Dietary Metabolites and Chronic Kidney Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
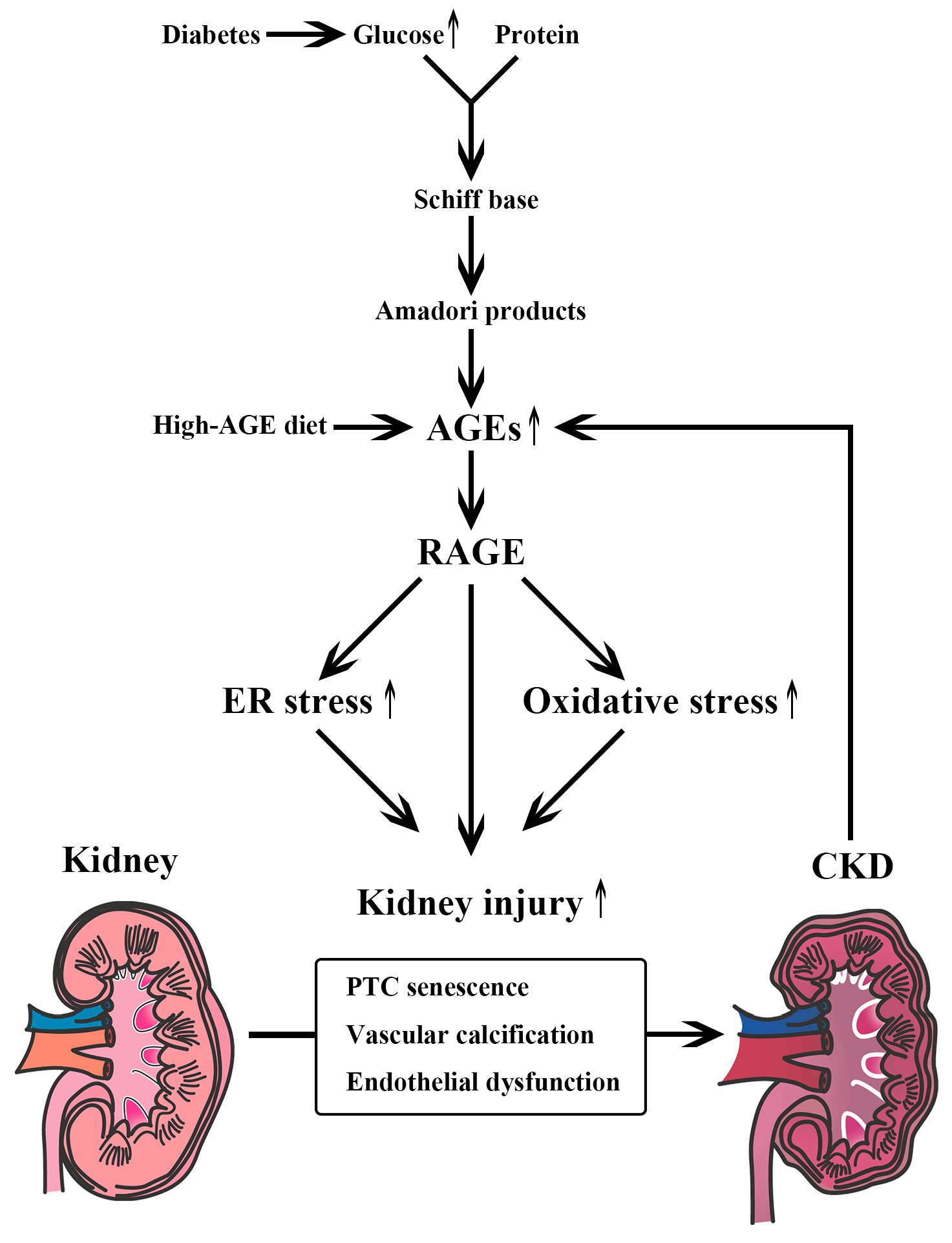
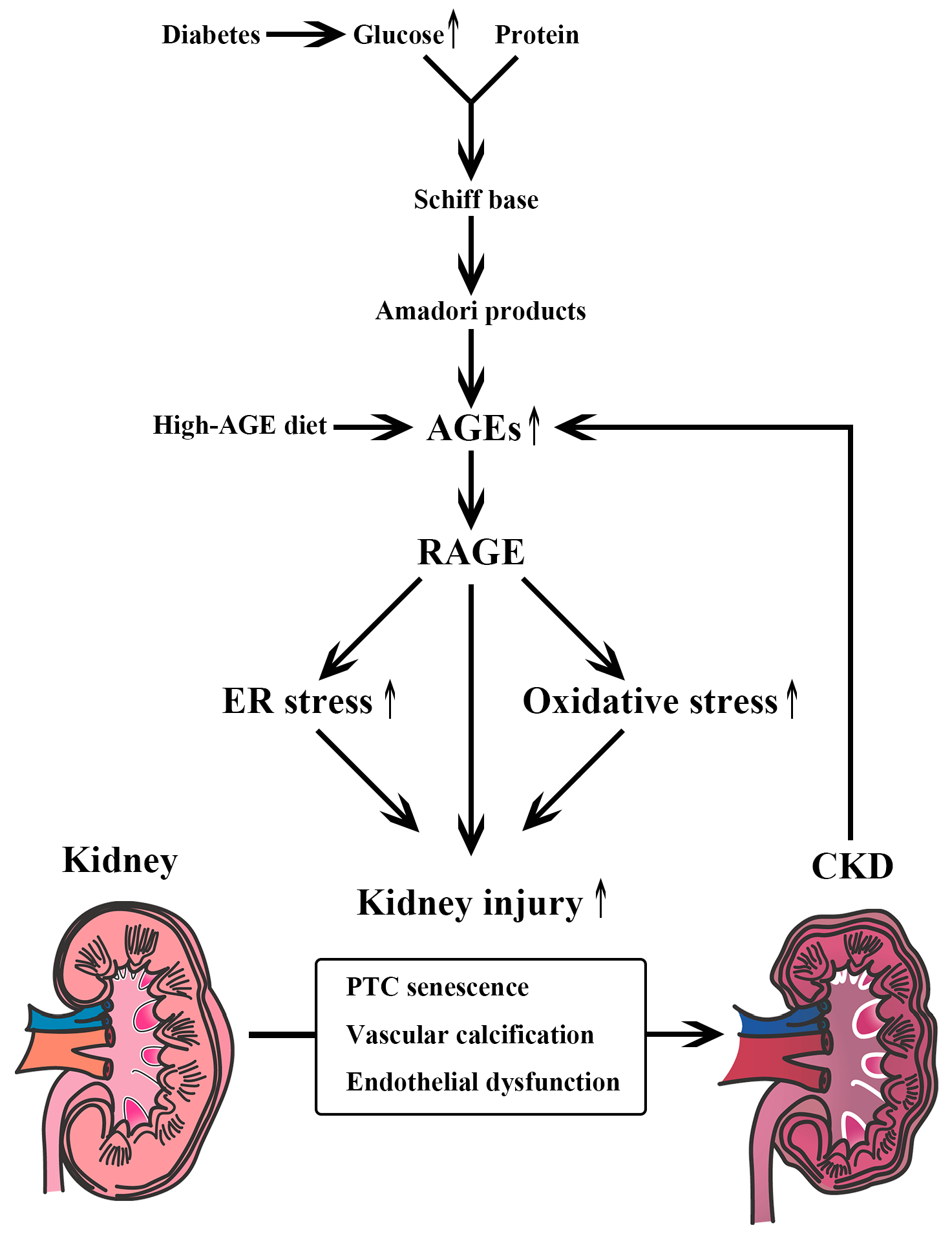
2. Carbohydrate Metabolism and CKD
3. Amino Acid Metabolism and CKD
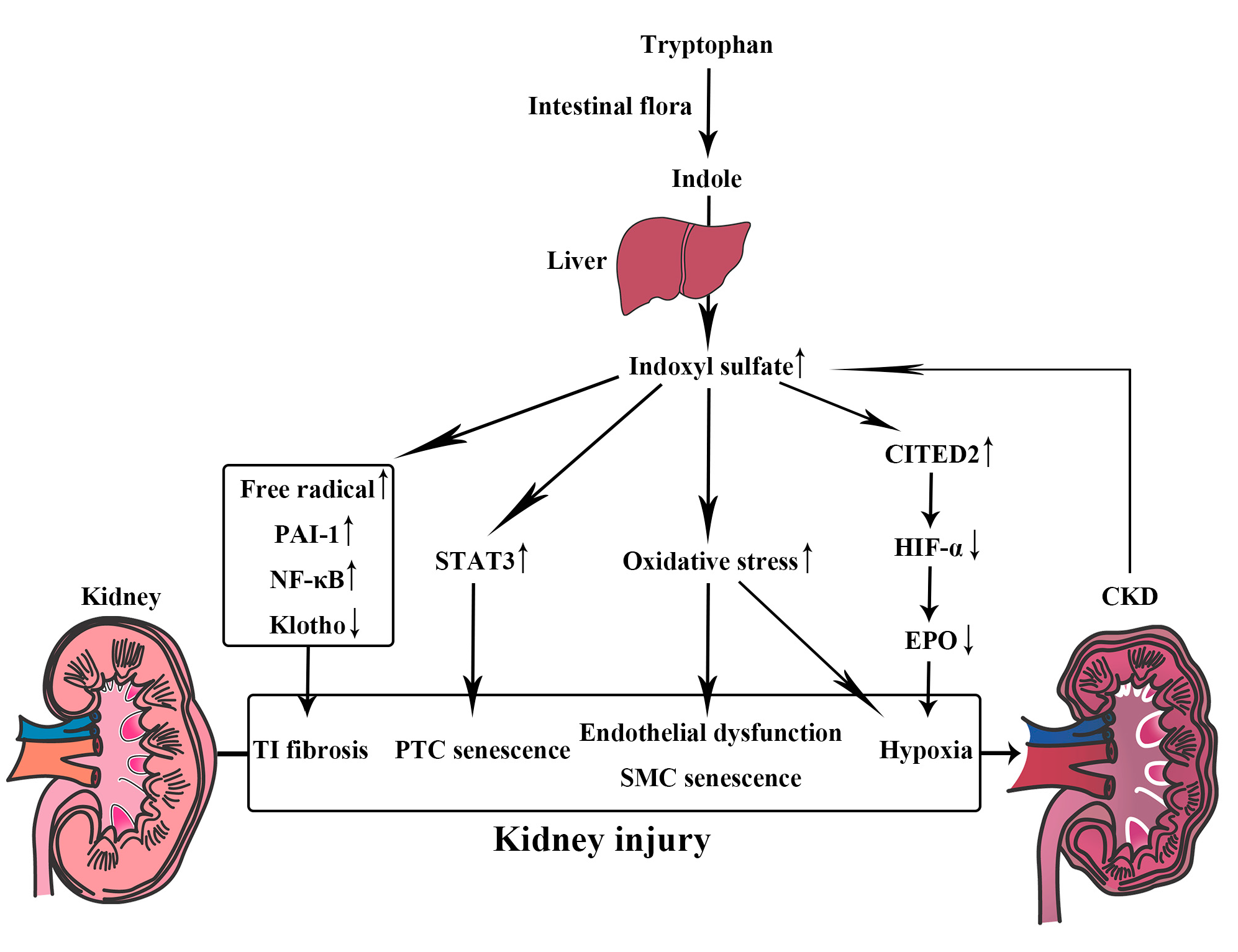
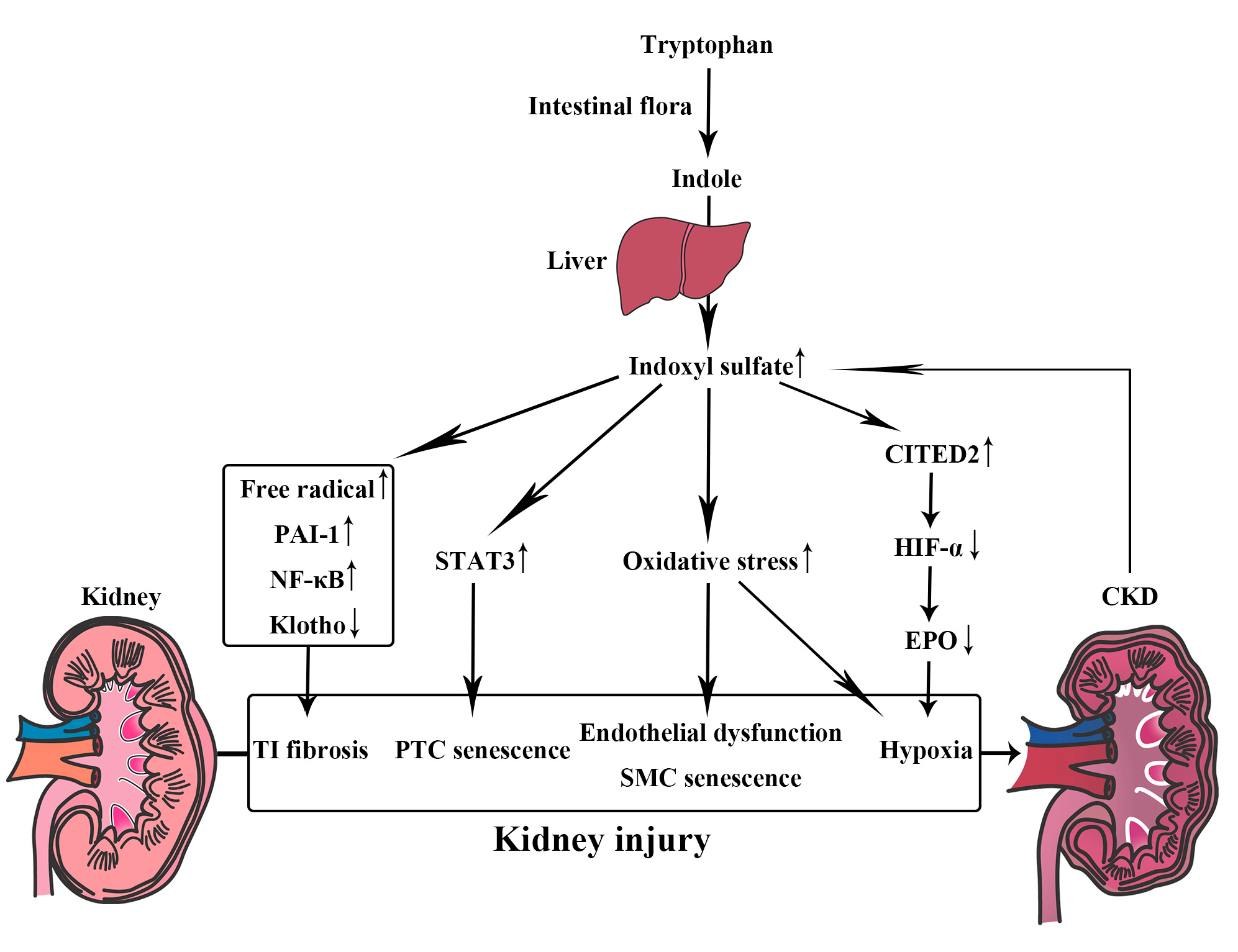
3.1. Indoxyl Sulfate, a Metabolite of Tryptophan
3.2. d-Amino Acids
4. Fatty Acid Profiles and CKD
5. Metabolic Acidosis and Nutrition
6. Therapeutic Targets Associated with Nutrition for Preventing CKD Progression
6.1. Dietary Intake Profile
6.2. Uremic Toxin Absorbent (AST-120)
6.3. AGE Formation Inhibitors
6.4. RAGE Inhibitors
6.5. Probiotics and Prebiotics
6.6. Nrf2 Pathway Activator
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Pollock, C.; Voss, D.; Hodson, E.; Crompton, C.; The CARI guidelines. Nutrition and growth in kidney disease. Nephrology 2005, 10, S177–S230. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Hayen, A.; Macaskill, P.; Pellegrini, F.; Craig, J.C.; Elder, G.J.; Strippoli, G.F. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA 2011, 305, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The gut as a source of inflammation in chronic kidney disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Maillard, L.C. Action of amino acids on sugars. Formation of melanoidins in a methodical way. Compt. Rend. 1912, 154, 66–68. [Google Scholar]
- Brownlee, M.; Vlassara, H.; Cerami, A. Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Ann. Intern. Med. 1984, 101, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.Y.; Cooper, M.E. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A.; Bendayan, M. Renal fate of circulating advanced glycated end products (AGE): Evidence for reabsorption and catabolism of age-peptides by renal proximal tubular cells. Diabetologia 1996, 39, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Ueda, Y.; Horie, K.; Nangaku, M.; Tanaka, S.; ele de Strihou, C.V.Y.; Kurokawa, K. Renal catabolism of advanced glycation end products: The fate of pentosidine. Kidney Int. 1998, 53, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic toxicity of advanced glycation end products in CKD. J. Am. Soc. Nephrol. 2016, 27, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Glycative stress and glyoxalase in kidney disease and aging. Biochem. Soc. Trans. 2014, 42, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Fuh, H.; Makita, Z.; Krungkrai, S.; Cerami, A.; Bucala, R. Exogenous advanced glycosylation end products induce complex vascular dysfunction in normal animals: A model for diabetic and aging complications. Proc. Natl. Acad. Sci. USA 1992, 89, 12043–12047. [Google Scholar] [CrossRef] [PubMed]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor rage in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [PubMed]
- Horie, K.; Miyata, T.; Maeda, K.; Miyata, S.; Sugiyama, S.; Sakai, H.; de Strihou, C.V.Y.; Monnier, V.M.; Witztum, J.L.; Kurokawa, K. Immunohistochemical colocalization of glycoxidation products and lipid peroxidation products in diabetic renal glomerular lesions. Implication for glycoxidative stress in the pathogenesis of diabetic nephropathy. J. Clin. Investig. 1997, 100, 2995–3004. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Yu, M.R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, S241–S245. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by age links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar] [PubMed]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, K.; Cai, G.Y.; Chen, X.M.; Yang, J.R.; Lin, L.R.; Yang, J.; Huo, B.G.; Zhan, J.; He, Y.N. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell. Signal. 2014, 26, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Inagi, R. Glycative stress and its defense machinery glyoxalase 1 in renal pathogenesis. Int. J. Mol. Sci. 2017, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Nangaku, M.; Kojima, I.; Nagai, R.; Ingelfinger, J.R.; Miyata, T.; Fujita, T.; Inagi, R. Glyoxalase I overexpression ameliorates renal ischemia-reperfusion injury in rats. Am. J. Physiol. Renal. Physiol. 2009, 296, F912–F921. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Ostergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Inagi, R.; Miyata, T.; Nagai, R.; Arai, M.; Miyashita, M.; Itokawa, M.; Fujita, T.; Nangaku, M. Glyoxalase I retards renal senescence. Am. J. Pathol. 2011, 179, 2810–2821. [Google Scholar] [CrossRef] [PubMed]
- Jo-Watanabe, A.; Ohse, T.; Nishimatsu, H.; Takahashi, M.; Ikeda, Y.; Wada, T.; Shirakawa, J.; Nagai, R.; Miyata, T.; Nagano, T.; et al. Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell 2014, 13, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. RAGE and glyoxalase in kidney disease. Glycoconj. J. 2016, 33, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.W.; Hsu, K.H.; Lee, C.C.; Sun, C.Y.; Hsu, H.J.; Tsai, C.J.; Tzen, C.Y.; Wang, Y.C.; Lin, C.Y.; Wu, M.S. P-cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Tanaka, T.; Nangaku, M. Dysregulated oxygen metabolism of the kidney by uremic toxins: Review. J. Ren. Nutr. 2012, 22, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Ohtsuki, S.; Otagiri, M.; Takanaga, H.; Asaba, H.; Mori, S.; Terasaki, T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002, 61, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar] [PubMed]
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-kappaB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Suppression of klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Stat3 contributes to indoxyl sulfate-induced inflammatory and fibrotic gene expression and cellular senescence. Am. J. Nephrol. 2012, 36, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol. Cell Physiol. 2012, 303, C126–C134. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Mimura, I.; Yamaguchi, J.; Higashijima, Y.; Wada, T.; Tanaka, T. Role of uremic toxins in erythropoiesis-stimulating agent resistance in chronic kidney disease and dialysis patients. J. Ren. Nutr. 2015, 25, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Tanaka, T.; Inagi, R.; Fujita, T.; Nangaku, M. Indoxyl sulfate, a representative uremic toxin, suppresses erythropoietin production in a HIF-dependent manner. Lab. Investig. 2011, 91, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Mothet, J.P.; Parent, A.T.; Wolosker, H.; Brady, R.O., Jr.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. d-serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Alt, J.; Ator, N.A.; Thomas, A.G.; Wu, Y.; Hin, N.; Wozniak, K.; Ferraris, D.; Rais, R.; Tsukamoto, T.; et al. d-amino-acid oxidase inhibition increases d-serine plasma levels in mouse but not in monkey or dog. Neuropsychopharmacology 2016, 41, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, P.K.; Chang, Y.C.; Chuo, L.J.; Chen, Y.S.; Tsai, G.E.; Lane, H.Y. Benzoate, a d-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: A randomized, double-blind, placebo-controlled trial. Biol. Psychiatry 2014, 75, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.W.; Volker, K.; Dantzler, W.H.; Silbernagl, S. Why is D-serine nephrotoxic and alpha-aminoisobutyric acid protective? Am. J. Physiol. Renal. Physiol. 2007, 293, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, J.; Suzuki, M.; Miyoshi, Y.; Tojo, Y.; Okamura, C.; Ito, S.; Konno, R.; Mita, M.; Hamase, K.; Aiso, S. Ischemic acute kidney injury perturbs homeostasis of serine enantiomers in the body fluid in mice: Early detection of renal dysfunction using the ratio of serine enantiomers. PLoS ONE 2014, 9, e86504. [Google Scholar] [CrossRef] [PubMed]
- Narayana, N.; Phillips, N.B.; Hua, Q.X.; Jia, W.; Weiss, M.A. Diabetes mellitus due to misfolding of a beta-cell transcription factor: Stereospecific frustration of a schellman motif in HNF-1alpha. J. Mol. Biol. 2006, 362, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Akino, T.; Ohno, K.; Kataoka, Y.; Ueda, T.; Sakurai, T.; Shiroshita, K.; Yasuda, T. Free d-amino acids in human plasma in relation to senescence and renal diseases. Clin. Sci. 1987, 73, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hamase, K.; Miyoshi, Y.; Yamamoto, R.; Yasuda, K.; Mita, M.; Rakugi, H.; Hayashi, T.; Isaka, Y. Chiral amino acid metabolomics for novel biomarker screening in the prognosis of chronic kidney disease. Sci. Rep. 2016, 6, 26137. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.V.; LeDoux, S.P.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA damage via augmented oxidative stress regulates endoplasmic reticulum stress and autophagy: Crosstalk, links and signaling. PLoS ONE 2013, 8, e83349. [Google Scholar] [CrossRef] [PubMed]
- Soumura, M.; Kume, S.; Isshiki, K.; Takeda, N.; Araki, S.; Tanaka, Y.; Sugimoto, T.; Chin-Kanasaki, M.; Nishio, Y.; Haneda, M.; et al. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem. Biophys. Res. Commun. 2010, 402, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Liu, J.; Kumar, G.; Skapek, S.X.; Falck, J.R.; Imig, J.D. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013, 27, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Kraut, J.A.; Madias, N.E. Metabolic acidosis of CKD: An update. Am. J. Kidney Dis. 2016, 67, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Bellasi, A.; Di Micco, L.; Santoro, D.; Marzocco, S.; De Simone, E.; Cozzolino, M.; Di Lullo, L.; Guastaferro, P.; Di Iorio, B. Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L.; Murphy, R.A.; Shlipak, M.G.; Satterfield, S.; Huston, H.K.; Sebastian, A.; Sellmeyer, D.E.; Patel, K.V.; Newman, A.B.; Sarnak, M.J.; et al. Bicarbonate concentration, acid-base status, and mortality in the health, aging, and body composition study. Clin. J. Am. Soc. Nephrol. 2016, 11, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E.; Remuzzi, G. Diets for patients with chronic kidney disease, should we reconsider? BMC Nephrol. 2016, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, C.M.; Coresh, J.; Grams, M.E.; Steffen, L.M.; Anderson, C.A.; Appel, L.J.; Crews, D.C. Dietary acid load and incident chronic kidney disease: Results from the ARIC study. Am. J. Nephrol. 2015, 42, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am. J. Kidney Dis. 2000, 35, S1–S140.
- Raphael, K.L. Approach to the treatment of chronic metabolic acidosis in CKD. Am. J. Kidney Dis. 2016, 67, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Di Micco, L.; Marzocco, S.; De Simone, E.; De Blasio, A.; Sirico, M.L.; Nardone, L.; On Behalf of Ubi Study Group. Very low-protein diet (VLPD) reduces metabolic acidosis in subjects with chronic kidney disease: The “nutritional light signal” of the renal acid load. Nutrients 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bugel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: Role of the antiinflammatory AGE receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483–4491. [Google Scholar] [CrossRef] [PubMed]
- Maschio, G.; Oldrizzi, L.; Tessitore, N.; D’Angelo, A.; Valvo, E.; Lupo, A.; Loschiavo, C.; Fabris, A.; Gammaro, L.; Rugiu, C.; et al. Effects of dietary protein and phosphorus restriction on the progression of early renal failure. Kidney Int. 1982, 22, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Zeller, K.; Whittaker, E.; Sullivan, L.; Raskin, P.; Jacobson, H.R. Effect of restricting dietary protein on the progression of renal failure in patients with insulin-dependent diabetes mellitus. N. Engl. J. Med. 1991, 324, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Dal Piaz, F.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Tartaglia, D.; Autore, G.; Di Iorio, B. Very low protein diet reduces indoxyl sulfate levels in chronic kidney disease. Blood Purif. 2013, 35, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Klahr, S.; Levey, A.S.; Beck, G.J.; Caggiula, A.W.; Hunsicker, L.; Kusek, J.W.; Striker, G. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of diet in renal disease study group. N. Engl. J. Med. 1994, 330, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Kopple, J.D.; Wang, X.; Beck, G.J.; Collins, A.J.; Kusek, J.W.; Greene, T.; Levey, A.S.; Sarnak, M.J. Effect of a very low-protein diet on outcomes: Long-term follow-up of the modification of diet in renal disease (mdrd) study. Am. J. Kidney Dis. 2009, 53, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Greene, T.; Beck, G.J.; Caggiula, A.W.; Kusek, J.W.; Hunsicker, L.G.; Klahr, S. Dietary protein restriction and the progression of chronic renal disease: What have all of the results of the mdrd study shown? Modification of diet in renal disease study group. J. Am. Soc. Nephrol. 1999, 10, 2426–2439. [Google Scholar] [PubMed]
- Friedman, A.N.; Moe, S.M.; Perkins, S.M.; Li, Y.; Watkins, B.A. Fish consumption and omega-3 fatty acid status and determinants in long-term hemodialysis. Am. J. Kidney Dis. 2006, 47, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Noori, N.; Dukkipati, R.; Kovesdy, C.P.; Sim, J.J.; Feroze, U.; Murali, S.B.; Bross, R.; Benner, D.; Kopple, J.D.; Kalantar-Zadeh, K. Dietary omega-3 fatty acid, ratio of omega-6 to omega-3 intake, inflammation, and survival in long-term hemodialysis patients. Am. J. Kidney Dis. 2011, 58, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Skeaff, C.M.; Fielding, B.A. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res. 2008, 47, 348–380. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sjogren, P.; Cederholm, T.; Arnlov, J.; Lindholm, B.; Riserus, U.; Carrero, J.J. Serum and adipose tissue fatty acid composition as biomarkers of habitual dietary fat intake in elderly men with chronic kidney disease. Nephrol. Dial. Transplant. 2014, 29, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Tanaka, T.; Inagi, R. Effect of AST-120 in chronic kidney disease treatment: Still a controversy. Nephron 2016, 135, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Yamagishi, S.; Takeuchi, M.; Kohno, K.; Shibata, R.; Matsumoto, Y.; Kaneyuki, U.; Fujimura, T.; Hayashida, A.; Okuda, S. Oral adsorbent AST-120 decreases serum levels of ages in patients with chronic renal failure. Mol. Med. 2006, 12, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl sulfate, a uremic toxin, downregulates renal expression of Nrf2 through activation of NF-kappaB. BMC Nephrol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Sanaka, T.; Sugino, N.; Teraoka, S.; Ota, K. Therapeutic effects of oral sorbent in undialyzed uremia. Am. J. Kidney Dis. 1988, 12, 97–103. [Google Scholar] [CrossRef]
- Ueda, H.; Shibahara, N.; Takagi, S.; Inoue, T.; Katsuoka, Y. AST-120, an oral adsorbent, delays the initiation of dialysis in patients with chronic kidney diseases. Ther. Apher. Dial. 2007, 11, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized placebo-controlled EPPIC trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Akizawa, T.; Asano, Y.; Morita, S.; Wakita, T.; Onishi, Y.; Fukuhara, S.; Gejyo, F.; Matsuo, S.; Yorioka, N.; Kurokawa, K.; et al. Effect of a carbonaceous oral adsorbent on the progression of CKD: A multicenter, randomized, controlled trial. Am. J. Kidney Dis. 2009, 54, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Shimizu, M.; Shobu, Y.; Kikuchi, M. The effects of AST-120 on chronic kidney disease progression in the United States of America: A post hoc subgroup analysis of randomized controlled trials. BMC Nephrol. 2016, 17, 141. [Google Scholar] [CrossRef] [PubMed]
- Nagai, R.; Murray, D.B.; Metz, T.O.; Baynes, J.W. Chelation: A fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 2012, 61, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Voziyan, P.A.; Hudson, B.G. Pyridoxamine: The many virtues of a maillard reaction inhibitor. Ann. N. Y. Acad. Sci. 2005, 1043, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Voziyan, P.A.; Hudson, B.G. Pyridoxamine as a multifunctional pharmaceutical: Targeting pathogenic glycation and oxidative damage. Cell. Mol. Life Sci. 2005, 62, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, M.; Gohda, T.; Kaneko, S.; Hagiwara, S.; Murakoshi, M.; Aoki, T.; Yamada, K.; Ito, T.; Matsumoto, M.; Horikoshi, S.; et al. Effect of pyridoxamine (K-163), an inhibitor of advanced glycation end products, on type 2 diabetic nephropathy in KK-A(y)/Ta mice. Metabolism 2007, 56, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; van Ypersele de Strihou, C.; Ueda, Y.; Ichimori, K.; Inagi, R.; Onogi, H.; Ishikawa, N.; Nangaku, M.; Kurokawa, K. Angiotensin II receptor antagonists and angiotensin-converting enzyme inhibitors lower in vitro the formation of advanced glycation end products: Biochemical mechanisms. J. Am. Soc. Nephrol. 2002, 13, 2478–2487. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; van Ypersele de Strihou, C. Angiotensin II receptor blockers and angiotensin converting enzyme inhibitors: Implication of radical scavenging and transition metal chelation in inhibition of advanced glycation end product formation. Arch. Biochem. Biophys. 2003, 419, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S.; Yamagishi, S. Irbesartan inhibits advanced glycation end product-induced increase in asymmetric dimethylarginine level in mesangial cells through its anti-oxidative properties. Int. J. Cardiol. 2014, 176, 1120–1122. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Miyata, T.; Sada, T.; Mizuno, M.; Inagi, R.; Ueda, Y.; Ishikawa, N.; Yuzawa, H.; Koike, H.; van Ypersele de Strihou, C.; et al. Anti-hypertensive agents inhibit in vivo the formation of advanced glycation end products and improve renal damage in a type 2 diabetic nephropathy rat model. J. Am. Soc. Nephrol. 2003, 14, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Flyvbjerg, A.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Mogensen, T.H.; Paludan, S.R.; Rasch, R. Long-term renal effects of a neutralizing rage antibody in obese type 2 diabetic mice. Diabetes 2004, 53, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lapidos, K.A.; Gal-Moscovici, A.; Sprague, S.M.; Ameer, G.A. A receptor-based bioadsorbent to target advanced glycation end products in chronic kidney disease. Artif. Organs 2014, 38, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. The gut-kidney connection in advanced chronic kidney disease. Kidney Res. Clin. Pract. 2015, 34, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Mafra, D.; Fouque, D. Probiotics and chronic kidney disease. Kidney Int. 2015, 88, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Furuse, S.U.; Ohse, T.; Jo-Watanabe, A.; Shigehisa, A.; Kawakami, K.; Matsuki, T.; Chonan, O.; Nangaku, M. Galacto-oligosaccharides attenuate renal injury with microbiota modification. Physiol. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.; Kelly, J.; Tapsell, L. Dietary modeling of foods for advanced ckd based on general healthy eating guidelines: What should be on the plate? Am. J. Kidney Dis. 2017, 69, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Montemurno, E.; Cosola, C.; Dalfino, G.; Daidone, G.; De Angelis, M.; Gobbetti, M.; Gesualdo, L. What would you like to eat, mr ckd microbiota? A mediterranean diet, please! Kidney Blood Press. Res. 2014, 39, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Di Daniele, N.; Di Renzo, L.; Noce, A.; Iacopino, L.; Ferraro, P.M.; Rizzo, M.; Sarlo, F.; Domino, E.; De Lorenzo, A. Effects of italian mediterranean organic diet vs. Low-protein diet in nephropathic patients according to mthfr genotypes. J. Nephrol. 2014, 27, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jimenez-Moleon, J.J.; Lindholm, B.; Cederholm, T.; Arnlov, J.; Riserus, U.; Sjogren, P.; Carrero, J.J. Mediterranean diet, kidney function, and mortality in men with ckd. Clin. J. Am. Soc. Nephrol. 2013, 8, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Khatri, M.; Moon, Y.P.; Scarmeas, N.; Gu, Y.; Gardener, H.; Cheung, K.; Wright, C.B.; Sacco, R.L.; Nickolas, T.L.; Elkind, M.S. The association between a mediterranean-style diet and kidney function in the northern manhattan study cohort. Clin. J. Am. Soc. Nephrol. 2014, 9, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney. Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, S.; Jao, T.-M.; Inagi, R. Dietary Metabolites and Chronic Kidney Disease. Nutrients 2017, 9, 358. https://doi.org/10.3390/nu9040358
Hasegawa S, Jao T-M, Inagi R. Dietary Metabolites and Chronic Kidney Disease. Nutrients. 2017; 9(4):358. https://doi.org/10.3390/nu9040358
Chicago/Turabian StyleHasegawa, Sho, Tzu-Ming Jao, and Reiko Inagi. 2017. "Dietary Metabolites and Chronic Kidney Disease" Nutrients 9, no. 4: 358. https://doi.org/10.3390/nu9040358
APA StyleHasegawa, S., Jao, T.-M., & Inagi, R. (2017). Dietary Metabolites and Chronic Kidney Disease. Nutrients, 9(4), 358. https://doi.org/10.3390/nu9040358