Zinc as a Gatekeeper of Immune Function


Abstract
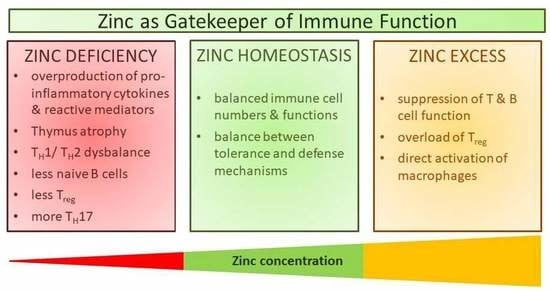
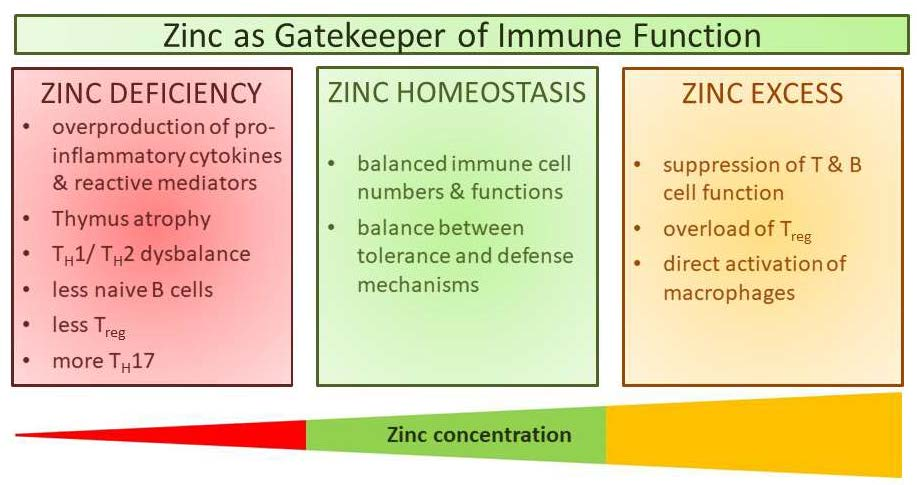{kind=link}
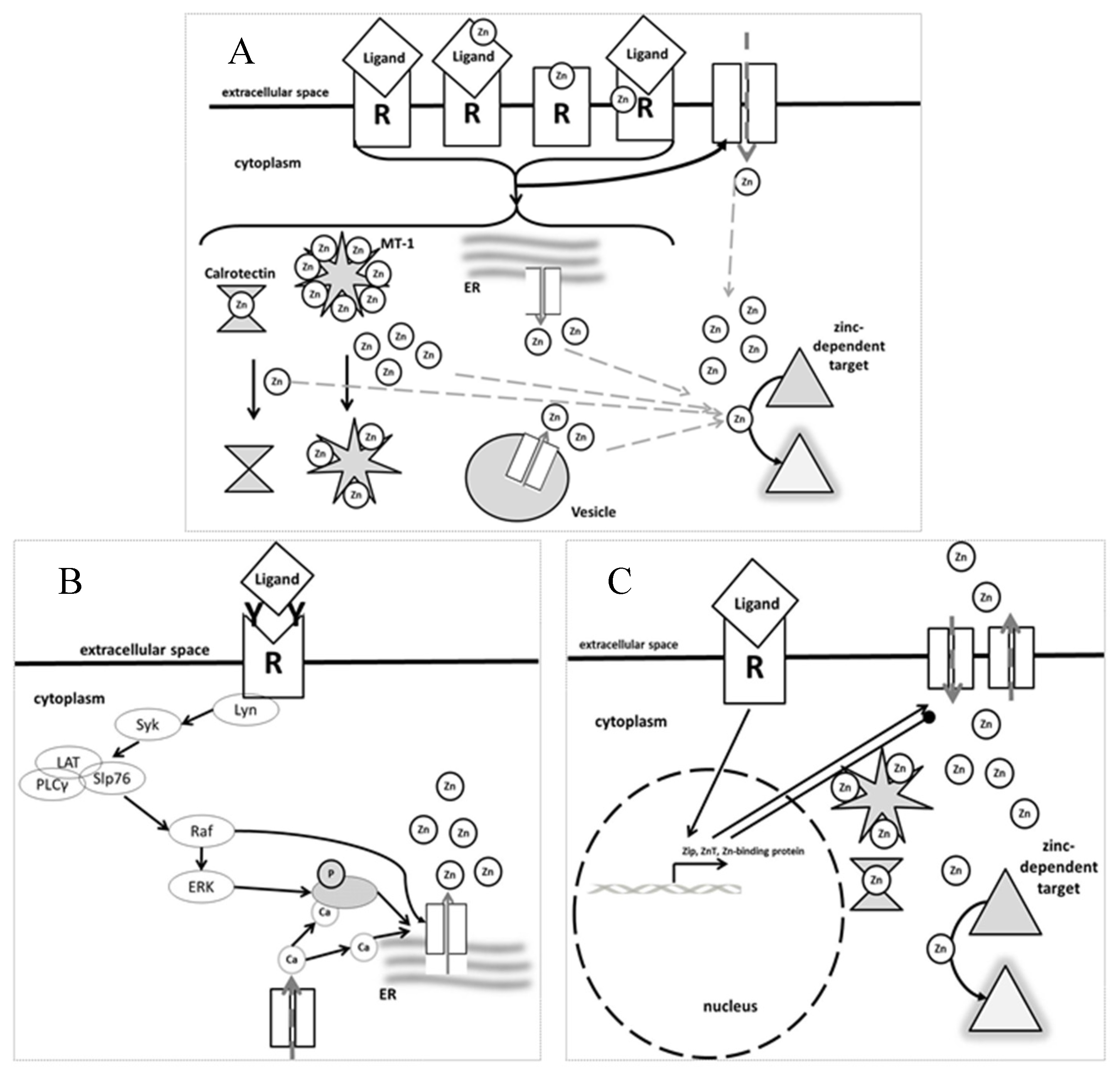{kind=link}
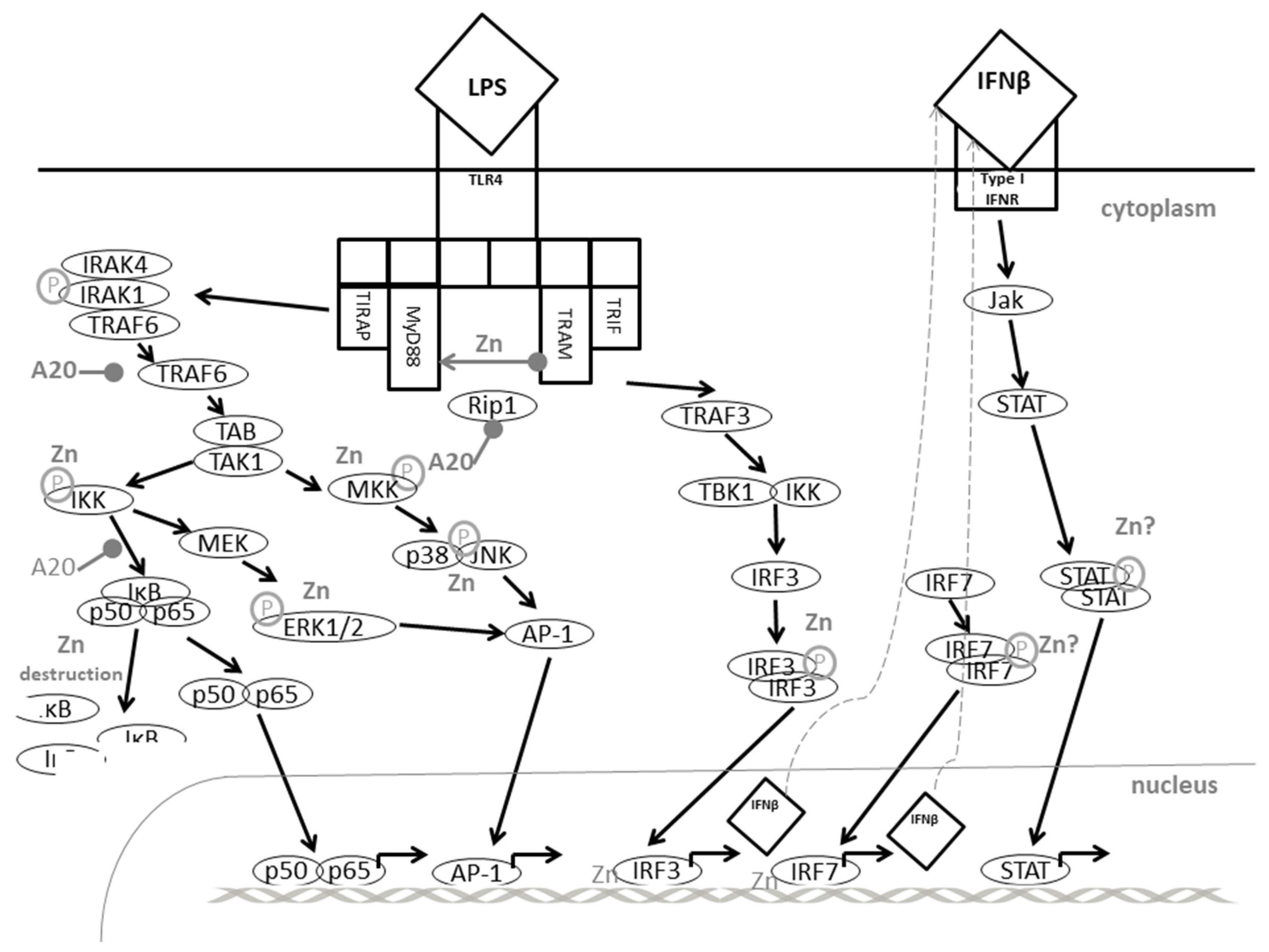{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Zinc Metabolism and Homeostasis
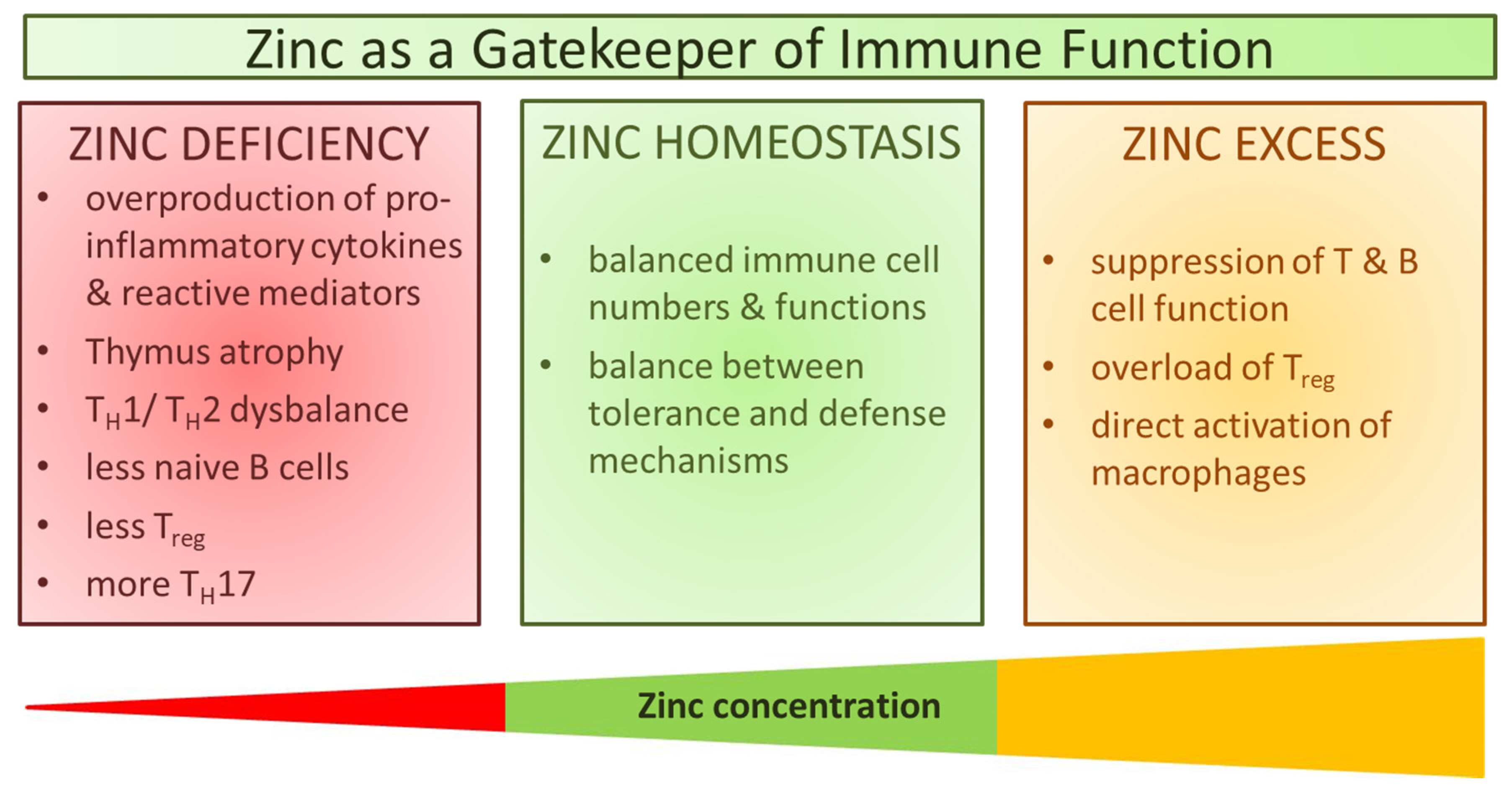
3. Immune Function during Zinc Deficiency
4. Types of Zinc Signals
5. Effects of Zinc in Immune Cell Signaling
5.1. Zinc Signals Can Regulate Phosphatase and Kinase Activities
5.2. Effects of Zinc Finger Proteins on Signal Transduction
5.3. Zinc Alters Hematopoiesis by Altering Intracellular Signaling
5.4. Zinc, Smad Signaling and the Development of Regulatory T Cells
5.5. PMA-Induced NET Formation in Granulocytes
5.6. TLR4-Induced Signaling: A Great Example of Fine-Tuning of Signaling Pathways by Zinc
5.7. Zinc Levels Regulate Fc Receptor-Induced Signaling
5.8. Killing Activity of NK Cells Varies with Zinc Availability
5.9. The Role of Zinc in IL-2-Induced Signaling in T Cells
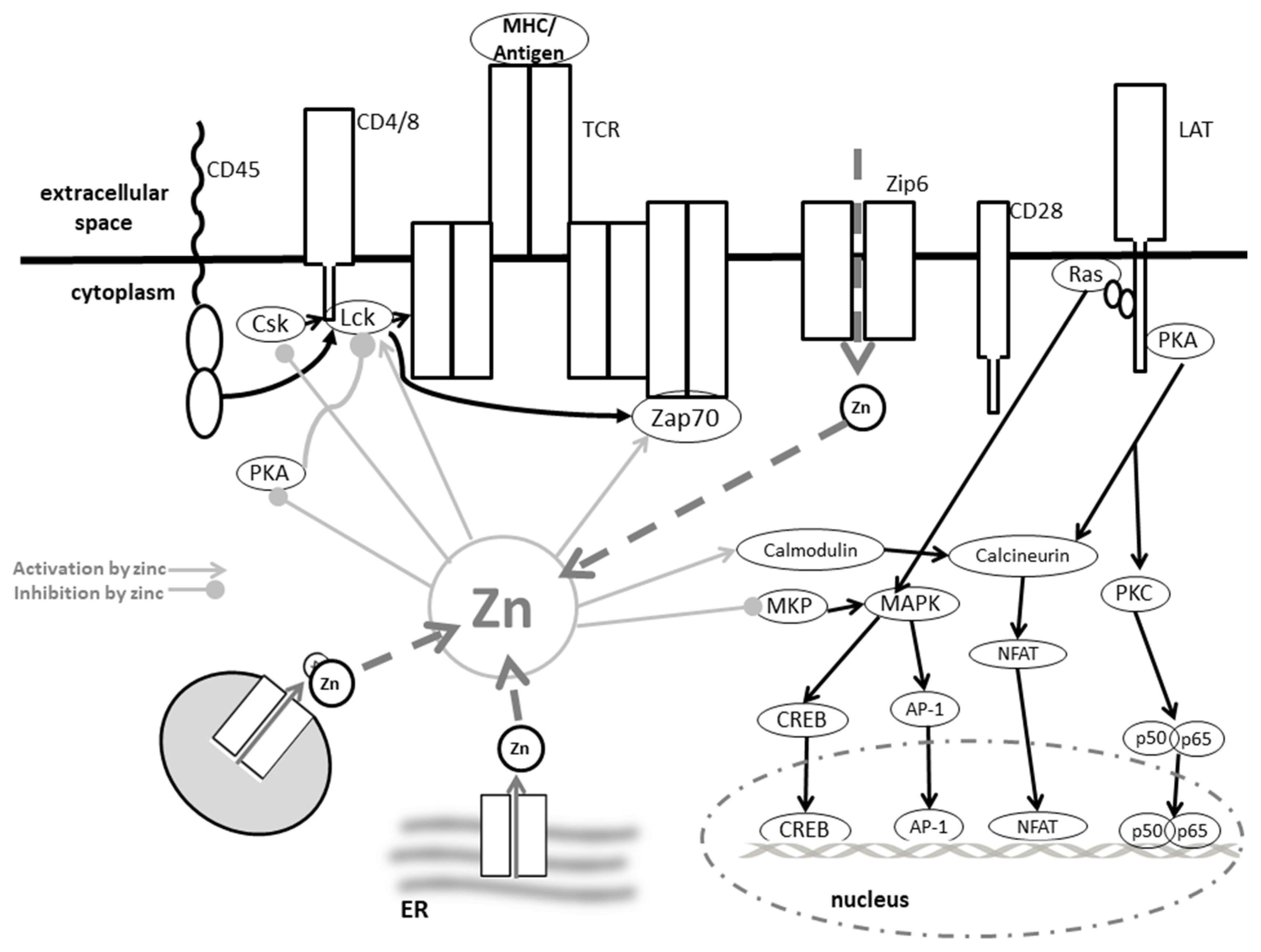
5.10. Zinc in T Cell Receptor Signaling
5.11. The Role of Zinc in IL-1 Receptor Signaling in T Cells
5.12. The Role of Zinc Signaling in B Cells
6. Zinc and Transcription Factors
7. Zinc, Epigenetics, and Immunity
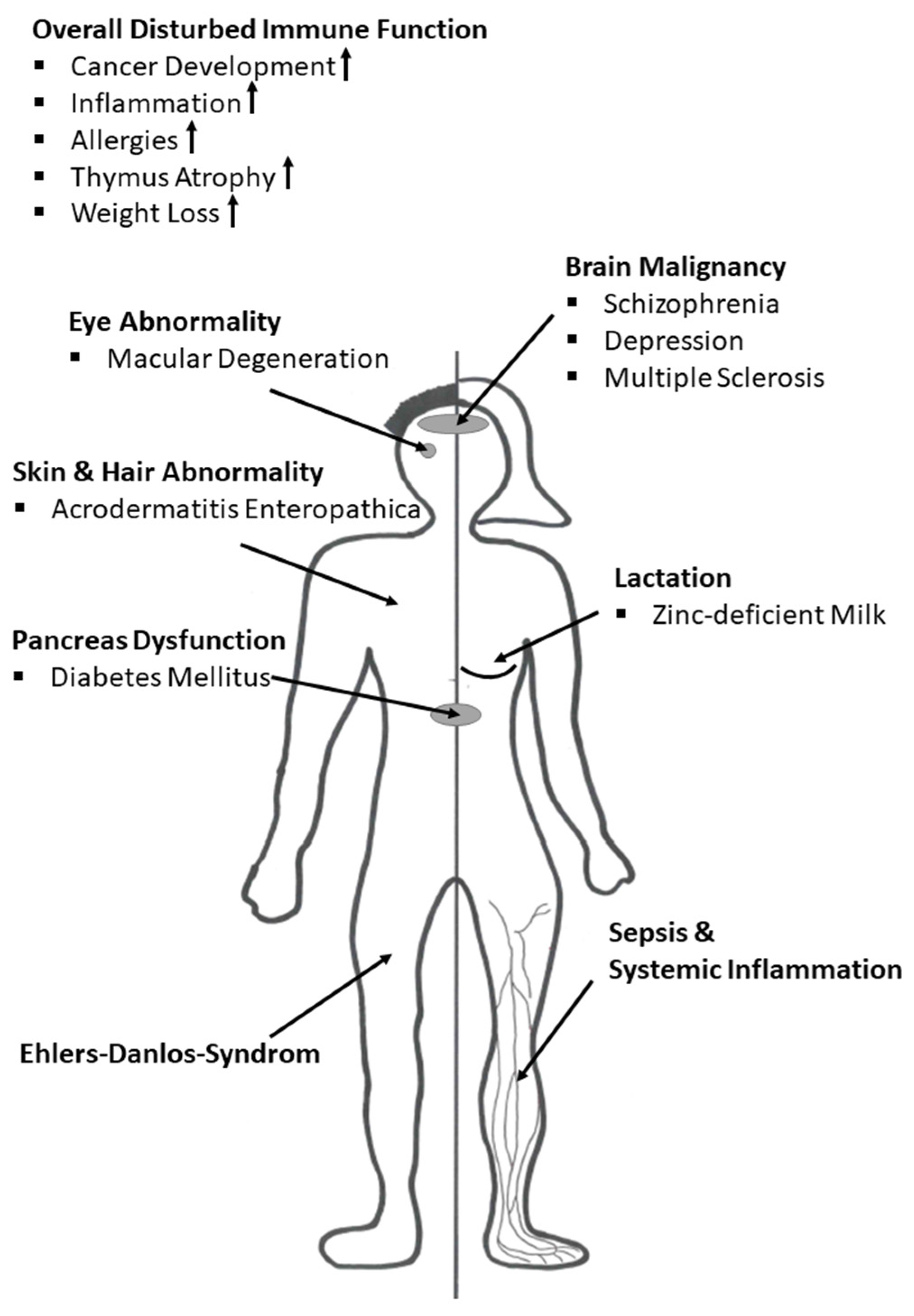
8. Zinc Deficiency in the Context of Disease
8.1. Zinc and Acrodermatitis Enteropathica
8.2. Zinc in Cancer Development
8.3. Zinc and Schizophrenia
8.4. Zinc and Depression
8.5. Zinc and Multiple Sclerosis
8.6. Zinc and Ehlers-Danlos Syndrome
8.7. Zinc in Inflammatory Disease and Sepsis
8.8. Zinc in Lactation
8.9. Zinc and Diabetes
8.10. Zinc and Alzheimer’s Disease
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Raulin, J. Chemical studies on vegetation. Ann. Sci. Nat. 1869, 11, 93–99. [Google Scholar]
- Todd, W.R.; Elvehjem, C.A.; Hart, E.B. Zinc in the nutrition of the rat. Am. J. Physiol. 1980, 107, 146–156. [Google Scholar] [CrossRef]
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Am. Coll. Nutr. 2009, 28, 257–265. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). The World Health Report. 83; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Rink, L. Zinc in Human Health; IOS Press: Amsterdam, The Netherlands, 2011; p. 596. [Google Scholar]
- Haase, H.; Mocchegiani, E.; Rink, L. Correlation between zinc status and immune function in the elderly. Biogerontology 2006, 7, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, H.; Luczkowski, M.; Remelli, M.; Valensin, D. Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord. Chem. Rev. 2012, 256, 2129–2141. [Google Scholar] [CrossRef]
- Chabosseau, P.; Rutter, G.A. Zinc and diabetes. Arch. Biochem. Biophys. 2016, 611, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Benedetti, G.; Albarede, F.; Miossec, P. Zinc and its role in immunity and inflammation. Autoimmun. Rev. 2015, 14, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Wapnir, R.A. Zinc deficiency, malnutrition and the gastrointestinal tract. J. Nutr. 2000, 130, 1388s–1392s. [Google Scholar] [PubMed]
- Bonomini, M.; Di Paolo, B.; De Risio, F.; Niri, L.; Klinkmann, H.; Ivanovich, P.; Albertazzi, A. Effects of zinc supplementation in chronic haemodialysis patients. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc. Eur. Renal Assoc. 1993, 8, 1166–1168. [Google Scholar]
- Lestienne, I.; Icard-Vernière, C.; Mouquet, C.; Picq, C.; Trèche, S. Effects of soaking whole cereal and legume seeds on iron, zinc and phytate contents. Food Chem. 2005, 89, 421–425. [Google Scholar] [CrossRef]
- Sandstead, H.H.; Prasad, A.S.; Schulert, A.R.; Farid, Z.; Miale, A.; Bassilly, S.; Darby, W.J. Human zinc deficiency, endocrine manifestations and response to treatment. Am. J. Clin. Nutr. 1967, 20, 422–442. [Google Scholar] [PubMed]
- Sandstead, H.H.; Penland, J.G.; Alcock, N.W.; Dayal, H.H.; Chen, X.C.; Li, J.S.; Zhao, F.; Yang, J.J. Effects of repletion with zinc and other micronutrients on neuropsychologic performance and growth of Chinese children. Am. J. Clin. Nutr. 1998, 68, 470S–475S. [Google Scholar] [PubMed]
- Prasad, A.S.; Miale, A., Jr.; Farid, Z.; Sandstead, H.H.; Schulert, A.R. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J. Lab. Clin. Med. 1963, 61, 537–549. [Google Scholar] [PubMed]
- Giacconi, R.; Costarelli, L.; Piacenza, F.; Basso, A.; Rink, L.; Mariani, E.; Fulop, T.; Dedoussis, G.; Herbein, G.; Provinciali, M.; et al. Main biomarkers associated with age-related plasma zinc decrease and copper/zinc ratio in healthy elderly from zincage study. Eur. J. Nutr. 2016, 58, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.K. Regulation and Function of ZIP4, the Acrodermatitis Enteropathica Gene; Portland Press Limited: London, UK, 2008. [Google Scholar]
- Wuehler, S.E.; Peerson, J.M.; Brown, K.H. Use of national food balance data to estimate the adequacy of zinc in national food supplies: Methodology and regional estimates. Public Health Nutr. 2005, 8, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Yang, X.; Cai, G.; Fan, D.; Xia, Q.; Liu, L.; Hu, Y.; Ding, N.; Xu, S.; Wang, L.; et al. Serum levels of copper and zinc in patients with rheumatoid arthritis: A meta-analysis. Biol. Trace Elem. Res. 2015, 168, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Lines, C.M.; McKay, E.C. Iron and zinc status in multiple sclerosis patients with pressure sores. Eur. J. Clin. Nutr. 1988, 42, 321–328. [Google Scholar] [PubMed]
- Socha, K.; Karpinska, E.; Kochanowicz, J.; Soroczynska, J.; Jakoniuk, M.; Wilkiel, M.; Mariak, Z.D.; Borawska, M.H. Dietary habits; concentration of copper, zinc, and cu-to-zn ratio in serum and ability status of patients with relapsing-remitting multiple sclerosis. Nutrition 2017, 39–40, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.; Frischer, J.M.; Webb, S.M.; Tham, M.; Adiele, R.C.; Robinson, C.A.; Fitz-Gibbon, P.D.; Weigand, S.D.; Metz, I.; Nehzati, S.; et al. Pathogenic implications of distinct patterns of iron and zinc in chronic ms lesions. Acta Neuropathol. 2017, 134, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Deibel, M.A.; Ehmann, W.D.; Markesbery, W.R. Copper, iron, and zinc imbalances in severely degenerated brain regions in alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 1996, 143, 137–142. [Google Scholar] [CrossRef]
- Flinn, J.M.; Kakalec, P.; Tappero, R.; Jones, B.; Lengyel, I. Correlations in distribution and concentration of calcium, copper and iron with zinc in isolated extracellular deposits associated with age-related macular degeneration. Metallomics Integr. Biomet. Sci. 2014, 6, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Lönnerdal, B. Dietary factors influencing zinc absorption. J. Nutr. 2000, 130, 1378S–1383S. [Google Scholar] [PubMed]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [PubMed]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 2001, 14, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L. Metallothionein: Historical review and perspectives. Exp. Suppl. 1979, 34, 19–39. [Google Scholar]
- Colvin, R.A.; Holmes, W.R.; Fontaine, C.P.; Maret, W. Cytosolic zinc buffering and muffling: Their role in intracellular zinc homeostasis. Metallomics Integr. Biomet. Sci. 2010, 2, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. The biological inorganic chemistry of zinc ions. Arch. Biochem. Biophys. 2016, 611, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.C.; Coles, J.A.; Deitmer, J.W. Homeostatic muffling. Nature 1991, 350, 564. [Google Scholar] [CrossRef] [PubMed]
- Colvin, R.A.; Bush, A.I.; Volitakis, I.; Fontaine, C.P.; Thomas, D.; Kikuchi, K.; Holmes, W.R. Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am. J. Physiol. Cell Physiol. 2008, 294, C726–C742. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. The regulatory and signaling functions of zinc ions in human cellular physiology. In Cellular and Molecular Biology of Metals; Zalups, R.K., Koropatnick, J., Eds.; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Kramer, K.K.; Zoelle, J.T.; Klaassen, C.D. Induction of metallothionein mrna and proteinin primary murine neuron cultures. Toxicol. Appl. Pharmacol. 1996, 141, 1–7. [Google Scholar] [CrossRef]
- Vasak, M.; Meloni, G. Mammalian metallothionein-3: New functional and structural insights. Int. J. Mol. Sci. 2017, 18, 1117. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, D.E.; Stillman, M.J. The “magic numbers” of metallothionein. Metallomics Integr. Biomet. Sci. 2011, 3, 444–463. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The functions of metallothionein and ZIP and ZnT transporters: An overview and perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Kambe, T. Molecular and genetic features of zinc transporters in physiology and pathogenesis. Metallomics Integr. Biomet. Sci. 2011, 3, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Kambe, T. Zinc Signals in Cellular Functions and Disorders; Springer: Tokyo, Japan, 2014. [Google Scholar]
- Vallee, B.L.; Galdes, A. The metallobiochemistry of zinc enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 1984, 56, 283–430. [Google Scholar] [PubMed]
- McCall, K.A.; Huang, C.; Fierke, C.A. Function and mechanism of zinc metalloenzymes. J. Nutr. 2000, 130, 1437S–1446S. [Google Scholar] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I. A bioinformatics view of zinc enzymes. J. Inorg. Biochem. 2012, 111, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A bioinformatic approach. Acc. Chem. Res. 2009, 42, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Rink, L. Functional significance of zinc-related signaling pathways in immune cells. Ann. Rev. Nutr. 2009, 29, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; Gershwin, M.E.; Good, R.A.; Prasad, A. Interrelationships between zinc and immune function. Fed. Proc. 1986, 45, 1474–1479. [Google Scholar] [PubMed]
- Prasad, A.S. Clinical manifestations of zinc deficiency. Ann. Rev. Nutr. 1985, 5, 341–363. [Google Scholar] [CrossRef] [PubMed]
- Bajait, C.; Thawani, V. Role of zinc in pediatric diarrhea. Indian J. Pharmacol. 2011, 43, 232. [Google Scholar] [PubMed]
- Bode, C.; Bode, J.C. Effect of alcohol consumption on the gut. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 575–592. [Google Scholar] [CrossRef]
- Küry, S.; Dréno, B.; Bézieau, S.; Giraudet, S.; Kharfi, M.; Kamoun, R.; Moisan, J.-P. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat. Genet. 2002, 31, 239. [Google Scholar] [CrossRef] [PubMed]
- Neldner, K.H.; Hambidge, K.M. Zinc therapy of acrodermatitis enteropathica. N. Engl. J. Med. 1975, 292, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.I.; Perri, R.T.; McClain, C.J.; Kay, N.E. Alterations in human natural killer cell activity and monocyte cytotoxicity induced by zinc deficiency. J. Lab. Clin. Med. 1983, 102, 577–589. [Google Scholar] [PubMed]
- Miller, L.V.; Krebs, N.F.; Hambidge, K.M. A mathematical model of zinc absorption in humans as a function of dietary zinc and phytate. J. Nutr. 2007, 137, 135–141. [Google Scholar] [PubMed]
- Maywald, M.; Rink, L. Zinc homeostasis and immunosenescence. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2015, 29, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Fitzgerald, J.; Hess, J.; Kaplan, J.; Pelen, F.; Dardenne, M. Zinc deficiency in elderly patients. Nutrition 1993, 9, 218–224. [Google Scholar] [PubMed]
- Weyand, C.M.; Goronzy, J.J. Aging of the immune system. Mechanisms and therapeutic targets. Ann. Am. Thorac. Soc. 2016, 13, S422–S428. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Hill, G.M.; Prasad, A.S.; Cossack, Z.T.; Rabbani, P. Oral zinc therapy for Wilson’s disease. Ann. Intern. Med. 1983, 99, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Newsome, D.A.; Miceli, M.V.; Tate, D.J.; Alcock, N.W.; Oliver, P.D. Zinc content of human retinal pigment epithelium decreases with age and macular degeneration, but superoxide dismutase activity increases. J. Trace Elem. Exp. Med. 1996, 8, 193–199. [Google Scholar] [CrossRef]
- Newsome, D.A.; Swartz, M.; Leone, N.C.; Elston, R.C.; Miller, E. Oral zinc in macular degeneration. Arch. Ophthalmol. 1988, 106, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Overbeck, S.; Rink, L.; Haase, H. Modulating the immune response by oral zinc supplementation: A single approach for multiple diseases. Arch. Immunol. Ther. Exp. 2008, 56, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Lowe, N.M.; Dykes, F.C.; Skinner, A.L.; Patel, S.; Warthon-Medina, M.; Decsi, T.; Fekete, K.; Souverein, O.W.; Dullemeijer, C.; Cavelaars, A.E.; et al. Eurreca-estimating zinc requirements for deriving dietary reference values. Crit. Rev. Food Sci. Nutr. 2013, 53, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Rink, L. Multiple impacts of zinc on immune function. Metallomics Integr. Biomet. Sci. 2014, 6, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Cousins, R.J. Zinc dyshomeostasis during polymicrobial sepsis in mice involves zinc transporter ZIP14 and can be overcome by zinc supplementation. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G768–G778. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Malavolta, M. Zinc dyshomeostasis, ageing and neurodegeneration: Implications of A2M and inflammatory gene polymorphisms. J. Alzheimer’s Dis. 2007, 12, 101–109. [Google Scholar] [CrossRef]
- Knoell, D.L.; Julian, M.W.; Bao, S.; Besecker, B.; Macre, J.E.; Leikauf, G.D.; DiSilvestro, R.A.; Crouser, E.D. Zinc deficiency increases organ damage and mortality in a murine model of polymicrobial sepsis. Crit. Care Med. 2009, 37, 1380. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Haase, H.; Engelhardt, G.; Rink, L.; Uciechowski, P. Zinc deficiency induces production of the proinflammatory cytokines IL-1β and TNFΑ in promyeloid cells via epigenetic and redox-dependent mechanisms. J. Nutr. Biochem. 2013, 24, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Effects of zinc deficiency on Th1 and Th2 cytokine shifts. J. Infect. Dis. 2000, 182, S62–S68. [Google Scholar] [CrossRef] [PubMed]
- Cossack, Z.T. T-lymphocyte dysfunction in the elderly associated with zinc deficiency and subnormal nucleoside phosphorylase activity: Effect of zinc supplementation. Eur. J. Cancer Clin. Oncol. 1989, 25, 973–976. [Google Scholar] [CrossRef]
- Uciechowski, P.; Kahmann, L.; Plumakers, B.; Malavolta, M.; Mocchegiani, E.; Dedoussis, G.; Herbein, G.; Jajte, J.; Fulop, T.; Rink, L. Th1 and TH2 cell polarization increases with aging and is modulated by zinc supplementation. Exp. Gerontol. 2008, 43, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Beck, F.W.; Bao, B.; Fitzgerald, J.T.; Snell, D.C.; Steinberg, J.D.; Cardozo, L.J. Zinc supplementation decreases incidence of infections in the elderly: Effect of zinc on generation of cytokines and oxidative stress. Am. J. Clin. Nutr. 2007, 85, 837–844. [Google Scholar] [PubMed]
- Kaltenberg, J.; Plum, L.M.; Ober-Blobaum, J.L.; Honscheid, A.; Rink, L.; Haase, H. Zinc signals promote il-2-dependent proliferation of t cells. Eur. J. Immunol. 2010, 40, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, E.; Metz, C.H.; Maywald, M.; Hilgers, R.D.; Wessels, I.; Senff, T.; Haase, H.; Jager, M.; Ott, M.; Aspinall, R.; et al. Zinc supplementation induces regulatory T cells by inhibition of Sirt-1 deacetylase in mixed lymphocyte cultures. Mol. Nutr. Food Res. 2016, 60, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, E.; Maywald, M.; Hilgers, R.D.; Brieger, A.; Clarner, T.; Kipp, M.; Plumakers, B.; Meyer, S.; Schwerdtle, T.; Rink, L. Induction of regulatory T cells in Th1-/Th17-driven experimental autoimmune encephalomyelitis by zinc administration. J. Nutr. Biochem. 2016, 29, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, C.; Fukada, T.; Kanamoto, M.; Ohashi, W.; Hojyo, S.; Atsumi, T.; Ueda, N.; Azuma, I.; Hirota, H.; Murakami, M.; et al. Zinc suppresses Th17 development via inhibition of STAT3 activation. Int. Immunol. 2010, 22, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Maywald, M.; Rink, L. Zinc supplementation dampens t helper 9 differentiation in allogeneic immune reactions in vitro. Unpublished work. 2017. [Google Scholar]
- George, M.M.; Vignesh, K.S.; Landero Figueroa, J.A.; Caruso, J.A.; Deepe, G.S. Zinc induces dendritic cell tolerogenic phenotype and skews regulatory T cell-Th17 balance. J. Immunol. 2016, 197, 1864–1876. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I. Epigenetics and metal deficiencies. Curr. Nutr. Rep. 2014, 3, 196–203. [Google Scholar] [CrossRef]
- Kahmann, L.; Uciechowski, P.; Warmuth, S.; Plümäkers, B.; Gressner, A.M.; Malavolta, M.; Mocchegiani, E.; Rink, L. Zinc supplementation in the elderly reduces spontaneous inflammatory cytokine release and restores t cell functions. Rejuv. Res. 2008, 11, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.; Jernigan, J.; Bailey, L.; Nickens, C.; Brazzi, G. Zinc nutritive and cell-mediated immunity in the aged. Int. J. Vitam. Nutr. Res. 1983, 53, 94–101. [Google Scholar] [PubMed]
- Dubben, S.; Honscheid, A.; Winkler, K.; Rink, L.; Haase, H. Cellular zinc homeostasis is a regulator in monocyte differentiation of HL-60 cells by 1 alpha,25-dihydroxyvitamin D3. J. Leukoc. Biol. 2010, 87, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Kehl-Fie, T.E.; Skaar, E.P. Nutritional immunity beyond iron: A role for manganese and zinc. Curr. Opin. Chem. Biol. 2010, 14, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Stříž, I.; Trebichavský, I. Calprotectin—A pleiotropic molecule in acute and chronic inflammation. Physiol. Res. 2004, 53, 245–253. [Google Scholar] [PubMed]
- Hasan, R.; Rink, L.; Haase, H. Zinc signals in neutrophil granulocytes are required for the formation of neutrophil extracellular traps. Innate Immun. 2013, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Rink, L.; Haase, H. Chelation of free Zn2+ impairs chemotaxis, phagocytosis, oxidative burst, degranulation, and cytokine production by neutrophil granulocytes. Biol. Trace Elem. Res. 2016, 171, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Suzuki, K.; Suzuki, K.; Nakaji, S.; Sugawara, K. Effects of zinc on the reactive oxygen species generating capacity of human neutrophils and on the serum opsonic activity in vitro. Luminescence 2000, 15, 321–327. [Google Scholar] [CrossRef]
- DeCoursey, T.E.; Morgan, D.; Cherny, V.V. The voltage dependence of nadph oxidase reveals why phagocytes need proton channels. Nature 2003, 422, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. Zinc bound to the killer cell-inhibitory receptor modulates the negative signal in human NK cells. J. Immunol. 1998, 161, 1299–1305. [Google Scholar] [PubMed]
- Rajagopalan, S.; Winter, C.C.; Wagtmann, N.; Long, E.O. The ig-related killer cell inhibitory receptor binds zinc and requires zinc for recognition of HLA-C on target cells. J. Immunol. 1995, 155, 4143–4146. [Google Scholar] [PubMed]
- Kumar, S.; Rajagopalan, S.; Sarkar, P.; Dorward, D.W.; Peterson, M.E.; Liao, H.-S.; Guillermier, C.; Steinhauser, M.L.; Vogel, S.S.; Long, E.O. Zinc-induced polymerization of killer-cell Ig-like receptor into filaments promotes its inhibitory function at cytotoxic immunological synapses. Mol. Cell 2016, 62, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Rolles, B.; Maywald, M.; Rink, L. Influence of zinc deficiency and supplementation on NK cell cytotoxicity. Unpublished work. 2017. [Google Scholar]
- Muzzioli, M.; Stecconi, R.; Moresi, R.; Provinciali, M. Zinc improves the development of human CD34+ cell progenitors towards NK cells and increases the expression of GATA-3 transcription factor in young and old ages. Biogerontology 2009, 10, 593–604. [Google Scholar] [CrossRef] [PubMed]
- King, K.L.; Cidlowski, J.A. Cell cycle regulation and apoptosis. Ann. Rev. Physiol. 1998, 60, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Hebel, S.; Engelhardt, G.; Rink, L. Flow cytometric measurement of labile zinc in peripheral blood mononuclear cells. Anal. Biochem. 2006, 352, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Ober-Blobaum, J.L.; Engelhardt, G.; Hebel, S.; Heit, A.; Heine, H.; Rink, L. Zinc signals are essential for lipopolysaccharide-induced signal transduction in monocytes. J. Immunol. 2008, 181, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Hirano, T. Intracellular zinc homeostasis and zinc signaling. Cancer Sci. 2008, 99, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Sakata-Sogawa, K.; Hasegawa, A.; Suzuki, T.; Kabu, K.; Sato, E.; Kurosaki, T.; Yamashita, S.; Tokunaga, M.; Nishida, K. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007, 177, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Brieger, A.; Rink, L.; Haase, H. Differential regulation of TLR-dependent MYD88 and TRIF signaling pathways by free zinc ions. J. Immunol. 2013, 191, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Hiscox, S.; Nicholson, R.I.; Hogstrand, C.; Kille, P. Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid. Redox Signal. 2006, 8, 1419–1441. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Morikawa, H.; Kamon, H.; Iguchi, M.; Hojyo, S.; Fukada, T.; Yamashita, S.; Kaisho, T.; Akira, S.; Murakami, M.; et al. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat. Immunol. 2006, 7, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Liuzzi, J.P.; McClellan, S.; Cousins, R.J. Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-γ expression in activated human T cells. J. Leukoc. Biol. 2009, 86, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Lee, W.W.; Tomar, D.; Sergey, P.; Czesnikiewicz-Guzik, M.; Lamar, D.L.; Li, G.; Singh, K.; Tian, L.; Weyand, C.M.; et al. Regulation of T cell receptor signaling by activation-induced zinc influx. J. Exp. Med. 2011, 208, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Fukada, T. Roles of zinc signaling in the immune system. J. Immunol. Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.D.; Gekonge, B.; Giri, M.S.; Hancock, A.; Papasavvas, E.; Chehimi, J.; Kossevkov, A.V.; Nicols, C.; Yousef, M.; Mounzer, K. Increased metallothionein gene expression, zinc, and zinc-dependent resistance to apoptosis in circulating monocytes during HIV viremia. J. Leukoc. Biol. 2010, 88, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Hashimoto, A.; Fujimoto, S. Current understanding of ZIP and ZnT zinc transporters in human health and diseases. Cell. Mol. Life Sci. 2014, 71, 3281–3295. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Hojyo, S.; Furuichi, T. Zinc signal: A new player in osteobiology. J. Bone Miner. Metab. 2013, 31, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Takeuchi, O. Pathogen recognition and toll-like receptor targeted therapeutics in innate immune cells. Int. Rev. Immunol. 2017, 36, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Ten Hacken, E.; Burger, J.A. Molecular pathways: Targeting the microenvironment in chronic lymphocytic leukemia—Focus on the B-cell receptor. Clin. Cancer Res. 2014, 20, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Mustelin, T.; Tasken, K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem. J. 2003, 371, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Krebs, E.G. Protein phosphorylation and signal transduction. Pharmacol. Ther. 1999, 82, 111–121. [Google Scholar] [CrossRef]
- Lee, E.J.; Kim, N.; Kang, K.H.; Kim, J.W. Phosphorylation/inactivation of PTEN by AKT-independent PI3K signaling in retinal pigment epithelium. Biochem. Biophys. Res. Commun. 2011, 414, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Vener, A.V.; Aksenova, M.V.; Burbaeva, G.S. Drastic reduction of the zinc-and magnesium-stimulated protein tyrosine kinase activities in Alzheimer’s disease hippocampus. FEBS Lett. 1993, 328, 6–8. [Google Scholar] [CrossRef]
- Baraldi, E.; Carugo, K.D.; Hyvönen, M.; Surdo, P.L.; Riley, A.M.; Potter, B.V.; O’Brien, R.; Ladbury, J.E.; Saraste, M. Structure of the PH domain from Bruton’s tyrosine kinase in complex with inositol 1, 3, 4, 5-tetrakisphosphate. Structure 1999, 7, 449–460. [Google Scholar] [CrossRef]
- Arbibe, L.; Jean-Paul, M.; Teusch, N.; Kline, L.; Guha, M.; Mackman, N.; Godowski, P.J.; Ulevitch, R.J.; Knaus, U.G. Toll-like receptor 2-mediated NF-[kappa] B activation requires a Rac1-dependent pathway. Nat. Immunol. 2000, 1, 533. [Google Scholar] [CrossRef] [PubMed]
- Bennasroune, A.; Mazot, P.; Boutterin, M.-C.; Vigny, M. Activation of the orphan receptor tyrosine kinase alk by zinc. Biochem. Biophys. Res. Commun. 2010, 398, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xing, F.; Zheng, H.; Xi, J.; Cui, X.; Xu, Z. Roles of mitochondrial src tyrosine kinase and zinc in nitric oxide-induced cardioprotection against ischemia/reperfusion injury. Free Radic. Res. 2013, 47, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Quest, A.F.; Bloomenthal, J.; Bardes, E.S.; Bell, R.M. The regulatory domain of protein kinase C coordinates four atoms of zinc. J. Biol. Chem. 1992, 267, 10193–10197. [Google Scholar] [PubMed]
- Korichneva, I.; Hoyos, B.; Chua, R.; Levi, E.; Hammerling, U. Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J. Biol. Chem. 2002, 277, 44327–44331. [Google Scholar] [CrossRef] [PubMed]
- Forbes, I.J.; Zalewski, P.D.; Giannakis, C.; Petkoff, H.S.; Cowled, P.A. Interaction between protein kinase C and regulatory ligand is enhanced by a chelatable pool of cellular zinc. Biochim. Biophys. Acta 1990, 1053, 113–117. [Google Scholar] [CrossRef]
- Castrillo, A.; Pennington, D.J.; Otto, F.; Parker, P.J.; Owen, M.J.; Boscá, L. Protein kinase Cϵ is required for macrophage activation and defense against bacterial infection. J. Exp. Med. 2001, 194, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Korichneva, I. Redox regulation of cardiac protein kinase C. Exp. Clin. Cardiol. 2005, 10, 256–261. [Google Scholar] [PubMed]
- Krężel, A.; Hao, Q.; Maret, W. The zinc/thiolate redox biochemistry of metallothionein and the control of zinc ion fluctuations in cell signaling. Arch. Biochem. Biophys. 2007, 463, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, P.; Forbes, I.; Giannakis, C.; Cowled, P.; Betts, W. Synergy between zinc and phorbol ester in translocation of protein kinase C to cytoskeleton. FEBS Lett. 1990, 273, 131–134. [Google Scholar] [CrossRef]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. In Zinc Biochemistry, Physiology, and Homeostasis; Springer: Berlin, Germany, 2001; pp. 145–155. [Google Scholar]
- Lindahl, M.; Leanderson, P.; Tagesson, C. Novel aspect on metal fume fever: Zinc stimulates oxygen radical formation in human neutrophils. Hum. Exp. Toxicol. 1998, 17, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.R. The antioxidant properties of zinc. J. Nutr. 2000, 130, 1447s–1454s. [Google Scholar] [PubMed]
- Freitas, M.; Porto, G.; Lima, J.L.; Fernandes, E. Zinc activates neutrophils’ oxidative burst. Biometals 2010, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Percival, M.D.; Yeh, B.; Falgueyret, J.-P. Zinc dependent activation of camp-specific phosphodiesterase (PDE4A). Biochem. Biophys. Res. Commun. 1997, 241, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Londesborough, J.; Suoranta, K. Zinc-containing cyclic nucleotide phosphodiesterases from bakers’ yeast. Methods Enzymol. 1988, 159, 777–785. [Google Scholar] [PubMed]
- Von Bülow, V.; Rink, L.; Haase, H. Zinc-mediated inhibition of cyclic nucleotide phosphodiesterase activity and expression suppresses TNF-α and il-1β production in monocytes by elevation of guanosine 3′, 5′-cyclic monophosphate. J. Immunol. 2005, 175, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Heyduk, T.; Sunahara, R.K. Zinc inhibition of adenylyl cyclase correlates with conformational changes in the enzyme. Cell. Signal. 2004, 16, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Du, Z.; Patel, T.B. Copper and zinc inhibit Gαs function: A nucleotide-free state of Gαs induced by Cu2+ and Zn2+. J. Biol. Chem. 2005, 280, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Von Bülow, V.; Dubben, S.; Engelhardt, G.; Hebel, S.; Plümäkers, B.; Heine, H.; Rink, L.; Haase, H. Zinc-dependent suppression of TNF-α production is mediated by protein kinase a-induced inhibition of Raf-1, IκB kinase β, and NF-κB. J. Immunol. 2007, 179, 4180–4186. [Google Scholar] [CrossRef] [PubMed]
- Medgyesi, D.; Hobeika, E.; Biesen, R.; Kollert, F.; Taddeo, A.; Voll, R.E.; Hiepe, F.; Reth, M. The protein tyrosine phosphatase PTP1B is a negative regulator of CD40 and BAFF-R signaling and controls B cell autoimmunity. J. Exp. Med. 2014, 211, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.M.; Brieger, A.; Engelhardt, G.; Hebel, S.; Nessel, A.; Arlt, M.; Kaltenberg, J.; Schwaneberg, U.; Huber, M.; Rink, L.; et al. Pten-inhibition by zinc ions augments interleukin-2-mediated akt phosphorylation. Metallomics Integr. Biomet. Sci. 2014, 6, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Protein tyrosine phosphatases as targets of the combined insulinomimetic effects of zinc and oxidants. Biometals 2005, 18, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Jacob, C.; Vallee, B.L.; Fischer, E.H. Inhibitory sites in enzymes: Zinc removal and reactivation by thionein. Proc. Natl. Acad. Sci. USA 1999, 96, 1936–1940. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.; Samarasinghe, R.; Knoch, M.E.; Lewis, M.; Aizenman, E.; DeFranco, D.B. Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellular signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol. Pharmacol. 2008, 74, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Akaishi, E.; Abe, Y.; Ishikawa, R.; Tanaka, S.; Hosaka, K.; Kubohara, Y. Zinc inhibits calcineurin activity in vitro by competing with nickel. Biochem. Biophys. Res. Commun. 2003, 307, 64–68. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, D.; Xing, W.; Ma, X.; Yin, Y.; Wei, Q.; Li, G. An approach to assay calcineurin activity and the inhibitory effect of zinc ion. Anal. Biochem. 2008, 375, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.; Massarotti, A.; Hogstrand, C.; Maret, W. Zinc ions modulate protein tyrosine phosphatase 1B activity. Metallomics Integr. Biomet. Sci. 2014, 6, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-J.; Bao, S.; Napolitano, J.R.; Burris, D.L.; Yu, L.; Tridandapani, S.; Knoell, D.L. Zinc regulates the acute phase response and serum amyloid a production in response to sepsis through JAK-STAT3 signaling. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yang, Z.; Wang, J.; Yu, J.; Guo, J.; Liu, S.; Qian, C.; Song, L.; Wu, Y.; Cheng, J. Zinc chloride transiently maintains mouse embryonic stem cell pluripotency by activating stat3 signaling. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Azriel-Tamir, H.; Sharir, H.; Schwartz, B.; Hershfinkel, M. Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J. Biol. Chem. 2004, 279, 51804–51816. [Google Scholar] [CrossRef] [PubMed]
- Gruber, K.; Maywald, M.; Rosenkranz, E.; Haase, H.; Plumakers, B.; Rink, L. Zinc deficiency adversely influences interleukin-4 and interleukin-6 signaling. J. Biol. Regul. Homeost. Agents 2013, 27, 661–671. [Google Scholar] [PubMed]
- Dierichs, L.; Kloubert, V.; Rink, L. Cellular zinc homeostasis modulates polarization of THP-1-derived macrophages. Eur. J. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aster, I.; Engelhardt, G.; Rink, L.; Weßels, I. The influence of zinc on granulocyte-macrophage colony stimulating factor-induced signaling in u937 cells. Unpublished work. 2017. [Google Scholar]
- Prasad, A.S.; Bao, B.; Beck, F.W.; Kucuk, O.; Sarkar, F.H. Antioxidant effect of zinc in humans. Free Radic. Biol. Med. 2004, 37, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Bao, B.; Beck, F.W.; Sarkar, F.H. Zinc-suppressed inflammatory cytokines by induction of A20-mediated inhibition of nuclear factor-kappaB. Nutrition 2011, 27, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Prasad, A.S.; Beck, F.W.; Fitzgerald, J.T.; Snell, D.; Bao, G.W.; Singh, T.; Cardozo, L.J. Zinc decreases c-reactive protein, lipid peroxidation, and inflammatory cytokines in elderly subjects: A potential implication of zinc as an atheroprotective agent. Am. J. Clin. Nutr. 2010, 91, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.I.; Ledford, J.R.; Zhou, P.; Page, K. Zinc supplementation alters airway inflammation and airway hyperresponsiveness to a common allergen. J. Inflamm. 2011, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Guo, S.; Gao, J.; Guo, Y.; Du, E.; Lv, Z.; Zhang, B. Maternal high-zinc diet attenuates intestinal inflammation by reducing DNA methylation and elevating H3K9 acetylation in the A20 promoter of offspring chicks. J. Nutr. Biochem. 2015, 26, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-W.; Fan, J.; Bai, S.-L.; Hou, W.-J.; Li, X.; Tong, H. Zinc prevents abdominal aortic aneurysm formation by induction of A20-mediated suppression of NF-κB pathway. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.; King, L. Changes in regulation of lymphopoiesis and myelopoiesis in the zinc-deficient mouse. Nutr. Rev. 1998, 56, S65–S69. [Google Scholar] [CrossRef] [PubMed]
- Supasai, S.; Aimo, L.; Adamo, A.; Mackenzie, G.; Oteiza, P. Zinc deficiency affects the stat1/3 signaling pathways in part through redox-mediated mechanisms. Redox Biol. 2017, 11, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Fluctuations of cellular, available zinc modulate insulin signaling via inhibition of protein tyrosine phosphatases. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2005, 19, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.M.; Lee, M.Y.; Yun, S.P.; Han, H.J. Zinc chloride stimulates DNA synthesis of mouse embryonic stem cells: Involvement of PI3K/Akt, MAPKs, and mTOR. J. Cell. Physiol. 2009, 218, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.W.; Kaplan, J.; Fine, N.; Handschu, W.; Prasad, A.S. Decreased expression of CD73 (ecto-5′-nucleotidase) in the CD8+ subset is associated with zinc deficiency in human patients. J. Lab. Clin. Med. 1997, 130, 147–156. [Google Scholar] [CrossRef]
- Fraker, P.J. Roles for cell death in zinc deficiency. J. Nutr. 2005, 135, 359–362. [Google Scholar] [PubMed]
- Chai, F.; Truong-Tran, A.Q.; Evdokiou, A.; Young, G.P.; Zalewski, P.D. Intracellular zinc depletion induces caspase activation and p21 Waf1/Cip1 cleavage in human epithelial cell lines. J. Infect. Dis. 2000, 182 (Suppl. 1), S85–S92. [Google Scholar] [CrossRef] [PubMed]
- Coto, J.A.; Hadden, E.M.; Sauro, M.; Zorn, N.; Hadden, J.W. Interleukin 1 regulates secretion of zinc-thymulin by human thymic epithelial cells and its action on t-lymphocyte proliferation and nuclear protein kinase C. Proc. Natl. Acad. Sci. USA 1992, 89, 7752–7756. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, M.; Savino, W.; Wade, S.; Kaiserlian, D.; Lemonnier, D.; Bach, J.F. In vivo and in vitro studies of thymulin in marginally zinc-deficient mice. Eur. J. Immunol. 1984, 14, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Dowd, P.S.; Kelleher, J.; Guillou, P.J. T-lymphocyte subsets and interleukin-2 production in zinc-deficient rats. Br. J. Nutr. 1986, 55, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Meftah, S.; Abdallah, J.; Kaplan, J.; Brewer, G.J.; Bach, J.F.; Dardenne, M. Serum thymulin in human zinc deficiency. J. Clin. Investig. 1988, 82, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, K.; Brunner, C. The role of transcription factors in the guidance of granulopoiesis. Am. J. Blood Res. 2012, 2, 57. [Google Scholar] [PubMed]
- Garg, G.; Nikolouli, E.; Hardtke-Wolenski, M.; Toker, A.; Ohkura, N.; Beckstette, M.; Miyao, T.; Geffers, R.; Floess, S.; Gerdes, N. Unique properties of thymic antigen-presenting cells promote epigenetic imprinting of alloantigen-specific regulatory T cells. Oncotarget 2017, 8, 35542–35557. [Google Scholar] [CrossRef] [PubMed]
- Maywald, M.; Meurer, S.K.; Weiskirchen, R.; Rink, L. Zinc supplementation augments tgf-beta1-dependent regulatory T cell induction. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Boskovic, Z.V.; Kemp, M.M.; Freedy, A.M.; Viswanathan, V.S.; Pop, M.S.; Fuller, J.H.; Martinez, N.M.; Figueroa Lazú, S.O.; Hong, J.A.; Lewis, T.A. Inhibition of zinc-dependent histone deacetylases with a chemically triggered electrophile. ACS Chem. Biol. 2016, 11, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Maywald, M.; Rink, L. Zinc supplementation induces CD4+CD25+Foxp3+ antigen-specific regulatory T cells and suppresses IFN-gamma production by upregulation of Foxp3 and KLF-10 and downregulation of IRF-1. Eur. J. Nutr. 2016, 56, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Hogstrand, C.; Kille, P.; Nicholson, R.I.; Taylor, K.M. Zinc transporters and cancer: A potential role for ZIP7 as a hub for tyrosine kinase activation. Trends Mol. Med. 2009, 15, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.M.; Zweifach, A.; Lynes, M.A. Metallothionein regulates intracellular zinc signaling during CD4(+) T cell activation. BMC Immunol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Lienau, S.; Engelardt, G.; Rink, L.; Weßels, I. The role of zinc in calprotectin expression in human monocytic cells. Unpublished work. 2017. [Google Scholar]
- Chasapis, C.T.; Loutsidou, A.C.; Spiliopoulou, C.A.; Stefanidou, M.E. Zinc and human health: An update. Arch. Toxicol. 2012, 86, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Tsichlis, P.N. Phosphorylation at Thr-290 regulates Tpl2 binding to NF-κB1/p105 and Tpl2 activation and degradation by lipopolysaccharide. Proc. Natl. Acad. Sci. USA 2005, 102, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Petris, M.J.; Peck, S.C. Separation of zinc-dependent and zinc-independent events during early LPS-stimulated TLR4 signaling in macrophage cells. FEBS Lett. 2014, 588, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenes Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.Z.; Rink, L. Zinc in infection and inflammation. Nutrients 2017, 9. [Google Scholar] [CrossRef]
- Denk, A.; Wirth, T.; Baumann, B. NF-κB transcription factors: Critical regulators of hematopoiesis and neuronal survival. Cytokine Growth Factor Rev. 2000, 11, 303–320. [Google Scholar] [CrossRef]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.-I.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346. [Google Scholar] [CrossRef] [PubMed]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Regina-Celeste, A.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O. The ubiquitin-modifying enzyme A20 is required for termination of toll-like receptor responses. Nat. Immunol. 2004, 5, 1052. [Google Scholar] [CrossRef] [PubMed]
- Aude-Garcia, C.; Dalzon, B.; Ravanat, J.-L.; Collin-Faure, V.; Diemer, H.; Strub, J.M.; Cianferani, S.; Van Dorsselaer, A.; Carrière, M.; Rabilloud, T. A combined proteomic and targeted analysis unravels new toxic mechanisms for zinc oxide nanoparticles in macrophages. J. Proteom. 2016, 134, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.H.; Ruffin, R.E.; Murgia, C.; Li, L.; Krilis, S.A.; Zalewski, P.D. Labile zinc and zinc transporter ZNT4 in mast cell granules: Role in regulation of caspase activation and NF-κB translocation. J. Immunol. 2004, 172, 7750–7760. [Google Scholar] [CrossRef] [PubMed]
- Kabu, K.; Yamasaki, S.; Kamimura, D.; Ito, Y.; Hasegawa, A.; Sato, E.; Kitamura, H.; Nishida, K.; Hirano, T. Zinc is required for FcεRI-mediated mast cell activation. J. Immunol. 2006, 177, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Kessels, J.E.; Wessels, I.; Haase, H.; Rink, L.; Uciechowski, P. Influence of DNA-methylation on zinc homeostasis in myeloid cells: Regulation of zinc transporters and zinc binding proteins. J. Trace Elem. Med. Biol. 2016, 37, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Ollig, J.; Kloubert, V.; Weßels, I.; Haase, H.; Rink, L. Parameters influencing zinc in experimental systems in vivo and in vitro. Metals 2016, 6, 71. [Google Scholar] [CrossRef]
- Topham, N.J.; Hewitt, E.W. Natural killer cell cytotoxicity: How do they pull the trigger? Immunology 2009, 128, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Rolles, B.; Maywald, M.; Rink, L. Intracellular zinc homeostasis during cell activation and zinc deficiency. Unpublished work. 2017. [Google Scholar]
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Hasegawa, A.; Hojyo, S.; Ohashi, W.; Fukada, T.; Nishida, K.; Hirano, T. A novel role of the L-type calcium channel alpha1D subunit as a gatekeeper for intracellular zinc signaling: Zinc wave. PLoS ONE 2012, 7, e39654. [Google Scholar] [CrossRef] [PubMed]
- Alder, H.; Taccioli, C.; Chen, H.; Jiang, Y.; Smalley, K.J.; Fadda, P.; Ozer, H.G.; Huebner, K.; Farber, J.L.; Croce, C.M.; et al. Dysregulation of miR-31 and miR-21 induced by zinc deficiency promotes esophageal cancer. Carcinogenesis 2012, 33, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.W.; Sun, Z.Y.; Blacklow, S.C.; Wagner, G.; Eck, M.J. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science 2003, 301, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Romir, J.; Lilie, H.; Egerer-Sieber, C.; Bauer, F.; Sticht, H.; Muller, Y.A. Crystal structure analysis and solution studies of human Lck-SH3; zinc-induced homodimerization competes with the binding of proline-rich motifs. J. Mol. Biol. 2007, 365, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Mustelin, T.; Vang, T.; Bottini, N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.; Hogstrand, C.; Maret, W. Redox and zinc signalling pathways converging on protein tyrosine phosphatases. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S9. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Budde, R.J. Affinity purification of Csk protein tyrosine kinase based on its catalytic requirement for divalent metal cations. Protein Expr. Purif. 2001, 21, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Honscheid, A.; Rink, L.; Haase, H. T-lymphocytes: A target for stimulatory and inhibitory effects of zinc ions. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.J.; Hodes, R.J. T-cell development is regulated by the coordinated function of proximal and distal lck promoters active at different developmental stages. Eur. J. Immunol. 2016, 46, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-W.; Cui, D.; Czesnikiewicz-Guzik, M.; Vencio, R.Z.; Shmulevich, I.; Aderem, A.; Weyand, C.M.; Goronzy, J.J. Age-dependent signature of metallothionein expression in primary CD4 T cell responses is due to sustained zinc signaling. Rejuv. Res. 2008, 11, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Devadas, S.; Zaritskaya, L.; Rhee, S.G.; Oberley, L.; Williams, M.S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation. J. Exp. Med. 2002, 195, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-J.; Bao, S.; Gálvez-Peralta, M.; Pyle, C.J.; Rudawsky, A.C.; Pavlovicz, R.E.; Killilea, D.W.; Li, C.; Nebert, D.W.; Wewers, M.D. ZIP8 regulates host defense through zinc-mediated inhibition of NF-κB. Cell Rep. 2013, 3, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, B.; Choi, Y.H.; Hwang, Y.; Kim, D.H.; Cho, S.; Hong, S.J.; Lee, W.W. Inhibition of interleukin-1β-mediated interleukin-1 receptor-associated kinase 4 phosphorylation by zinc leads to repression of memory T helper type 17 response in humans. Immunology 2015, 146, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Daaboul, D.; Rosenkranz, E.; Uciechowski, P.; Rink, L. Repletion of zinc in zinc-deficient cells strongly up-regulates IL-1beta-induced IL-2 production in T-cells. Metallomics Integr. Biomet. Sci. 2012, 4, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; Telford, W.G. A reappraisal of the role of zinc in life and death decisions of cells. Proc. Soc. Exp. Biol. Med. 1997, 215, 229–236. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Frentzel, J.W.; Mann, J.J.; Fraker, P.J. Chronic zinc deficiency in mice disrupted T cell lymphopoiesis and erythropoiesis while B cell lymphopoiesis and myelopoiesis were maintained. J. Am. Coll. Nutr. 2005, 24, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; King, L.E. Reprogramming of the immune system during zinc deficiency. Annu. Rev. Nutr. 2004, 24, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.J.; Metzler, G.; Wray-Dutra, M.; Jackson, S.W. Altered B cell signalling in autoimmunity. Nat. Rev. Immunol. 2017, 17, 421. [Google Scholar] [CrossRef] [PubMed]
- Moulder, K.; Steward, M.W. Experimental zinc deficiency: Effects on cellular responses and the affinity of humoral antibody. Clin. Exp. Immunol. 1989, 77, 269–274. [Google Scholar] [PubMed]
- Cook-Mills, J.M.; Fraker, P.J. Functional capacity of the residual lymphocytes from zinc-deficient adult mice. Br. J. Nutr. 1993, 69, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Miyai, T.; Fujishiro, H.; Kawamura, M.; Yasuda, T.; Hijikata, A.; Bin, B.H.; Irie, T.; Tanaka, J.; Atsumi, T.; et al. Zinc transporter SLC39A10/ZIP10 controls humoral immunity by modulating B-cell receptor signal strength. Proc. Natl. Acad. Sci. USA 2014, 111, 11786–11791. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Sandstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2006, 20, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Lue, C.; Kiyono, H.; McGhee, J.R.; Fujihashi, K.; Kishimoto, T.; Hirano, T.; Mestecky, J. Recombinant human interleukin 6 (rhiL-6) promotes the terminal differentiation of in vivo-activated human B cells into antibody-secreting cells. Cell. Immunol. 1991, 132, 423–432. [Google Scholar] [CrossRef]
- Truong-Tran, A.Q.; Carter, J.; Ruffin, R.E.; Zalewski, P.D. The role of zinc in caspase activation and apoptotic cell death. Biometals 2001, 14, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Kreft, B.; Fischer, A.; Krüger, S.; Sack, K.; Kirchner, H.; Rink, L. The impaired immune response to diphtheria vaccinationin elderly chronic hemodialysis patients is related to zinc deficiency. Biogerontology 2000, 1, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Afsharian, M.; Vaziri, S.; Janbakhsh, A.R.; Sayad, B.; Mansouri, F.; Nourbakhsh, J.; Qadiri, K.; Najafi, F.; Shirvanii, M. The effect of zinc sulfate on immunologic response to recombinant hepatitis b vaccine in elderly: Zinc sulfate and immunologic response to recombinant hepatitis B vaccine. Hepat. Mon. 2011, 11, 32. [Google Scholar] [PubMed]
- Miyai, T.; Hojyo, S.; Ikawa, T.; Kawamura, M.; Irie, T.; Ogura, H.; Hijikata, A.; Bin, B.H.; Yasuda, T.; Kitamura, H.; et al. Zinc transporter SLC39A10/ZIP10 facilitates antiapoptotic signaling during early B-cell development. Proc. Natl. Acad. Sci. USA 2014, 111, 11780–11785. [Google Scholar] [CrossRef] [PubMed]
- Schrantz, N.; Auffredou, M.; Bourgeade, M.; Besnault, L.; Leca, G.; Vazquez, A. Zinc-mediated regulation of caspases activity: Dose-dependent inhibition or activation of caspase-3 in the human Burkitt lymphoma B cells (Ramos). Cell Death Differ. 2001, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.; Bhattacharya, N. Learning to live together: Harnessing regulatory t cells to induce organ transplant tolerance. Yale J Biol Med. 2011, 84, 345–351. [Google Scholar] [PubMed]
- Stennicke, H.R.; Salvesen, G.S. Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol. Chem. 1997, 272, 25719–25723. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D. The zinc finger-containing transcription factors gata-4,-5, and-6 ubiquitously expressed regulators of tissue-specific gene expression. J. Biol. Chem. 2000, 275, 38949–38952. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Nandan, M.O.; Chanchevalap, S.; Dalton, W.B.; Hisamuddin, I.M. Krüppel-like factors 4 and 5: The yin and yang regulators of cellular proliferation. Cell Res. 2005, 15, 92. [Google Scholar] [CrossRef] [PubMed]
- Staitieh, B.S.; Fan, X.; Neveu, W.; Guidot, D.M. Nrf2 regulates PU. 1 expression and activity in the alveolar macrophage. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1086–L1093. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Hoyos, G.; Anderson, M.K.; Wang, C.; Rothenberg, E.V.; Alberola-Ila, J. Gata-3 expression is controlled by tcr signals and regulates CD4/CD8 differentiation. Immunity 2003, 19, 83–94. [Google Scholar] [CrossRef]
- Aliahmad, P.; Kaye, J. Development of all CD4 T lineages requires nuclear factor tox. J. Exp. Med. 2008, 205, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.C.; Tai, T.S.; Pai, S.Y. GATA3 and the T-cell lineage: Essential functions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 2009, 9, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Min, B.; Hu-Li, J.; Watson, C.J.; Grinberg, A.; Wang, Q.; Killeen, N.; Urban, J.F.; Guo, L.; Paul, W.E. Conditional deletion of gata3 shows its essential function in Th1-Th2 responses. Nat. Immunol. 2004, 5, 1157. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.T.; Hogquist, K.A.; Jameson, S.C. Krüppel-like factors in lymphocyte biology. J. Immunol. 2012, 188, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Laity, J.H.; Andrews, G.K. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1). Arch. Biochem. Biophys. 2007, 463, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C. Organizers and Genes Cambridge; Cambridge University Press: Cambridge, UK, 1940. [Google Scholar]
- Sharma, A.; Nguyen, H.; Geng, C.; Hinman, M.N.; Luo, G.; Lou, H. Calcium-mediated histone modifications regulate alternative splicing in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2014, 111, E4920–E4928. [Google Scholar] [CrossRef] [PubMed]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, U.; Oikarinen, S.; Hyoty, H.; Ludvigsson, J. Low zinc in drinking water is associated with the risk of type 1 diabetes in children. Pediatr. Diabetes 2011, 12, 156–164. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.O.; Suderman, M.; Sasaki, A.; Huang, T.C.; Hallett, M.; Meaney, M.J.; Szyf, M. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Rinaldi, N.A.; Ho, E. Zinc deficiency enhanced inflammatory response by increasing immune cell activation and inducing IL6 promoter demethylation. Mol. Nutr. Food Res. 2015, 59, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Diaz, F. Acute dietary zinc deficiency before conception compromises oocyte epigenetic programming and disrupts embryonic development. Dev. Biol. 2013, 376, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Valera, P.; Zavattari, P.; Albanese, S.; Cicchella, D.; Dinelli, E.; Lima, A.; De Vivo, B. A correlation study between multiple sclerosis and type 1 diabetes incidences and geochemical data in europe. Environ. Geochem. Health 2014, 36, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I. Epigenetics and minerals. In Handbook of Nutrition, Diet, and Epigenetics; Patel, V., Preedy, V., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–19. [Google Scholar]
- Schapira, M. Structural biology of human metal-dependent histone deacetylases. In Histone Deacetylases: The Biology and Clinical Implication; Springer: Berlin, Germany, 2011; pp. 225–240. [Google Scholar]
- Davis, C.D.; Uthus, E.O. DNA methylation, cancer susceptibility, and nutrient interactions. Exp. Biol. Med. 2004, 229, 988–995. [Google Scholar] [CrossRef]
- Wong, C.P.; Magnusson, K.R.; Ho, E. Increased inflammatory response in aged mice is associated with age-related zinc deficiency and zinc transporter dysregulation. J. Nutr. Biochem. 2013, 24, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Wuelling, M.; Pasdziernik, M.; Moll, C.N.; Thiesen, A.M.; Schneider, S.; Johannes, C.; Vortkamp, A. The multi zinc-finger protein Trps1 acts as a regulator of histone deacetylation during mitosis. Cell Cycle 2013, 12, 2219–2232. [Google Scholar] [CrossRef]
- Apgar, J. Zinc and reproduction. Ann. Rev. Nutr. 1985, 5, 43–68. [Google Scholar] [CrossRef] [PubMed]
- Uriu-Adams, J.Y.; Keen, C.L. Zinc and reproduction: Effects of zinc deficiency on prenatal and early postnatal development. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2010, 89, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Beach, R.S.; Gershwin, M.E.; Hurley, L.S. Gestational zinc deprivation in mice: Persistence of immunodeficiency for three generations. Science 1982, 218, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Tomat, A.L.; Inserra, F.; Veiras, L.; Vallone, M.C.; Balaszczuk, A.M.; Costa, M.A.; Arranz, C. Moderate zinc restriction during fetal and postnatal growth of rats: Effects on adult arterial blood pressure and kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R543–R549. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Yoshida, K.; Yasuda, Y.; Tsutsui, T. Infantile zinc deficiency: Association with autism spectrum disorders. Sci. Rep. 2011, 1, 129. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.Y.; Bernhardt, M.L.; Kim, A.M.; O’Halloran, T.V.; Woodruff, T.K. Zinc maintains prophase I arrest in mouse oocytes through regulation of the MOS-MAPK pathway. Biol. Reprod. 2012, 87, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kurita, H.; Ohsako, S.; Hashimoto, S.-I.; Yoshinaga, J.; Tohyama, C. Prenatal zinc deficiency-dependent epigenetic alterations of mouse metallothionein-2 gene. J. Nutr. Biochem. 2013, 24, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Girgenti, M.J.; LoTurco, J.J.; Maher, B.J. ZNF804A regulates expression of the schizophrenia-associated genes PRSS16, COMT, PDE4B, and DRD2. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.-J.; Chen, C.-H.; Cheng, C.-Y.; Chen, S.-Y.; Chen, T.-J.; Younger, W.; Nian, F.-S.; Tsai, S.-J.; Hong, C.-J. Convergent evidence from mouse and human studies suggests the involvement of zinc finger protein 326 gene in antidepressant treatment response. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Pan, E.; Xiong, Z.-Q.; McNamara, J.O. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 2008, 57, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Dufault, R.; Lukiw, W.J.; Crider, R.; Schnoll, R.; Wallinga, D.; Deth, R. A macroepigenetic approach to identify factors responsible for the autism epidemic in the united states. Clin. Epigenet. 2012, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Tsutsui, T. Assessment of infantile mineral imbalances in autism spectrum disorders (ASDs). Int. J. Environ. Res. Public Health 2013, 10, 6027–6043. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M. A role for synaptic zinc in ProSAP/Shank PSD scaffold malformation in autism spectrum disorders. Dev. Neurobiol. 2014, 74, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Coneyworth, L.; Mathers, J.; Ford, D. Does promoter methylation of the SLC30A5 (ZNT5) zinc transporter gene contribute to the ageing-related decline in zinc status?: Conference on ‘multidisciplinary approaches to nutritional problems’. Proc. Nutr. Soc. 2009, 68, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, Z.; Li, D.; Li, N.; Dindot, S.V.; Satterfield, M.C.; Bazer, F.W.; Wu, G. Nutrition, epigenetics, and metabolic syndrome. Antioxid. Redox Signal. 2012, 17, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Dufner-Beattie, J.; Weaver, B.P.; Geiser, J.; Bilgen, M.; Larson, M.; Xu, W.; Andrews, G.K. The mouse acrodermatitis enteropathica gene SLC39A4 (ZIP4) is essential for early development and heterozygosity causes hypersensitivity to zinc deficiency. Hum. Mol. Genet. 2007, 16, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Geiser, J.; Venken, K.J.; De Lisle, R.C.; Andrews, G.K. A mouse model of acrodermatitis enteropathica: Loss of intestine zinc transporter ZIP4 (SLC39A4) disrupts the stem cell niche and intestine integrity. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Simeone, D.M.; Binkley, C.; Arumugam, T.; Greenson, J.K.; Giordano, T.J.; Misek, D.E.; Hanash, S. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003, 63, 2649–2657. [Google Scholar] [PubMed]
- Eom, S.J.; Kim, E.Y.; Lee, J.E.; Kang, H.J.; Shim, J.; Kim, S.U.; Gwag, B.J.; Choi, E.J. Zn(2+) induces stimulation of the c-Jun N-terminal kinase signaling pathway through phosphoinositide 3-Kinase. Mol. Pharmacol. 2001, 59, 981–986. [Google Scholar] [PubMed]
- Li, M.; Zhang, Y.; Liu, Z.; Bharadwaj, U.; Wang, H.; Wang, X.; Zhang, S.; Liuzzi, J.P.; Chang, S.-M.; Cousins, R.J. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc. Natl. Acad. Sci. USA 2007, 104, 18636–18641. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.; Bharadwaj, U.; Zhai, Q.J.; Ahern, C.H.; Fisher, W.E.; Brunicardi, F.C.; Logsdon, C.D.; Chen, C.; Yao, Q. Down-regulation of ZIP4 by RNA interference inhibits pancreatic cancer growth and increases the survival of nude mice with pancreatic cancer xenografts. Clin. Cancer Res. 2009, 15, 5993–6001. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.P.; Zhang, Y.; Hiscox, S.; Guo, G.L.; Apte, U.; Taylor, K.M.; Sheline, C.T.; Wang, L.; Andrews, G.K. ZIP4 (SLC39A4) expression is activated in hepatocellular carcinomas and functions to repress apoptosis, enhance cell cycle and increase migration. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bharadwaj, U.; Logsdon, C.D.; Chen, C.; Yao, Q.; Li, M. ZIP4 regulates pancreatic cancer cell growth by activating IL-6/STAT3 pathway through zinc finger transcription factor creb. Clin. Cancer Res. 2010, 16, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, C.; Yao, Q.; Li, M. ZIP4 upregulates the expression of neuropilin-1, vascular endothelial growth factor, and matrix metalloproteases in pancreatic cancer cell lines and xenografts. Cancer Biol. Ther. 2010, 9, 235–241. [Google Scholar] [CrossRef]
- Schneider, J.; Ruschhaupt, M.; Buneß, A.; Asslaber, M.; Regitnig, P.; Zatloukal, K.; Schippinger, W.; Ploner, F.; Poustka, A.; Sültmann, H. Identification and meta-analysis of a small gene expression signature for the diagnosis of estrogen receptor status in invasive ductal breast cancer. Int. J. Cancer 2006, 119, 2974–2979. [Google Scholar] [CrossRef] [PubMed]
- Tozlu, S.; Girault, I.; Vacher, S.; Vendrell, J.; Andrieu, C.; Spyratos, F.; Cohen, P.; Lidereau, R.; Bieche, I. Identification of novel genes that co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-pcr approach. Endocr. Relat. Cancer 2006, 13, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M. A distinct role in breast cancer for two LIV-1 family zinc transporters. Biochem. Soc. Trans. 2008, 36, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Vichova, P.; Jordan, N.; Hiscox, S.; Hendley, R.; Nicholson, R.I. ZIP7-mediated intracellular zinc transport contributes to aberrant growth factor signaling in antihormone-resistant breast cancer cells. Endocrinology 2008, 149, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Kagara, N.; Tanaka, N.; Noguchi, S.; Hirano, T. Zinc and its transporter ZIP10 are involved in invasive behavior of breast cancer cells. Cancer Sci. 2007, 98, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Nicholson, R.I. The LZT proteins; the LIV-1 subfamily of zinc transporters. Biochim. Biophys. Acta 2003, 1611, 16–30. [Google Scholar] [CrossRef]
- McClelland, R.A.; Manning, D.; Gee, J.; Willsher, P.; Robertson, J.; Ellis, I.; Blamey, R.; Nicholson, R.I. Oestrogen-regulated genes in breast cancer: Association of pLIV1 with response to endocrine therapy. Br. J. Cancer 1998, 77, 1653. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Muraina, I.A.; Brethour, D.; Schmitt-Ulms, G.; Nimmanon, T.; Ziliotto, S.; Kille, P.; Hogstrand, C. Zinc transporter ZIP10 forms a heteromer with ZIP6 which regulates embryonic development and cell migration. Biochem. J. 2016, 473, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Nimmanon, T.; Ziliotto, S.; Morris, S.; Flanagan, L.; Taylor, K. Phosphorylation of zinc channel ZIP7 drives MAPK, PI3K and mTOR growth and proliferation signalling. Metallomics Integr. Biomet. Sci. 2017, 9, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, J.M.; Hutcheson, I.R.; Jones, H.E.; Madden, T.; Gee, J.M.; Harper, M.E.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003, 144, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.E.; Goddard, L.; Gee, J.M.W.; Hiscox, S.; Rubini, M.; Barrow, D.; Knowlden, J.M.; Williams, S.; Wakeling, A.; Nicholson, R.I. Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr. Relat. Cancer 2004, 11, 793–814. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory t cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Costas, J. The highly pleiotropic gene SLC39A8 as an opportunity to gain insight into the molecular pathogenesis of schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chowanadisai, W. Comparative genomic analysis of SLC39A12/ZIP12: Insight into a zinc transporter required for vertebrate nervous system development. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Kumura, J. Preliminary reports on the metabolism of trace elements in neuro-psychiatric diseases. I. Proc. Jpn. Acad. 1965, 41, 943–947. [Google Scholar]
- Bly, M. Examination of the zinc transporter gene, SLC39A12. Schizophr. Res. 2006, 81, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Effects of zinc deficiency on TH1 and TH2 cytokine shifts. J. Infect. Dis. 2000, 182 (Suppl. 1), S62–S68. [Google Scholar] [CrossRef] [PubMed]
- Honscheid, A.; Dubben, S.; Rink, L.; Haase, H. Zinc differentially regulates mitogen-activated protein kinases in human T cells. J. Nutr. Biochem. 2012, 23, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, D.; Fornes, P. Suicide among youth and young adults, 15 through 24 years of age. A report of 392 cases from Paris, 1989–1996. J. Forensic Sci. 1998, 43, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Yary, T.; Aazami, S. Dietary intake of zinc was inversely associated with depression. Biol. Trace Elem. Res. 2012, 145, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G.; Szewczyk, B.; Pilc, A. Zinc and depression. An update. Pharmacol. Rep. 2005, 57, 713–718. [Google Scholar] [PubMed]
- Maes, M.; Bosmans, E.; De Jongh, R.; Kenis, G.; Vandoolaeghe, E.; Neels, H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 1997, 9, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989, 31, 145–238. [Google Scholar] [PubMed]
- Styczen, K.; Sowa-Kucma, M.; Siwek, M.; Dudek, D.; Reczynski, W.; Szewczyk, B.; Misztak, P.; Topor-Madry, R.; Opoka, W.; Nowak, G. The serum zinc concentration as a potential biological marker in patients with major depressive disorder. Metab. Brain Dis. 2017, 32, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Himmerich, H.; Milenovic, S.; Fulda, S.; Plumakers, B.; Sheldrick, A.J.; Michel, T.M.; Kircher, T.; Rink, L. Regulatory T cells increased while IL-1beta decreased during antidepressant therapy. J. Psychiatr. Res. 2010, 44, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.K., Jr.; Enomoto, H.; Leopold, I.H.; Fleischer, E.B.; Schoon, D.V.; Fender, D.; Tucker, H.G.; Adamson, B.; Kladde, L.; Kazan, D.; et al. Plasma zinc levels in multiple sclerosis. Metab. Pediatr. Ophthalmol. 1980, 4, 3–8. [Google Scholar] [PubMed]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Bredholt, M.; Frederiksen, J.L. Zinc in multiple sclerosis: A systematic review and meta-analysis. ASN Neuro 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Campo, C.A.; Wellinghausen, N.; Faber, C.; Fischer, A.; Rink, L. Zinc inhibits the mixed lymphocyte culture. Biol. Trace Elem. Res. 2001, 79, 15–22. [Google Scholar] [PubMed]
- Faber, C.; Gabriel, P.; Ibs, K.H.; Rink, L. Zinc in pharmacological doses suppresses allogeneic reaction without affecting the antigenic response. Bone Marrow Transplant. 2004, 33, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Kown, M.H.; van der Steenhoven, T.J.; Jahncke, C.L.; Mari, C.; Lijkwan, M.A.; Koransky, M.L.; Blankenberg, F.G.; Strauss, H.W.; Robbins, R.C. Zinc chloride-mediated reduction of apoptosis as an adjunct immunosuppressive modality in cardiac transplantation. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2002, 21, 360–365. [Google Scholar] [CrossRef]
- Schubert, C.; Guttek, K.; Grungreiff, K.; Thielitz, A.; Buhling, F.; Reinhold, A.; Brocke, S.; Reinhold, D. Oral zinc aspartate treats experimental autoimmune encephalomyelitis. Biometals 2014, 27, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Giunta, C.; Randolph, A.; Steinmann, B. Mutation analysis of the PLOD1 gene: An efficient multistep approach to the molecular diagnosis of the kyphoscoliotic type of ehlers-danlos syndrome (EDS VIA). Mol. Genet. Metab. 2005, 86, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Bin, B.-H.; Hojyo, S.; Ryong Lee, T.; Fukada, T. Spondylocheirodysplastic Ehlers-Danlos syndrome (SCD-EDS) and the mutant zinc transporter ZIP13. Rare Dis. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Asada, Y.; Mishima, K.; Shimoda, S.; Saito, I. SLC39A13/ZIP13: A crucial zinc transporter involved in tooth development and inherited disorders. J. Oral Biosci. 2011, 53, 1–12. [Google Scholar] [CrossRef]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-β signaling pathways. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Chai, J.; Wu, J.-W.; Yan, N.; Massagué, J.; Pavletich, N.P.; Shi, Y. Features of a Smad3 MH1-DNA complex roles of water and zinc in DNA binding. J. Biol. Chem. 2003, 278, 20327–20331. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Jeon, J.; Shin, M.; Won, Y.; Lee, M.; Kwak, J.-S.; Lee, G.; Rhee, J.; Ryu, J.-H.; Chun, C.-H. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 2014, 156, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Hogrebe, M.; Grüneberg, M.; DuChesne, I.; Ava, L.; Reunert, J.; Schlingmann, K.P.; Boycott, K.M.; Beaulieu, C.L.; Mhanni, A.A. SLC39A8 deficiency: A disorder of manganese transport and glycosylation. Am. J. Hum. Genet. 2015, 97, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Besecker, B.; Bao, S.; Bohacova, B.; Papp, A.; Sadee, W.; Knoell, D.L. The human zinc transporter SLC39A8 (ZIP8) is critical in zinc-mediated cytoprotection in lung epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1127–L1136. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, J.R.; Liu, M.-J.; Bao, S.; Crawford, M.; Nana-Sinkam, P.; Cormet-Boyaka, E.; Knoell, D.L. Cadmium-mediated toxicity of lung epithelia is enhanced through NF-κB-mediated transcriptional activation of the human zinc transporter ZIP8. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L909–L918. [Google Scholar] [CrossRef] [PubMed]
- Aggett, P.; Atherton, D.; More, J.; Davey, J.; Delves, H.; Harries, J. Symptomatic zinc deficiency in a breast-fed preterm infant. Arch. Dis. Child. 1980, 55, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Chowanadisai, W.; Lönnerdal, B.; Kelleher, S.L. Identification of a mutation in SLC30A2 (ZNT-2) in women with low milk zinc concentration that results in transient neonatal zinc deficiency. J. Biol. Chem. 2006, 281, 39699–39707. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.C.; Udagani, M.M.; Badakali, A.V.; Yelameli, B.C. Symptomatic zinc deficiency in a full-term breast-fed infant. Dermatol. Online J. 2010, 16, 307–308. [Google Scholar]
- Itsumura, N.; Inamo, Y.; Okazaki, F.; Teranishi, F.; Narita, H.; Kambe, T.; Kodama, H. Compound heterozygous mutations in SLC30A2/ZNT2 results in low milk zinc concentrations: A novel mechanism for zinc deficiency in a breast-fed infant. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Itsumura, N.; Kibihara, Y.; Fukue, K.; Miyata, A.; Fukushima, K.; Tamagawa-Mineoka, R.; Katoh, N.; Nishito, Y.; Ishida, R.; Narita, H. Novel mutations in SLC30A2 involved in the pathogenesis of transient neonatal zinc deficiency. Pediatr. Res. 2016, 80, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Gitschier, J. A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nat. Genet. 1997, 17, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Ackland, M.L.; Mercer, J. The murine mutation, lethal milk, results in production of zinc-deficient milk. J. Nutr. 1992, 122, 1214. [Google Scholar] [PubMed]
- McCormick, N.H.; Lee, S.; Hennigar, S.R.; Kelleher, S.L. Znt4 (SLC30a4)-null (“lethal milk”) mice have defects in mammary gland secretion and hallmarks of precocious involution during lactation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R33–R40. [Google Scholar] [CrossRef] [PubMed]
- Ackland, M.L.; Michalczyk, A. Zinc deficiency and its inherited disorders—A review. Genes Nutr. 2006, 1, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J.; Lin, J.K.; Farzadfar, F.; Khang, Y.-H.; Stevens, G.A. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011, 378, 31–40. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Prim. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Wenzlau, J.M.; Liu, Y.; Yu, L.; Moua, O.; Fowler, K.T.; Rangasamy, S.; Walters, J.; Eisenbarth, G.S.; Davidson, H.W.; Hutton, J.C. A common nonsynonymous single nucleotide polymorphism in the SLC30A8 gene determines ZNT8 autoantibody specificity in type 1 diabetes. Diabetes 2008, 57, 2693–2697. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Christian, D.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881. [Google Scholar] [CrossRef] [PubMed]
- Pound, L.D.; Sarkar, S.A.; Benninger, R.K.; Wang, Y.; Suwanichkul, A.; Shadoan, M.K.; Printz, R.L.; Oeser, J.K.; Lee, C.E.; Piston, D.W. Deletion of the mouse SLC30A8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem. J. 2009, 421, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Ravier, M.; Schraenen, A.; Creemers, J.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.; et al. Insulin crystallization depends on zinc transporter ZNT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef] [PubMed]
- Trabucchi, A.; Faccinetti, N.I.; Guerra, L.L.; Puchulu, F.M.; Frechtel, G.D.; Poskus, E.; Valdez, S.N. Detection and characterization of ZNT8 autoantibodies could help to screen latent autoimmune diabetes in adult-onset patients with type 2 phenotype. Autoimmunity 2012, 45, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Vaziri-Sani, F.; Oak, S.; Radtke, J.; Lernmark, Å.; Lynch, K.; Agardh, C.-D.; Cilio, C.M.; Lethagen, Å.L.; Örtqvist, E.; Landin-Olsson, M. ZNT8 autoantibody titers in type 1 diabetes patients decline rapidly after clinical onset. Autoimmunity 2010, 43, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Rockell, J.; Wagner, R.; Wenzlau, J.M.; Yu, L.; Hutton, J.C.; Gottlieb, P.A.; Davidson, H.W. Human type 1 diabetes is associated with T cell autoimmunity to zinc transporter 8. J. Immunol. 2011, 186, 6056–6063. [Google Scholar] [CrossRef] [PubMed]
- Lampasona, V.; Petrone, A.; Tiberti, C.; Capizzi, M.; Spoletini, M.; Di Pietro, S.; Songini, M.; Bonicchio, S.; Giorgino, F.; Bonifacio, E. Zinc transporter 8 antibodies complement GAD and IA-2 antibodies in the identification and characterization of adult-onset autoimmune diabetes. Diabetes Care 2010, 33, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Rungby, J. Zinc, zinc transporters and diabetes. Diabetologia 2010, 53, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; Karges, W.; Rink, L. Zinc and diabetes—Clinical links and molecular mechanisms. J. Nutr. Biochem. 2009, 20, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter Znt8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58, 2070–2083. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Dai, F.; Hardy, A.; Giglou, P.; Bhattacharjee, A.; Koshkin, V.; Chimienti, F.; Gaisano, H.; Rutter, G.; Wheeler, M. Beta cell-specific ZNT8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 2010, 53, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Franklin, I.; Gromada, J.; Gjinovci, A.; Theander, S.; Wollheim, C.B. Β-cell secretory products activate α-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005, 54, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; Rosenkranz, E.; Overbeck, S.; Warmuth, S.; Mocchegiani, E.; Giacconi, R.; Weiskirchen, R.; Karges, W.; Rink, L. Disturbed zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of zinc. J. Nutr. Biochem. 2012, 23, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, R.; Ranasinghe, P.; Galappatthy, P.; Malkanthi, R.; Constantine, G.; Katulanda, P. Effects of zinc supplementation on diabetes mellitus: A systematic review and meta-analysis. Diabetol. Metab. Syndr. 2012, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Valera, P.; Zavattari, P.; Sanna, A.; Pretti, S.; Marcello, A.; Mannu, C.; Targhetta, C.; Bruno, G.; Songini, M. Zinc and other metals deficiencies and risk of type 1 diabetes: An ecological study in the high risk sardinia island. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Taga, T.; Yasukawa, K.; Nakajima, K.; Nakano, N.; Takatsuki, F.; Shimizu, M.; Murashima, A.; Tsunasawa, S.; Sakiyama, F.; et al. Human B-cell differentiation factor defined by an anti-peptide antibody and its possible role in autoantibody production. Proc. Natl. Acad. Sci. USA 1987, 84, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Daviglus, M.L.; Bell, C.C.; Berrettini, W.; Bowen, P.E.; Connolly, E.S., Jr.; Cox, N.J.; Dunbar-Jacob, J.M.; Granieri, E.C.; Hunt, G.; McGarry, K.; et al. Nih state-of-the-science conference statement: Preventing alzheimer’s disease and cognitive decline. NIH Consens. State Sci. Statements 2010, 27, 1–30. [Google Scholar] [PubMed]
- Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 2013, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Lyubartseva, G.; Smith, J.L.; Markesbery, W.R.; Lovell, M.A. Alterations of zinc transporter proteins ZNT-1, ZNT-4 and ZNT-6 in preclinical alzheimer’s disease brain. Brain Pathol. 2010, 20, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Xiong, S.; Markesbery, W.; Lovell, M. Altered expression of zinc transporters-4 and-6 in mild cognitive impairment, early and late Alzheimer’s disease brain. Neuroscience 2006, 140, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Smith, J.L.; Xiong, S.; Markesbery, W.R. Alterations in zinc transporter protein-1 (ZNT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage Alzheimer’s disease. Neurotox. Res. 2005, 7, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Lukiw, W.J.; Bergeron, C.; Niznik, H.B.; Fraser, P.E. Metallothionein iii is reduced in Alzheimer’s disease. Brain Res. 2001, 894, 37–45. [Google Scholar] [CrossRef]
- Uchida, Y.; Takio, K.; Titani, K.; Ihara, Y.; Tomonaga, M. The growth inhibitory factor that is deficient in the alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron 1991, 7, 337–347. [Google Scholar] [CrossRef]
- Erickson, J.C.; Sewell, A.K.; Jensen, L.T.; Winge, D.R.; Palmiter, R.D. Enhanced neurotrophic activity in Alzheimer’s disease cortex is not associated with down-regulation of metallothionein-III (GIF). Brain Res. 1994, 649, 297–304. [Google Scholar] [CrossRef]
- Wu, H.Y.; Hudry, E.; Hashimoto, T.; Kuchibhotla, K.; Rozkalne, A.; Fan, Z.; Spires-Jones, T.; Xie, H.; Arbel-Ornath, M.; Grosskreutz, C.L.; et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 2636–2649. [Google Scholar] [CrossRef] [PubMed]
- Reese, L.C.; Zhang, W.; Dineley, K.T.; Kayed, R.; Taglialatela, G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell 2008, 7, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Panayi, A.E.; Spyrou, N.M.; Iversen, B.S.; White, M.A.; Part, P. Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J. Neurol. Sci. 2002, 195, 1–10. [Google Scholar] [CrossRef]
- Samudralwar, D.L.; Diprete, C.C.; Ni, B.F.; Ehmann, W.D.; Markesbery, W.R. Elemental imbalances in the olfactory pathway in Alzheimer’s disease. J. Neurol. Sci. 1995, 130, 139–145. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Faget, K.Y. Zinc takes the center stage: Its paradoxical role in Alzheimer’s disease. Brain Res. Brain Res. Rev. 2003, 41, 44–56. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Protection against amyloid beta peptide toxicity by zinc. Brain Res. 1999, 823, 88–95. [Google Scholar] [CrossRef]
- Garai, K.; Sengupta, P.; Sahoo, B.; Maiti, S. Selective destabilization of soluble amyloid beta oligomers by divalent metal ions. Biochem. Biophys. Res. Commun. 2006, 345, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Atrian-Blasco, E.; Conte-Daban, A.; Hureau, C. Mutual interference of Cu and Zn ions in Alzheimer’s disease: Perspectives at the molecular level. Dalton Trans. 2017, 46, 12750–12759. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wessels, I.; Maywald, M.; Rink, L. Zinc as a Gatekeeper of Immune Function. Nutrients 2017, 9, 1286. https://doi.org/10.3390/nu9121286
Wessels I, Maywald M, Rink L. Zinc as a Gatekeeper of Immune Function. Nutrients. 2017; 9(12):1286. https://doi.org/10.3390/nu9121286
Chicago/Turabian StyleWessels, Inga, Martina Maywald, and Lothar Rink. 2017. "Zinc as a Gatekeeper of Immune Function" Nutrients 9, no. 12: 1286. https://doi.org/10.3390/nu9121286
APA StyleWessels, I., Maywald, M., & Rink, L. (2017). Zinc as a Gatekeeper of Immune Function. Nutrients, 9(12), 1286. https://doi.org/10.3390/nu9121286