossRef] [PubMed]
- Lozano, I.; Van der Werf, R.; Bietiger, W.; Seyfritz, E.; Peronet, C.; Pinget, M.; Jeandidier, N.; Maillard, E.; Marchioni, E.; Sigrist, S.; et al. High-fructose and high-fat diet-induced disorders in rats: Impact on diabetes risk, hepatic and vascular complications. Nutr. Metab. Lond. 2016, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Cydylo, M.A.; Davis, A.T.; Kavanagh, K. Fatty liver promotes fibrosis in monkeys consuming high fructose. Obes. Silver Spring 2017, 25, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Goran, M.I.; Ulijaszek, S.J.; Ventura, E.E. High fructose corn syrup and diabetes prevalence: A global perspective. Glob. Public Health 2013, 8, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-T.; Chan, T.-F.; Huang, H.-L.; Lee, C.-Y.; Tsai, S.; Wu, P.-W.; Yang, Y.-C.; Wang, T.-N.; Lee, C.-H. Fructose-Rich Beverage Intake and Central Adiposity, Uric Acid, and Pediatric Insulin Resistance. J. Pediatr. 2016, 171, 90–96.e1. [Google Scholar] [CrossRef] [PubMed]
- Silbernagel, G.; Machann, J.; Unmuth, S.; Schick, F.; Stefan, N.; Häring, H.U.; Fritsche, A. Effects of 4-week very-high-fructose/glucose diets on insulin sensitivity, visceral fat and intrahepatic lipids: An exploratory trial. Br. J. Nutr. 2011, 106, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Sayed, A.; Binnert, C.; Lê, K.-A.; Bortolotti, M.; Schneiter, P.; Tappy, L. A high-fructose diet impairs basal and stress-mediated lipid metabolism in healthy male subjects. Br. J. Nutr. 2008, 100, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Lê, K.-A.; Faeh, D.; Stettler, R.; Ith, M.; Kreis, R.; Vermathen, P.; Boesch, C.; Ravussin, E.; Tappy, L. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am. J. Clin. Nutr. 2006, 84, 1374–1379. [Google Scholar] [PubMed]
- Melanson, K.J.; Zukley, L.; Lowndes, J.; Nguyen, V.; Angelopoulos, T.J.; Rippe, J.M. Effects of high-fructose corn syrup and sucrose consumption on circulating glucose, insulin, leptin, and ghrelin and on appetite in normal-weight women. Nutrition 2007, 23, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.-M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Sobrecases, H.; Lê, K.-A.; Bortolotti, M.; Schneiter, P.; Ith, M.; Kreis, R.; Boesch, C.; Tappy, L. Effects of short-term overfeeding with fructose, fat and fructose plus fat on plasma and hepatic lipids in healthy men. Diabetes Metab. 2010, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, S. Worldwide production of high-fructose syrup and crystalline fructose. Am. J. Clin. Nutr. 1993, 58, 733S–736S. [Google Scholar] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [PubMed]
- Johnson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J. Dietary sugars intake and cardiovascular health a scientific statement from the american heart association. Circulation 2009, 120, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H. Fructose: It’s “alcohol without the buzz”. Adv. Nutr. 2013, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Dulloo, A.G.; Montani, J.-P. Sugary drinks in the pathogenesis of obesity and cardiovascular diseases. Int. J. Obes. Lond. 2008, 32 (Suppl. 6), 28–34. [Google Scholar]
- Uldry, M.; Thorens, B. The SLC2 family of facilitated hexose and polyol transporters. Pflug. Arch. 2004, 447, 480–489. [Google Scholar] [CrossRef] [PubMed]
- SLC2A2 Solute Carrier Family 2 Member 2 [Homo sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/6514 (accessed on 2 April 2017).
- Teff, K.L.; Elliott, S.S.; Tschöp, M.; Kieffer, T.J.; Rader, D.; Heiman, M.; Townsend, R.R.; Keim, N.L.; D’Alessio, D.; Havel, P.J. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972. [Google Scholar] [PubMed]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.-Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [PubMed]
- Abizaid, A.; Horvath, T.L. Ghrelin and the central regulation of feeding and energy balance. Indian J. Endocrinol. Metab. 2012, 16, S617–S626. [Google Scholar] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Tanaka, K.; Fukamizu, A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. USA 2003, 100, 11285–11290. [Google Scholar] [CrossRef] [PubMed]
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.C.; Martin, R.J.; Whitney, M.L.; Edwards, G.L. Intracerebroventricular injection of fructose stimulates feeding in rats. Nutr. Neurosci. 2002, 5, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Nayuni, N.K.; Khan, N.Q.; Wood, E.G.; Corder, R. Fructose induces gluconeogenesis and lipogenesis through a SIRT1-dependent mechanism. J. Endocrinol. 2011, 208, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Valenti, L.; Accili, D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005, 280, 20589–20595. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Puigserver, P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. USA 2007, 104, 12861–12866. [Google Scholar] [CrossRef] [PubMed]
- Gersch, C.; Palii, S.P.; Kim, K.M.; Angerhofer, A.; Johnson, R.J.; Henderson, G.N. Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids 2008, 27, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Qi, L.; Qiao, N.; Choi, H.K.; Curhan, G.; Tucker, K.L.; Ascherio, A. Intake of added sugar and sugar-sweetened drink and serum uric acid concentration in US men and women. Hypertension 2007, 50, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M.; Fatty Liver Subgroup of Look AHEAD Research Group. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Löffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754S–765S. [Google Scholar] [PubMed]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [PubMed]
- Cooney, G.J.; Thompson, A.L.; Furler, S.M.; Ye, J.; Kraegen, E.W. Muscle long-chain acyl CoA esters and insulin resistance. Ann. N. Y. Acad. Sci. 2002, 967, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Kelpe, C.L.; Johnson, L.M.; Poitout, V. Increasing triglyceride synthesis inhibits glucose-induced insulin secretion in isolated rat islets of langerhans: A study using adenoviral expression of diacylglycerol acyltransferase. Endocrinology 2002, 143, 3326–3332. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Bezerra, R.M.; Silva, M.S.; Tavares, D.Q.; Carvalho, C.R.; Saad, M.J. A high-fructose diet induces changes in pp185 phosphorylation in muscle and liver of rats. Braz. J. Med. Biol. Res. 2000, 33, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Mellor, K.; Ritchie, R.H.; Meredith, G.; Woodman, O.L.; Morris, M.J.; Delbridge, L.M. High-fructose diet elevates myocardial superoxide generation in mice in the absence of cardiac hypertrophy. Nutrition 2010, 26, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef] [PubMed]
- García, O.P.; Long, K.Z.; Rosado, J.L. Impact of micronutrient deficiencies on obesity. Nutr. Rev. 2009, 67, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic syndrome: A comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Botezelli, J.D.; Cambri, L.T.; Ghezzi, A.C.; Dalia, R.A.; Voltarelli, F.A.; de Mello, M.A. Fructose-rich diet leads to reduced aerobic capacity and to liver injury in rats. Lipids Health Dis. 2012, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.; Bruce, W.R.; Dong, Q.; Bruce, J.; Mehta, R.; O’Brien, P.J. Fructose and carbonyl metabolites as endogenous toxins. Chem. Biol. Interact. 2009, 178, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Botezelli, J.D.; Coope, A.; Ghezzi, A.C.; Cambri, L.T.; Moura, L.P.; Scariot, P.P.M.; Gaspar, R.S.; Mekary, R.A.; Ropelle, E.R.; Pauli, J.R. Strength Training Prevents Hyperinsulinemia, Insulin Resistance, and Inflammation Independent of Weight Loss in Fructose-Fed Animals. Sci. Rep. 2016, 6, 31106. [Google Scholar] [CrossRef] [PubMed]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Volynets, V.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Toll-like receptors 1-9 are elevated in livers with fructose-induced hepatic steatosis. Br. J. Nutr. 2012, 107, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Kelley, G.L.; Allan, G.; Azhar, S. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinology 2004, 145, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Sentürk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Levine, R. Monosaccharides in health and disease. Annu. Rev. Nutr. 1986, 6, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Hallal, P.C.; Andersen, L.B.; Bull, F.C.; Guthold, R.; Haskell, W.; Ekelund, U.; Lancet Physical Activity Series Working Group. Global physical activity levels: Surveillance progress, pitfalls, and prospects. Lancet 2012, 380, 247–257. [Google Scholar] [CrossRef]
- Lawrence, R.D. The Effect of Exercise on Insulin Action in Diabetes. Br. Med. J. 1926, 1, 648–650. [Google Scholar] [CrossRef] [PubMed]
- Bradley, H.; Shaw, C.S.; Bendtsen, C.; Worthington, P.L.; Wilson, O.J.; Strauss, J.A.; Wallis, G.A.; Turner, A.M.; Wagenmakers, A.J. Visualization and quantitation of GLUT4 translocation in human skeletal muscle following glucose ingestion and exercise. Physiol. Rep. 2015, 3, e12375. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Treebak, J.T.; Glund, S.; Deshmukh, A.; Klein, D.K.; Long, Y.C.; Jensen, T.E.; Jørgensen, S.B.; Viollet, B.; Andersson, L.; Neumann, D.; et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 2006, 55, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Mîinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peränen, J.; Lane, W.S.; Lienhard, G.E. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 2005, 391, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Matos, A.; Ropelle, E.R.; Pauli, J.R.; Frederico, M.J.; de Pinho, R.A.; Velloso, L.A.; De Souza, C.T. Acute exercise reverses TRB3 expression in the skeletal muscle and ameliorates whole body insulin sensitivity in diabetic mice. Acta Physiol. Oxf. 2010, 198, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Pauli, J.R.; Ropelle, E.R.; Cintra, D.E.; De Souza, C.T.; da Silva, A.S.R.; Moraes, J.C.; Prada, P.O.; de Almeida Leme, J.A.C.; Luciano, E.; Velloso, L.A.; et al. Acute exercise reverses aged-induced impairments in insulin signaling in rodent skeletal muscle. Mech. Ageing Dev. 2010, 131, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, N.R.C.; Marques, S.O.; Luciano, T.F.; Pauli, J.R.; Moura, L.P.; Caperuto, E.; Pieri, B.L.S.; Engelmann, J.; Scaini, G.; Streck, E.L.; et al. Treadmill training increases SIRT-1 and PGC-1 α protein levels and AMPK phosphorylation in quadriceps of middle-aged rats in an intensity-dependent manner. Mediat. Inflamm. 2014, 2014, 987017. [Google Scholar] [CrossRef] [PubMed]
- De Moura, L.P.; Souza Pauli, L.S.; Cintra, D.E.; de Souza, C.T.; da Silva, A.S.R.; Marinho, R.; de Melo, M.A.R.; Ropelle, E.R.; Pauli, J.R. Acute exercise decreases PTP-1B protein level and improves insulin signaling in the liver of old rats. Immun. Ageing 2013, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Da Luz, G.; Frederico, M.J.S.; da Silva, S.; Vitto, M.F.; Cesconetto, P.A.; de Pinho, R.A.; Pauli, J.R.; Silva, A.S.R.; Cintra, D.E.; Ropelle, E.R.; et al. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur. J. Appl. Physiol. 2011, 111, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Ropelle, E.R.; Flores, M.B.; Cintra, D.E.; Rocha, G.Z.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Moraes, J.C.; Prada, P.O.; Guadagnini, D.; et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol. 2010, 8, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.D.A.; Pauli, L.S.S.; de Souza, C.T.; da Silva, A.S.R.; Cintra, D.E.C.; Marinho, R.; de Moura, L.P.; Ropelle, E.C.C.; Botezelli, J.D.; Ropelle, E.R.; Pauli, J.R. Acute Exercise Decreases Tribbles Homolog 3 Protein Levels in the Hypothalamus of Obese Rats. Med. Sci. Sports Exerc. 2015, 47, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, A.; Couturier, K.; Gauthier, M.-S.; Lavoie, J.-M. Evidence of hepatic glucagon resistance associated with hepatic steatosis: Reversal effect of training. Int. J. Sports Med. 2005, 26, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xu, X.; Yue, K.; Xu, G. Effect of different exercise protocols on metabolic profiles and fatty acid metabolism in skeletal muscle in high-fat diet-fed rats. Obes. Silver Spring 2015, 23, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Jorge, M.L.M.P.; de Oliveira, V.N.; Resende, N.M.; Paraiso, L.F.; Calixto, A.; Diniz, A.L.D.; Resende, E.S.; Ropelle, E.R.; Carvalheira, J.B.; Espindola, F.S.; et al. The effects of aerobic, resistance, and combined exercise on metabolic control, inflammatory markers, adipocytokines, and muscle insulin signaling in patients with type 2 diabetes mellitus. Metabolism 2011, 60, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.-S.; Couturier, K.; Latour, J.-G.; Lavoie, J.-M. Concurrent exercise prevents high-fat-diet-induced macrovesicular hepatic steatosis. J. Appl. Physiol. 2003, 94, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Ntambi, J.M.; Friedman, J.M. Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2003, 3, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, R.B.; Orthen-Gambill, N. Differential effects of sucrose, fructose and glucose on carbohydrate-induced obesity in rats. J. Nutr. 1982, 112, 1546–1554. [Google Scholar] [PubMed]
- Kok, N.; Roberfroid, M.; Delzenne, N. Dietary oligofructose modifies the impact of fructose on hepatic triacylglycerol metabolism. Metabolism 1996, 45, 1547–1550. [Google Scholar] [CrossRef]
- Catena, C.; Giacchetti, G.; Novello, M.; Colussi, G.; Cavarape, A.; Sechi, L.A. Cellular mechanisms of insulin resistance in rats with fructose-induced hypertension. Am. J. Hypertens. 2003, 16, 973–978. [Google Scholar] [CrossRef]
- Mann, S.; Beedie, C.; Jimenez, A. Differential effects of aerobic exercise, resistance training and combined exercise modalities on cholesterol and the lipid profile: Review, synthesis and recommendations. Sports Med. 2014, 44, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Leon, A.S.; Sanchez, O.A. Response of blood lipids to exercise training alone or combined with dietary intervention. Med. Sci. Sports Exerc. 2001, 33, S502–S515. [Google Scholar] [CrossRef] [PubMed]
- Botezelli, J.D.; Mora, R.F.; Dalia, R.A.; Moura, L.P.; Cambri, L.T.; Ghezzi, A.C.; Voltarelli, F.A.; Mello, M.A.R. Exercise counteracts fatty liver disease in rats fed on fructose-rich diet. Lipids Health Dis. 2010, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Stanišić, J.; Korićanac, G.; Ćulafić, T.; Romić, S.; Stojiljković, M.; Kostić, M.; Pantelić, M.; Tepavčević, S. Low intensity exercise prevents disturbances in rat cardiac insulin signaling and endothelial nitric oxide synthase induced by high fructose diet. Mol. Cell. Endocrinol. 2016, 420, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Farah, D.; Nunes, J.; Sartori, M.; Dias, D.D.S.; Sirvente, R.; Silva, M.B.; Fiorino, P.; Morris, M.; Llesuy, S.; Farah, V.; et al. Exercise Training Prevents Cardiovascular Derangements Induced by Fructose Overload in Developing Rats. PLoS ONE 2016, 11, e0167291. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [PubMed]
- Karaca, A.; Palabıyık, O.; Taştekin, E.; Turan, F.N.; Arzu Vardar, S. High fructose diet suppresses exercise-induced increase in AQP7 expression in the in vivo rat heart. Anatol. J. Cardiol. 2016, 16, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, A.J.; Fairchild, T.J.; Redmond, J.; Wang, L.; Keslacy, S.; Kanaley, J.A. Physical activity offsets the negative effects of a high-fructose diet. Med. Sci. Sports Exerc. 2014, 46, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Egli, L.; Lecoultre, V.; Theytaz, F.; Campos, V.; Hodson, L.; Schneiter, P.; Mittendorfer, B.; Patterson, B.W.; Fielding, B.A.; Gerber, P.A.; et al. Exercise prevents fructose-induced hypertriglyceridemia in healthy young subjects. Diabetes 2013, 62, 2259–2265. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.J.; Sherman, W.M.; Ivy, J.L. Effect of strength training on glucose tolerance and post-glucose insulin response. Med. Sci. Sports Exerc. 1984, 16, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Treuth, M.S.; Hunter, G.R.; Kekes-Szabo, T.; Weinsier, R.L.; Goran, M.I.; Berland, L. Reduction in intra-abdominal adipose tissue after strength training in older women. J. Appl. Physiol. 1995, 78, 1425–1431. [Google Scholar] [PubMed]
- Cheon, D.Y.; Kang, J.G.; Lee, S.J.; Ihm, S.H.; Lee, E.J.; Choi, M.G.; Yoo, H.J.; Kim, C.S. Serum Chemerin Levels are Associated with Visceral Adiposity, Independent of Waist Circumference, in Newly Diagnosed Type 2 Diabetic Subjects. Yonsei Med. J. 2017, 58, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Luo, K.; Liu, C.; Wang, X.; Zhang, D.; Chi, A.; Zhang, J.; Sun, L. Decrease in myostatin by ladder-climbing training is associated with insulin resistance in diet-induced obese rats. Chin. Med. J. Engl. 2014, 127, 2342–2349. [Google Scholar] [PubMed]
- Tang, L.; Gao, X.; Yang, X.; Liu, C.; Wang, X.; Han, Y.; Zhao, X.; Chi, A.; Sun, L. Ladder-Climbing Training Prevents Bone Loss and Microarchitecture Deterioration in Diet-Induced Obese Rats. Calcif. Tissue Int. 2016, 98, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Nurnazahiah, A.; Lua, P.L.; Shahril, M.R. Adiponectin, Leptin and Objectively Measured Physical Activity in Adults: A Narrative Review. Malays. J. Med. Sci. 2016, 23, 7–24. [Google Scholar] [PubMed]
- Klimcakova, E.; Polak, J.; Moro, C.; Hejnova, J.; Majercik, M.; Viguerie, N.; Berlan, M.; Langin, D.; Stich, V. Dynamic strength training improves insulin sensitivity without altering plasma levels and gene expression of adipokines in subcutaneous adipose tissue in obese men. J. Clin. Endocrinol. Metab. 2006, 91, 5107–5112. [Google Scholar] [CrossRef] [PubMed]
- Gippini, A.; Mato, A.; Pazos, R.; Suarez, B.; Vila, B.; Gayoso, P.; Lage, M.; Casanueva, F.F. Effect of long-term strength training on glucose metabolism. Implications for individual impact of high lean mass and high fat mass on relationship between BMI and insulin sensitivity. J. Endocrinol. Investig. 2002, 25, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.H.; Jensen, M.D.; Kugler, K.C.; Jeffery, R.W.; Leon, A.S. Strength training for obesity prevention in midlife women. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.H.; Hannan, P.J.; Stovitz, S.D.; Bryan, C.J.; Warren, M.; Jensen, M.D. Strength training and adiposity in premenopausal women: Strong, healthy, and empowered study. Am. J. Clin. Nutr. 2007, 86, 566–572. [Google Scholar] [PubMed]
- Lira, F.S.; Yamashita, A.S.; Uchida, M.C.; Zanchi, N.E.; Gualano, B.; Martins, E.; Caperuto, E.C.; Seelaender, M. Low and moderate, rather than high intensity strength exercise induces benefit regarding plasma lipid profile. Diabetol. Metab. Syndr. 2010, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, G.-J.; Wang, Z.J.; Chu, Z.; Toffolo, G.; Manesso, E.; Sauer, P.J.J.; Sunehag, A.L. Strength exercise improves muscle mass and hepatic insulin sensitivity in obese youth. Med. Sci. Sports Exerc. 2010, 42, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.Y.; Lee, S.R.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Humanized animal exercise model for clinical implication. Pflug. Arch. 2014, 466, 1673–1687. [Google Scholar] [CrossRef] [PubMed]
- Wilburn, J.R.; Bourquin, J.; Wysong, A.; Melby, C.L. Resistance Exercise Attenuates High-Fructose, High-Fat-Induced Postprandial Lipemia. Nutr. Metab. Insights 2015, 8, 29–35. [Google Scholar] [PubMed]
- Hickson, R.C. Interference of strength development by simultaneously training for strength and endurance. Eur. J. Appl. Physiol. Occup. Physiol. 1980, 45, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, W.J.; Patton, J.F.; Gordon, S.E.; Harman, E.A.; Deschenes, M.R.; Reynolds, K.; Newton, R.U.; Triplett, N.T.; Dziados, J.E. Compatibility of high-intensity strength and endurance training on hormonal and skeletal muscle adaptations. J. Appl. Physiol. 1995, 78, 976–989. [Google Scholar] [PubMed]
- McCarthy, J.P.; Pozniak, M.A.; Agre, J.C. Neuromuscular adaptations to concurrent strength and endurance training. Med. Sci. Sports Exerc. 2002, 34, 511–519. [Google Scholar] [CrossRef] [PubMed]
- De Souza, E.O.; Tricoli, V.; Aoki, M.S.; Roschel, H.; Brum, P.C.; Bacurau, A.V.N.; Silva-Batista, C.; Wilson, J.M.; Neves, M.; Soares, A.G.; et al. Effects of concurrent strength and endurance training on genes related to myostatin signaling pathway and muscle fiber responses. J. Strength Cond. Res. 2014, 28, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Apró, W.; Moberg, M.; Hamilton, D.L.; Ekblom, B.; van Hall, G.; Holmberg, H.-C.; Blomstrand, E. Resistance exercise-induced S6K1 kinase activity is not inhibited in human skeletal muscle despite prior activation of AMPK by high-intensity interval cycling. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E470–E481. [Google Scholar] [CrossRef] [PubMed]
- Pauli, J.R.; Ropelle, E.R.; Cintra, D.E.; Souza, C.T.D. Effects of Physical Exercise in the Ampkα Expression and Activity in High-fat Diet Induced Obese Rats. Rev. Bras. Med. Esporte 2009, 15, 98–103. [Google Scholar] [CrossRef]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef] [PubMed]
- Garber, C.E.; Blissmer, B.; Deschenes, M.R.; Franklin, B.A.; Lamonte, M.J.; Lee, I.-M.; Nieman, D.C.; Swain, D.P.; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: Guidance for prescribing exercise. Med. Sci. Sports Exerc. 2011, 43, 1334–1359. [Google Scholar] [PubMed]
- Monteiro, P.A.; Chen, K.Y.; Lira, F.S.; Saraiva, B.T.C.; Antunes, B.M.M.; Campos, E.Z.; Freitas, I.F. Concurrent and aerobic exercise training promote similar benefits in body composition and metabolic profiles in obese adolescents. Lipids Health Dis. 2015, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, N.D.S.; de Abreu, F.G.; Colato, A.S.; de Lemos, L.S.; Ramis, T.R.; Dorneles, G.P.; Funchal, C.; Dani, C. Effects of concurrent training on oxidative stress and insulin resistance in obese individuals. Oxid. Med. Cell. Longev. 2015, 2015, 697181. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid. Redox Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, R.M.; Botezelli, J.D.; Da Cruz Rodrigues, K.C.; Mekary, R.A.; Cintra, D.E.; Pauli, J.R.; Da Silva, A.S.R.; Ropelle, E.R.; De Moura, L.P. Fructose Consumption in the Development of Obesity and the Effects of Different Protocols of Physical Exercise on the Hepatic Metabolism. Nutrients 2017, 9, 405. https://doi.org/10.3390/nu9040405
Pereira RM, Botezelli JD, Da Cruz Rodrigues KC, Mekary RA, Cintra DE, Pauli JR, Da Silva ASR, Ropelle ER, De Moura LP. Fructose Consumption in the Development of Obesity and the Effects of Different Protocols of Physical Exercise on the Hepatic Metabolism. Nutrients. 2017; 9(4):405. https://doi.org/10.3390/nu9040405
Chicago/Turabian StylePereira, Rodrigo Martins, José Diego Botezelli, Kellen Cristina Da Cruz Rodrigues, Rania A. Mekary, Dennys Esper Cintra, José Rodrigo Pauli, Adelino Sanchez Ramos Da Silva, Eduardo Rochete Ropelle, and Leandro Pereira De Moura. 2017. "Fructose Consumption in the Development of Obesity and the Effects of Different Protocols of Physical Exercise on the Hepatic Metabolism" Nutrients 9, no. 4: 405. https://doi.org/10.3390/nu9040405
APA StylePereira, R. M., Botezelli, J. D., Da Cruz Rodrigues, K. C., Mekary, R. A., Cintra, D. E., Pauli, J. R., Da Silva, A. S. R., Ropelle, E. R., & De Moura, L. P. (2017). Fructose Consumption in the Development of Obesity and the Effects of Different Protocols of Physical Exercise on the Hepatic Metabolism. Nutrients, 9(4), 405. https://doi.org/10.3390/nu9040405