Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review





{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Search Strategy (Methods)
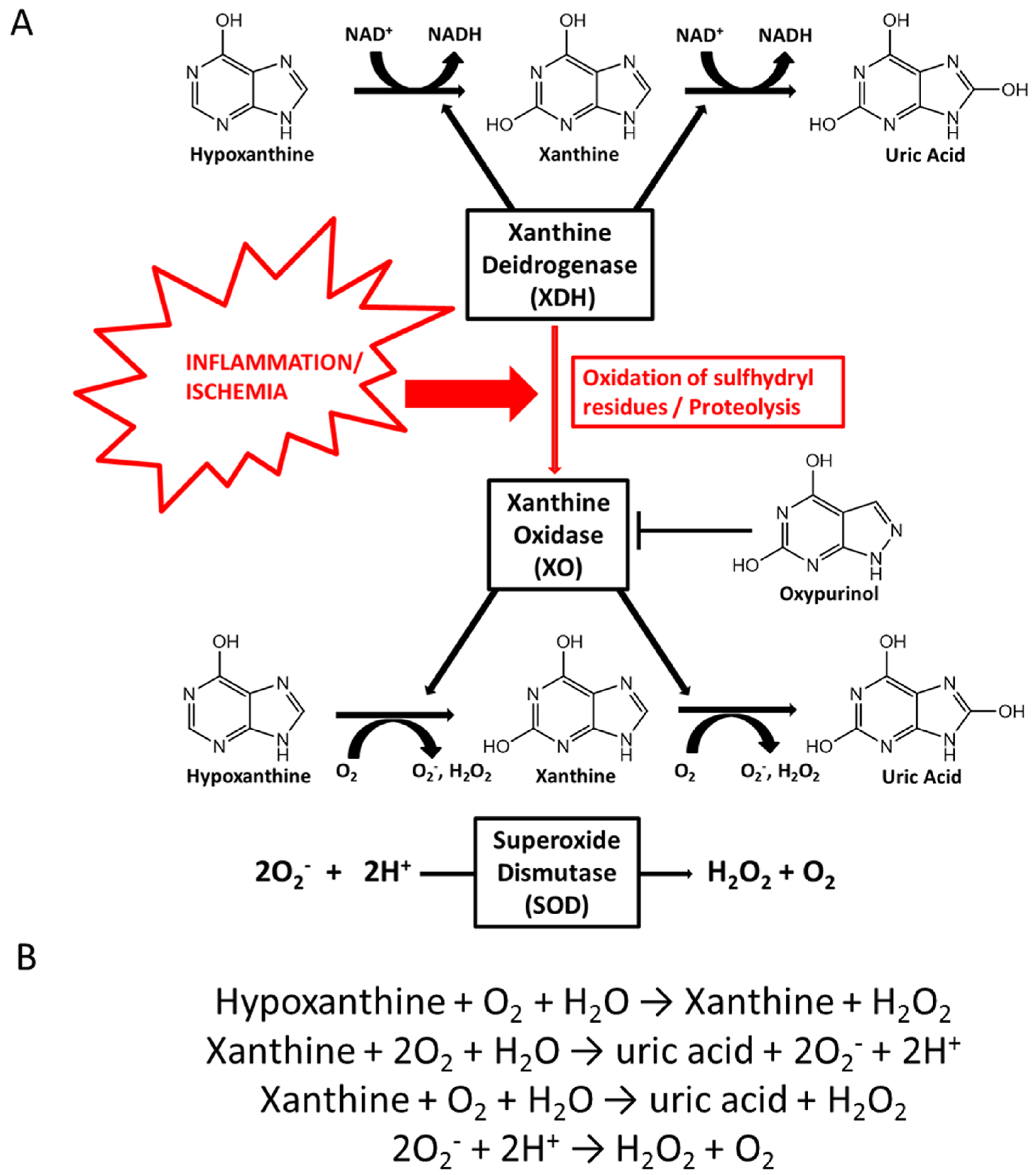
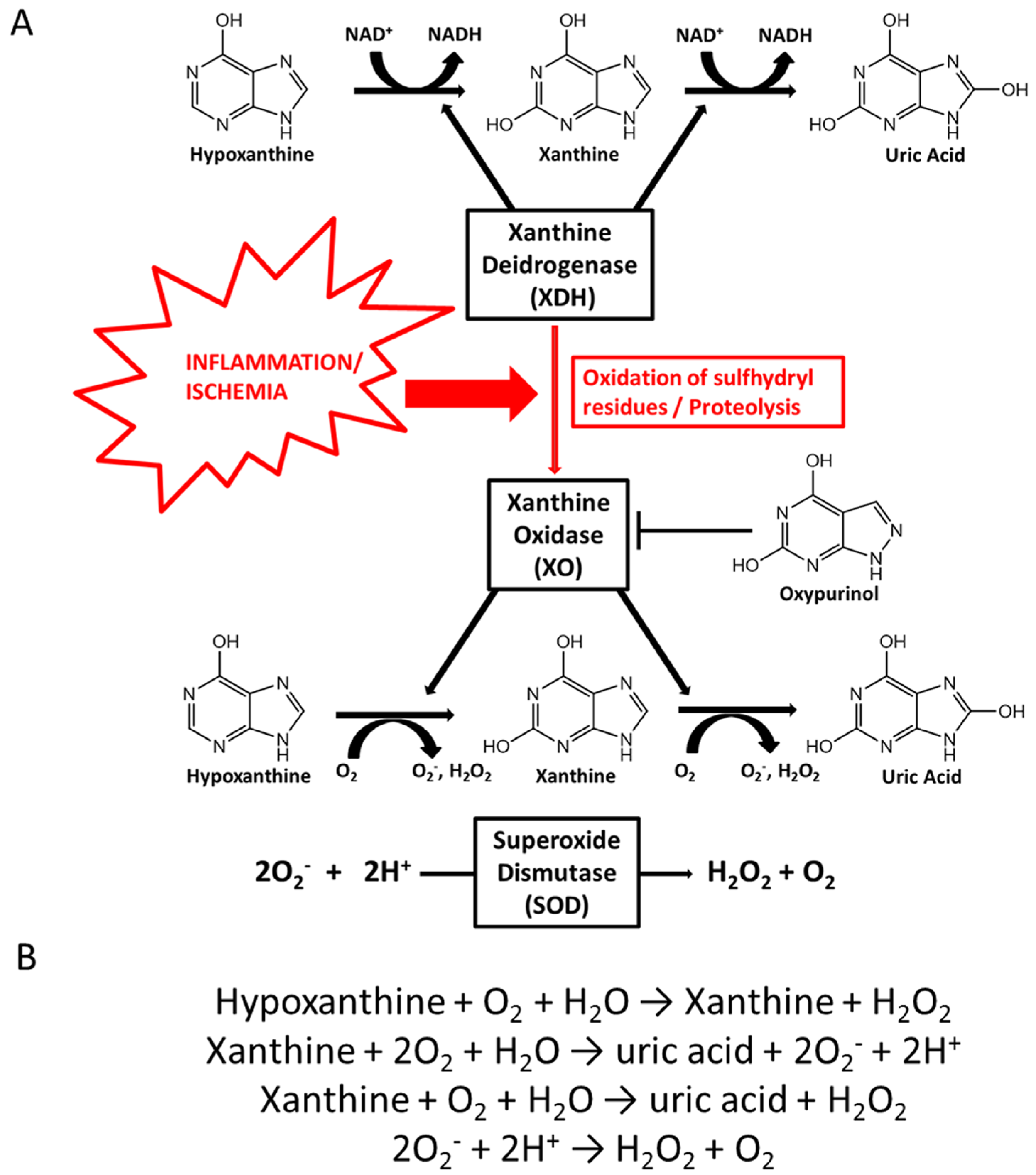
3. Purine Metabolism and Uric Acid Physiology
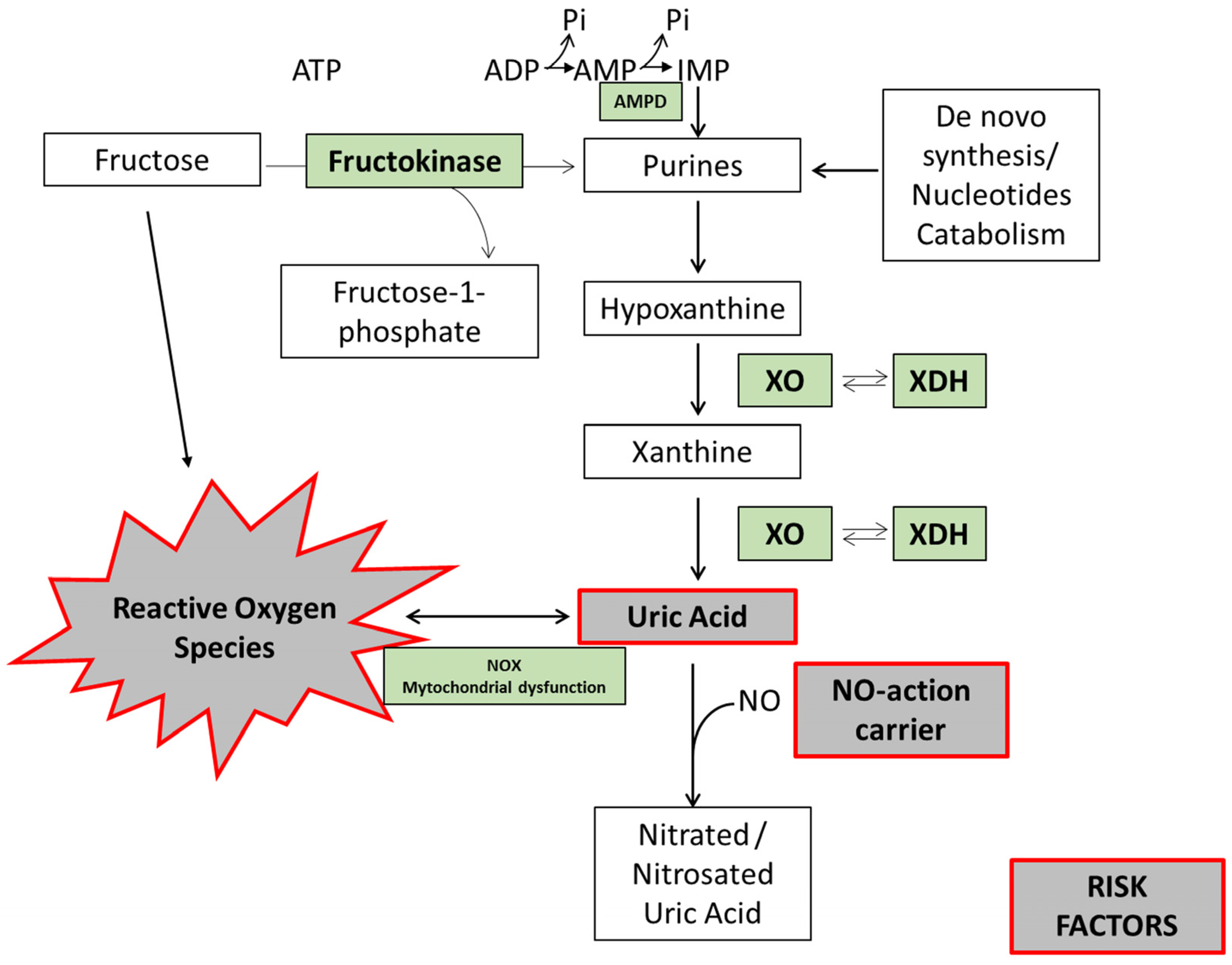
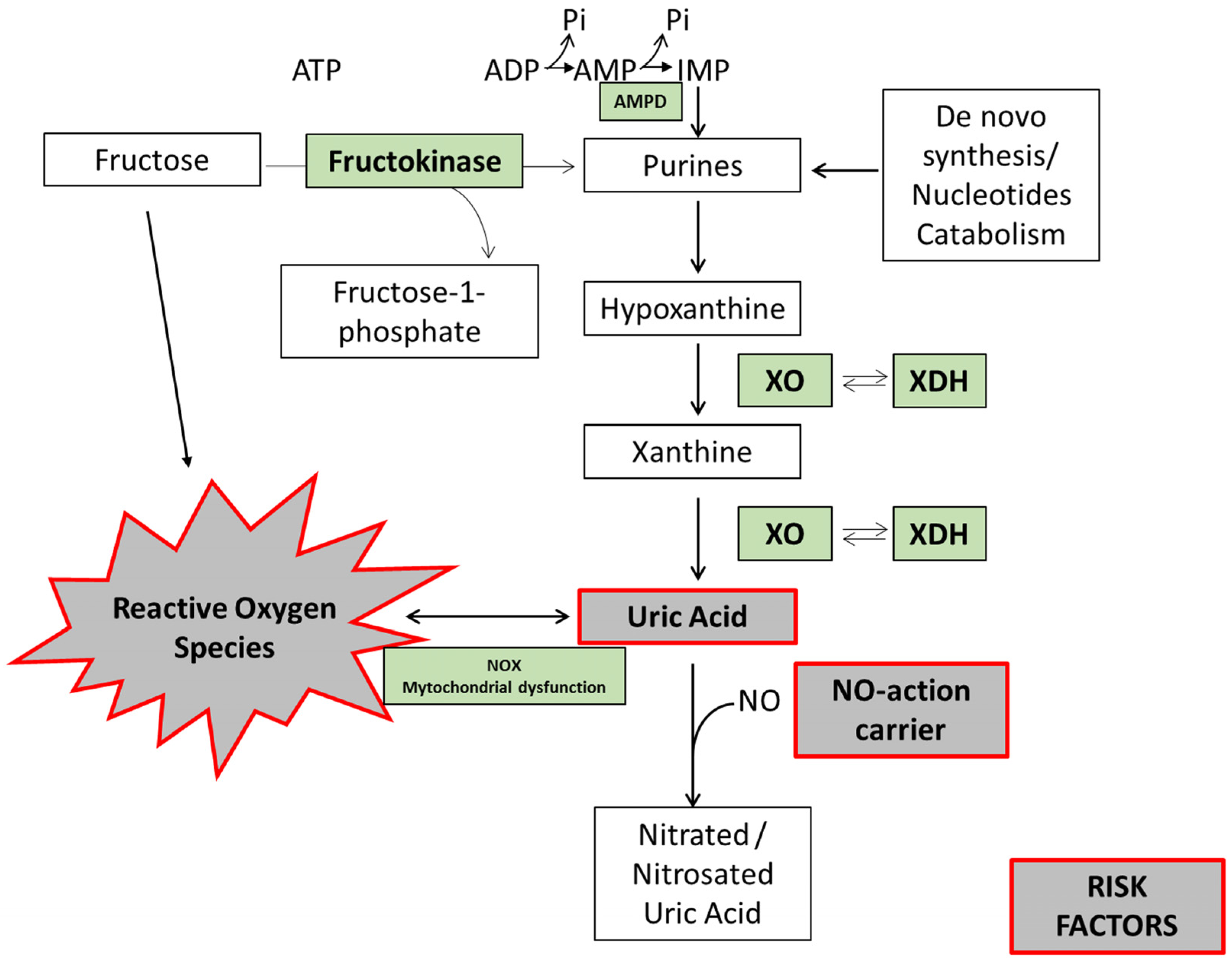
4. Fructose Metabolism and the Mechanisms by Which Fructose May Contribute to Uric Acid Production
5. Mechanisms by Which Fructose-Uric Acid May Contribute to Cardiometabolic Disorders
6. Evidence from Clinical Studies, Relevance for Humans
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zamudio-Cuevas, Y.; Hernandez-Diaz, C.; Pineda, C.; Reginato, A.M.; Cerna-Cortes, J.F.; Ventura-Rios, L.; Lopez-Reyes, A. Molecular basis of oxidative stress in gouty arthropathy. Clin. Rheumatol. 2015, 34, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Stack, A.G.; Hanley, A.; Casserly, L.F.; Cronin, C.J.; Abdalla, A.A.; Kiernan, T.J.; Murthy, B.V.; Hegarty, A.; Hannigan, A.; Nguyen, H.T. Independent and conjoint associations of gout and hyperuricaemia with total and cardiovascular mortality. QJM 2013, 106, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Church, T.S.; Meriwether, R.A.; Lobelo, F.; Blair, S.N. Uric acid and the development of metabolic syndrome in women and men. Metabolism 2008, 57, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Renal. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.S.; Mu, W.; Cirillo, P.; Reungjui, S.; Zhang, L.; Roncal, C.; Sautin, Y.Y.; Johnson, R.J.; Nakagawa, T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2007, 293, F1256–F1261. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [PubMed]
- Johnson, R.J.; Titte, S.; Cade, J.R.; Rideout, B.A.; Oliver, W.J. Uric acid, evolution and primitive cultures. Semin. Nephrol. 2005, 25, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Borghi, C. The role of uric acid in the development of cardiovascular disease. Curr. Med. Res. Opin. 2015, 31 (Suppl. S2), 1–2. [Google Scholar] [CrossRef] [PubMed]
- Iwama, M.; Kondo, Y.; Shimokado, K.; Maruyama, N.; Ishigami, A. Uric acid levels in tissues and plasma of mice during aging. Biol. Pharm. Bull. 2012, 35, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Andrews, P.; Benner, S.A.; Oliver, W.; Theodore, E. Woodward award. The evolution of obesity: Insights from the mid-Miocene. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 295–305. [Google Scholar] [PubMed]
- Johnson, R.J.; Sautin, Y.Y.; Oliver, W.J.; Roncal, C.; Mu, W.; Gabriela Sanchez-Lozada, L.; Rodriguez-Iturbe, B.; Nakagawa, T.; Benner, S.A. Lessons from comparative physiology: Could uric acid represent a physiologic alarm signal gone awry in western society? J. Comp. Physiol. B 2009, 179, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.; Dawson, J.; Lees, K.R.; McArthur, K.; Quinn, T.J.; Walters, M.R. Xanthine oxidase inhibition for the treatment of cardiovascular disease: A systematic review and meta-analysis. Cardiovasc. Ther. 2012, 30, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.B. Mechanism of excessive purine biosynthesis in hypoxanthine-guanine phosphoribosyltransferase deficiency. J. Clin. Investig. 1970, 49, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Pritsos, C.A. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem. Biol. Interact. 2000, 129, 195–208. [Google Scholar] [CrossRef]
- Vorbach, C.; Harrison, R.; Capecchi, M.R. Xanthine oxidoreductase is central to the evolution and function of the innate immune system. Trends Immunol. 2003, 24, 512–517. [Google Scholar] [CrossRef]
- Friedl, H.P.; Till, G.O.; Ryan, U.S.; Ward, P.A. Mediator-induced activation of xanthine oxidase in endothelial cells. FASEB J. 1989, 3, 2512–2518. [Google Scholar] [PubMed]
- Doehner, W.; Schoene, N.; Rauchhaus, M.; Leyva-Leon, F.; Pavitt, D.V.; Reaveley, D.A.; Schuler, G.; Coats, A.J.; Anker, S.D.; Hambrecht, R. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: Results from 2 placebo-controlled studies. Circulation 2002, 105, 2619–2624. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ruiz, F.; Calabozo, M.; Erauskin, G.G.; Ruibal, A.; Herrero-Beites, A.M. Renal underexcretion of uric acid is present in patients with apparent high urinary uric acid output. Arthritis Rheum. 2002, 47, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Anzai, N.; Ichida, K.; Jutabha, P.; Kimura, T.; Babu, E.; Jin, C.J.; Srivastava, S.; Kitamura, K.; Hisatome, I.; Endou, H.; et al. Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J. Biol. Chem. 2008, 283, 26834–26838. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Iharada, M.; Miyaji, T.; Fujimoto, T.; Hiasa, M.; Anzai, N.; Omote, H.; Moriyama, Y. Type 1 sodium-dependent phosphate transporter (SLC17A1 Protein) is a Cl(-)-dependent urate exporter. J. Biol. Chem. 2010, 285, 26107–26113. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Tamai, I.; Yamashita, K.; Minemoto, Y.; Sai, Y.; Yabuuchi, H.; Miyamoto, K.; Takeda, E.; Tsuji, A. p-Aminohippuric acid transport at renal apical membrane mediated by human inorganic phosphate transporter NPT1. Biochem. Biophys. Res. Commun. 2000, 270, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Bardin, T. Gout. Lancet 2010, 375, 318–328. [Google Scholar] [CrossRef]
- Hosomi, A.; Nakanishi, T.; Fujita, T.; Tamai, I. Extra-renal elimination of uric acid via intestinal efflux transporter BCRP/ABCG2. PLoS ONE 2012, 7, e30456. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Jensen, U.; Hansen, A.; Covitz, K.M.; Zhan, Z.; Fetsch, P.; Abati, A.; Hansen, P.R.; Horn, T.; Skovsgaard, T.; et al. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim. Biophys. Acta 2002, 1565, 6–16. [Google Scholar] [CrossRef]
- Kage, K.; Tsukahara, S.; Sugiyama, T.; Asada, S.; Ishikawa, E.; Tsuruo, T.; Sugimoto, Y. Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int. J. Cancer 2002, 97, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Suzuki, H.; Sugiyama, Y. Characterization of polarized expression of point- or deletion-mutated human BCRP/ABCG2 in LLC-PK1 cells. Pharm. Res. 2005, 22, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Ogura, J.; Kuwayama, K.; Sasaki, S.; Kaneko, C.; Koizumi, T.; Yabe, K.; Tsujimoto, T.; Takeno, R.; Takaya, A.; Kobayashi, M.; et al. Reactive oxygen species derived from xanthine oxidase interrupt dimerization of breast cancer resistance protein, resulting in suppression of uric acid excretion to the intestinal lumen. Biochem. Pharmacol. 2015, 97, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ogura, J.; Kuwayama, K.; Takaya, A.; Terada, Y.; Tsujimoto, T.; Koizumi, T.; Maruyama, H.; Fujikawa, A.; Takahashi, N.; Kobayashi, M.; et al. Intestinal ischemia-reperfusion increases efflux for uric acid via paracellular route in the intestine, but decreases that via transcellular route mediated by BCRP. J. Pharm. Pharm. Sci. 2012, 15, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Johnson, R.J.; Miyazaki, H.; Endou, H. Molecular physiology of urate transport. Physiology (Bethesda) 2005, 20, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.W.; Muzny, D.M.; Lee, C.C.; Caskey, C.T. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J. Mol. Evol. 1992, 34, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Gao, X.; Curhan, G. Vitamin C intake and the risk of gout in men: A prospective study. Arch. Intern. Med. 2009, 169, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Berger, L.; Gerson, C.D.; Yu, T.F. The effect of ascorbic acid on uric acid excretion with a commentary on the renal handling of ascorbic acid. Am. J. Med. 1977, 62, 71–76. [Google Scholar] [CrossRef]
- Stein, H.B.; Hasan, A.; Fox, I.H. Ascorbic acid-induced uricosuria. A consequency of megavitamin therapy. Ann. Intern. Med. 1976, 84, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Shin, H.S.; Choi, H.S.; Park, J.W.; Jo, I.; Oh, E.S.; Lee, K.Y.; Lee, B.H.; Johnson, R.J.; Kang, D.H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig. 2014, 94, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Bedir, A.; Topbas, M.; Tanyeri, F.; Alvur, M.; Arik, N. Leptin might be a regulator of serum uric acid concentrations in humans. Jpn. Heart J. 2003, 44, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Tzameli, I.; Pissios, P.; Rovira, I.; Gavrilova, O.; Ohtsubo, T.; Chen, Z.; Finkel, T.; Flier, J.S.; Friedman, J.M. Xanthine oxidoreductase is a regulator of adipogenesis and PPARgamma activity. Cell Metab. 2007, 5, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Fruehwald-Schultes, B.; Peters, A.; Kern, W.; Beyer, J.; Pfutzner, A. Serum leptin is associated with serum uric acid concentrations in humans. Metabolism 1999, 48, 677–680. [Google Scholar] [CrossRef]
- Matsuura, F.; Yamashita, S.; Nakamura, T.; Nishida, M.; Nozaki, S.; Funahashi, T.; Matsuzawa, Y. Effect of visceral fat accumulation on uric acid metabolism in male obese subjects: Visceral fat obesity is linked more closely to overproduction of uric acid than subcutaneous fat obesity. Metabolism 1998, 47, 929–933. [Google Scholar] [CrossRef]
- Romao, I.; Roth, J. Genetic and environmental interactions in obesity and type 2 diabetes. J. Am. Diet. Assoc. 2008, 108 (4 Suppl. 1S), S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Atkinson, K.; Karlson, E.W.; Willett, W.; Curhan, G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N. Engl. J. Med. 2004, 350, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Ford, E.S.; Gao, X.; Choi, H.K. Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: The Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2008, 59, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Bleich, S.N.; Gortmaker, S.L. Increasing caloric contribution from sugar-sweetened beverages and 100% fruit juices among US children and adolescents, 1988–2004. Pediatrics 2008, 121, e1604–e1614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; van Eys, J. Nutritional significance of fructose and sugar alcohols. Annu. Rev. Nutr. 1981, 1, 437–475. [Google Scholar] [CrossRef] [PubMed]
- Hanover, L.M.; White, J.S. Manufacturing, composition, and applications of fructose. Am. J. Clin. Nutr. 1993, 58 (Suppl. S5), 724S–732S. [Google Scholar] [PubMed]
- Lustig, R.H. Fructose: Metabolic, hedonic, and societal parallels with ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, A.; Niquet-Leridon, C.; Boulanger, E.; Tessier, F.J. How Can Diet Affect the Accumulation of Advanced Glycation End-Products in the Human Body? Foods 2016, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Bunn, H.F.; Higgins, P.J. Reaction of monosaccharides with proteins: Possible evolutionary significance. Science 1981, 213, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Suarez, G.; Rajaram, R.; Oronsky, A.L.; Gawinowicz, M.A. Nonenzymatic glycation of bovine serum albumin by fructose (fructation). Comparison with the Maillard reaction initiated by glucose. J. Biol. Chem. 1989, 264, 3674–3679. [Google Scholar] [PubMed]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660. [Google Scholar] [PubMed]
- Kaneko, C.; Ogura, J.; Sasaki, S.; Okamoto, K.; Kobayashi, M.; Kuwayama, K.; Narumi, K.; Iseki, K. Fructose suppresses uric acid excretion to the intestinal lumen as a result of the induction of oxidative stress by NADPH oxidase activation. Biochim. Biophys. Acta 2017, 1861, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G.; Bronfman, M.; Vanneste, R.; Hers, H.G. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem. J. 1977, 162, 601–609. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.P.; Burini, R.C. High plasma uric acid concentration: Causes and consequences. Diabetol. Metab. Syndr. 2012, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Brosh, S.; Boer, P.; Sperling, O. Effects of fructose on synthesis and degradation of purine nucleotides in isolated rat hepatocytes. Biochim. Biophys. Acta 1982, 717, 459–464. [Google Scholar] [CrossRef]
- Emmerson, B.T. Effect of oral fructose on urate production. Ann. Rheum. Dis. 1974, 33, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: Implications for uncoupling endothelial nitric oxide synthase. Biochem. Pharmacol. 2005, 70, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Morre, J.T.; Beckman, J.S. Triuret: A novel product of peroxynitrite-mediated oxidation of urate. Arch Biochem. Biophys. 2004, 423, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Holme, I.; Aastveit, A.H.; Hammar, N.; Jungner, I.; Walldius, G. Uric acid and risk of myocardial infarction, stroke and congestive heart failure in 417,734 men and women in the Apolipoprotein MOrtality RISk study (AMORIS). J. Intern. Med. 2009, 266, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Park, S.K.; Lee, I.K.; Johnson, R.J. Uric acid-induced C-reactive protein expression: Implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 2005, 16, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.A.; Sanchez-Lozada, L.G.; Johnson, R.J.; Kang, D.H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Ratto, E.; Villaggio, B.; Parodi, E.L.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS ONE 2014, 9, e115210. [Google Scholar] [CrossRef] [PubMed]
- De Souto Padron, F.A.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J. Biol. Chem. 2015, 290, 13427–13439. [Google Scholar] [CrossRef] [PubMed]
- Streeter, J.; Thiel, W.; Brieger, K.; Miller, F.J., Jr. Opportunity nox: The future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc. Ther. 2013, 31, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Kang, D.H. Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin. Nephrol. 2005, 25, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Prata, C.; Grasso, C.; Loizzo, S.; Vieceli Dalla Sega, F.; Caliceti, C.; Zambonin, L.; Fiorentini, D.; Hakim, G.; Berridge, M.V.; Landi, L. Inhibition of trans-plasma membrane electron transport: A potential anti-leukemic strategy. Leuk. Res. 2010, 34, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Hilgefort, J.; Banks, G.; Gilliam, C.; Stevens, S.; Ansinelli, H.A.; Getty, M.; Abraham, N.G.; Shapiro, J.I.; Khitan, Z. Uric Acid-Induced Adipocyte Dysfunction Is Attenuated by HO-1 Upregulation: Potential Role of Antioxidant Therapy to Target Obesity. Stem Cells Int. 2016, 2016, 8197325. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Koster, R.; Chumley, P.; Heitzer, T.; Rudolph, V.; Ostad, M.A.; Warnholtz, A.; Staude, H.J.; Thuneke, F.; Koss, K.; et al. Oxypurinol improves coronary and peripheral endothelial function in patients with coronary artery disease. Free Radic. Biol. Med. 2005, 39, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Price, K.L.; Sautin, Y.Y.; Long, D.A.; Zhang, L.; Miyazaki, H.; Mu, W.; Endou, H.; Johnson, R.J. Human vascular smooth muscle cells express a urate transporter. J. Am. Soc. Nephrol. 2006, 17, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, S.; Hisatome, I.; Kuwabara, M.; Niwa, K.; Maharani, N.; Kato, M.; Ogino, K.; Hamada, T.; Ninomiya, H.; Higashi, Y.; et al. Depletion of Uric Acid Due to SLC22A12 (URAT1) Loss-of-Function Mutation Causes Endothelial Dysfunction in Hypouricemia. Circ. J. 2015, 79, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.Y.; Zhu, X.Y.; Zhang, J.W.; Feng, X.R.; Wang, Y.C.; Liu, M.L. Uric acid promotes chemokine and adhesion molecule production in vascular endothelium via nuclear factor-kappa B signaling. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, B.; Brand, S.M. Uric acid and essential hypertension: The endothelial connection. J. Hypertens. 2016, 34, 2138–2139. [Google Scholar] [CrossRef] [PubMed]
- Puddu, P.; Puddu, G.M.; Cravero, E.; Vizioli, L.; Muscari, A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: Molecular mechanisms and clinical implications. J. Cardiol. 2012, 59, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Godber, B.L.; Doel, J.J.; Sapkota, G.P.; Blake, D.R.; Stevens, C.R.; Eisenthal, R.; Harrison, R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J. Biol. Chem. 2000, 275, 7757–7763. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Samouilov, A.; Liu, X.; Zweier, J.L. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrate reduction: Evaluation of its role in nitrite and nitric oxide generation in anoxic tissues. Biochemistry 2003, 42, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Millar, T.M.; Stevens, C.R.; Benjamin, N.; Eisenthal, R.; Harrison, R.; Blake, D.R. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998, 427, 225–228. [Google Scholar] [CrossRef]
- Hink, H.U.; Santanam, N.; Dikalov, S.; McCann, L.; Nguyen, A.D.; Parthasarathy, S.; Harrison, D.G.; Fukai, T. Peroxidase properties of extracellular superoxide dismutase: Role of uric acid in modulating in vivo activity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular superoxide dismutase. Int. J. Biochem. Cell Biol. 2005, 37, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.J.; Iribarren, C.; Gross, M.D.; Comstock, G.W.; Cutler, R.G. Uric acid and serum antioxidant capacity: A reaction to atherosclerosis? Atherosclerosis 2000, 148, 131–139. [Google Scholar] [CrossRef]
- Suzuki, T. Nitrosation of uric acid induced by nitric oxide under aerobic conditions. Nitric Oxide 2007, 16, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Fitzgerald, J.D.; Khanna, P.P.; Bae, S.; Singh, M.K.; Neogi, T.; Pillinger, M.H.; Merill, J.; Lee, S.; Prakash, S.; et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: Systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. (Hoboken) 2012, 64, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.I.; Soletsky, B.; Johnson, R.J. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: A randomized trial. JAMA 2008, 300, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pandya, B.J.; Choi, H.K. Prevalence of gout and hyperuricemia in the US general population: The National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011, 63, 3136–3141. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Keim, N.L.; et al. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr. Metab. 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.H.; Wang, C.; Li, J.M.; Zhang, D.M.; Kong, L.D. Allopurinol, rutin, and quercetin attenuate hyperuricemia and renal dysfunction in rats induced by fructose intake: Renal organic ion transporter involvement. Am. J. Physiol. Renal. Physiol. 2009, 297, F1080–F1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Sievenpiper, J.L.; de Souza, R.J.; Chiavaroli, L.; Ha, V.; Cozma, A.I.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Di Buono, M.; et al. The effects of fructose intake on serum uric acid vary among controlled dietary trials. J. Nutr. 2012, 142, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Bruun, J.M.; Maersk, M.; Belza, A.; Astrup, A.; Richelsen, B. Consumption of sucrose-sweetened soft drinks increases plasma levels of uric acid in overweight and obese subjects: A 6-month randomised controlled trial. Eur. J. Clin. Nutr. 2015, 69, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Medici, V.; Bremer, A.A.; Lee, V.; Lam, H.D.; Nunez, M.V.; Chen, G.X.; Keim, N.L.; Havel, P.J. A dose-response study of consuming high-fructose corn syrup-sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am. J. Clin. Nutr. 2015, 101, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Frye, R.F.; Rivard, C.J.; Cheng, J.; McFann, K.K.; Segal, M.S.; Johnson, R.J.; Johnson, J.A. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism 2012, 61, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Madero, M.; Rodríguez Castellanos, F.E.; Jalal, D.; Villalobos-Martín, M.; Salazar, J.; Vazquez-Rangel, A.; Johnson, R.J.; Sanchez-Lozada, L.G. A pilot study on the impact of a low fructose diet and allopurinol on clinic blood pressure among overweight and prehypertensive subjects: A randomized placebo controlled trial. J. Am. Soc. Hypertens. 2015, 9, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Moriarity, J.T.; Folsom, A.R.; Iribarren, C.; Nieto, F.J.; Rosamond, W.D. Serum uric acid and risk of coronary heart disease: Atherosclerosis Risk in Communities (ARIC) Study. Ann. Epidemiol. 2000, 10, 136–143. [Google Scholar] [CrossRef]
- Sakata, K.; Hashimoto, T.; Ueshima, H.; Okayama, A. Absence of an association between serum uric acid and mortality from cardiovascular disease: NIPPON DATA 80, 1980–1994. National Integrated Projects for Prospective Observation of Non-communicable Diseases and its Trend in the Aged. Eur. J. Epidemiol. 2001, 17, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Shaper, A.G.; Whincup, P.H. Serum urate and the risk of major coronary heart disease events. Heart 1997, 78, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.G.; Juzwishin, K.D.; Eiriksdottir, G.; Gudnason, V.; Danesh, J. Serum uric acid and coronary heart disease in 9,458 incident cases and 155,084 controls: Prospective study and meta-analysis. PLoS Med. 2005, 2, e76. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.D.; Brand, F.N.; Kannel, W.B.; Castelli, W.P. Gout and coronary heart disease: The Framingham Study. J. Clin. Epidemiol. 1988, 41, 237–242. [Google Scholar] [CrossRef]
- Waring, W.S.; Webb, D.J.; Maxwell, S.R. Uric acid as a risk factor for cardiovascular disease. QJM 2000, 93, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Pillinger, M.H.; Goldfarb, D.S.; Keenan, R.T. Gout and its comorbidities. Bull. NYU Hosp. Jt. Dis. 2010, 68, 199–203. [Google Scholar] [PubMed]
- Jayalath, V.H.; Sievenpiper, J.L.; de Souza, R.J.; Ha, V.; Mirrahimi, A.; Santaren, I.D.; Blanco, M.S.; Di, B.M.; Jenkins, A.L.; Leiter, L.A.; et al. Total fructose intake and risk of hypertension: A systematic review and meta-analysis of prospective cohorts. J. Am. Coll. Nutr. 2014, 33, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Jayalath, V.H.; de Souza, R.J.; Ha, V.; Mirrahimi, A.; Blanco-Mejia, S.; Di, B.M.; Jenkins, A.L.; Leiter, L.A.; Wolever, T.M.; Beyene, J.; et al. Sugar-sweetened beverage consumption and incident hypertension: A systematic review and meta-analysis of prospective cohorts. Am. J. Clin. Nutr. 2015, 102, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Dormi, A.; D’Addato, S.; Gaddi, A.V.; Borghi, C. Long-term effect of a dietary education program on postmenopausal cardiovascular risk and metabolic syndrome: The Brisighella Heart Study. J. Womens Health 2010, 19, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Rosticci, M.; Bove, M.; Fogacci, F.; Giovannini, M.; Urso, R.; D’Addato, S.; Borghi, C. Serum uric acid change and modification of blood pressure and fasting plasma glucose in an overall healthy population sample: Data from the Brisighella heart study. Ann. Med. 2017, 49, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Rosticci, M.; Cagnati, M.; Urso, R.; Scapagnini, G.; Morbini, M.; Grandi, E.; D’Addato, S.; Borghi, C. Serum uric acid and markers of low-density lipoprotein oxidation in nonsmoking healthy subjects: Data from the Brisighella Heart Study. Pol. Arch. Med. Wewn. 2014, 124, 661–668. [Google Scholar] [PubMed]
- Cicero, A.F.; Rosticci, M.; Fogacci, F.; Grandi, E.; D’Addato, S.; Borghi, C. High serum uric acid is associated to poorly controlled blood pressure and higher arterial stiffness in hypertensive subjects. Eur. J. Intern. Med. 2017, 37, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Desideri, G.; Grossi, G.; Urso, R.; Rosticci, M.; D’Addato, S.; Borghi, C. Serum uric acid and impaired cognitive function in a cohort of healthy young elderly: Data from the Brisighella Study. Intern. Emerg Med. 2015, 10, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Rosticci, M.; Parini, A.; Baronio, C.; D’Addato, S.; Borghi, C. Serum uric acid is inversely proportional to estimated stroke volume and cardiac output in a large sample of pharmacologically untreated subjects: Data from the Brisighella Heart Study. Intern. Emerg Med. 2014, 9, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Anderson, G.H.; Flickinger, B.D.; Williamson-Hughes, P.S.; Empie, M.W. Fructose and non-fructose sugar intakes in the US population and their associations with indicators of metabolic syndrome. Food Chem. Toxicol. 2011, 49, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Okafor, O.N.; Farrington, K.; Gorog, D.A. Allopurinol as a therapeutic option in cardiovascular disease. Pharmacol. Ther. 2016, 172, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Ghali, J.K.; Neuberg, G.W.; O’Connor, C.M.; Carson, P.E.; Levy, W.C. Uric acid level and allopurinol use as risk markers of mortality and morbidity in systolic heart failure. Am. Heart J. 2010, 160, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Mullerleile, K.; Chumley, P.; Steven, D.; Rudolph, V.; Lund, G.K.; Staude, H.J.; Stork, A.; Koster, R.; Kahler, J.; et al. Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic. Biol. Med. 2006, 41, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Ogino, K.; Kato, M.; Furuse, Y.; Kinugasa, Y.; Ishida, K.; Osaki, S.; Kinugawa, T.; Igawa, O.; Hisatome, I.; Shigemasa, C.; et al. Uric acid-lowering treatment with benzbromarone in patients with heart failure: A double-blind placebo-controlled crossover preliminary study. Circ. Heart Fail. 2010, 3, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, C.; Folsom, A.R.; Eckfeldt, J.H.; McGovern, P.G.; Nieto, F.J. Correlates of uric acid and its association with asymptomatic carotid atherosclerosis: The ARIC Study. Atherosclerosis Risk in Communities. Ann. Epidemiol. 1996, 6, 331–340. [Google Scholar] [CrossRef]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Borghi, C.; Cicero, A.F. Serum Uric Acid and Cardiometabolic Disease: Another Brick in the Wall? Hypertension 2017. [Google Scholar] [CrossRef] [PubMed]
- Bove, M.; Cicero, A.F.; Veronesi, M.; Borghi, C. An evidence-based review on urate-lowering treatments: Implications for optimal treatment of chronic hyperuricemia. Vasc. Health Risk Manag. 2017, 13, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, C.; Calabria, D.; Roda, A. A new sensitive and quantitative chemiluminescent assay to monitor intracellular xanthine oxidase activity for rapid screening of inhibitors in living endothelial cells. Anal. Bioanal. Chem. 2016, 408, 8755–8760. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caliceti, C.; Calabria, D.; Roda, A.; Cicero, A.F.G. Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients 2017, 9, 395. https://doi.org/10.3390/nu9040395
Caliceti C, Calabria D, Roda A, Cicero AFG. Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients. 2017; 9(4):395. https://doi.org/10.3390/nu9040395
Chicago/Turabian StyleCaliceti, Cristiana, Donato Calabria, Aldo Roda, and Arrigo F. G. Cicero. 2017. "Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review" Nutrients 9, no. 4: 395. https://doi.org/10.3390/nu9040395
APA StyleCaliceti, C., Calabria, D., Roda, A., & Cicero, A. F. G. (2017). Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients, 9(4), 395. https://doi.org/10.3390/nu9040395