Fatty Acids, Antioxidants and Physical Activity in Brain Aging

 ,
, 

Abstract
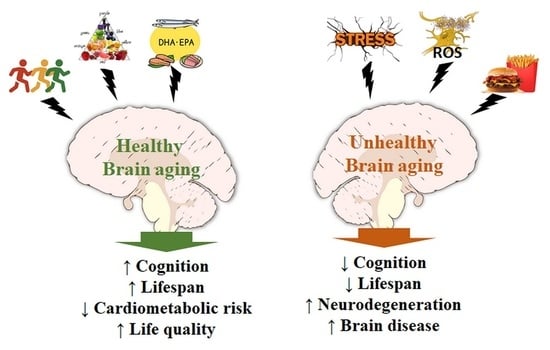
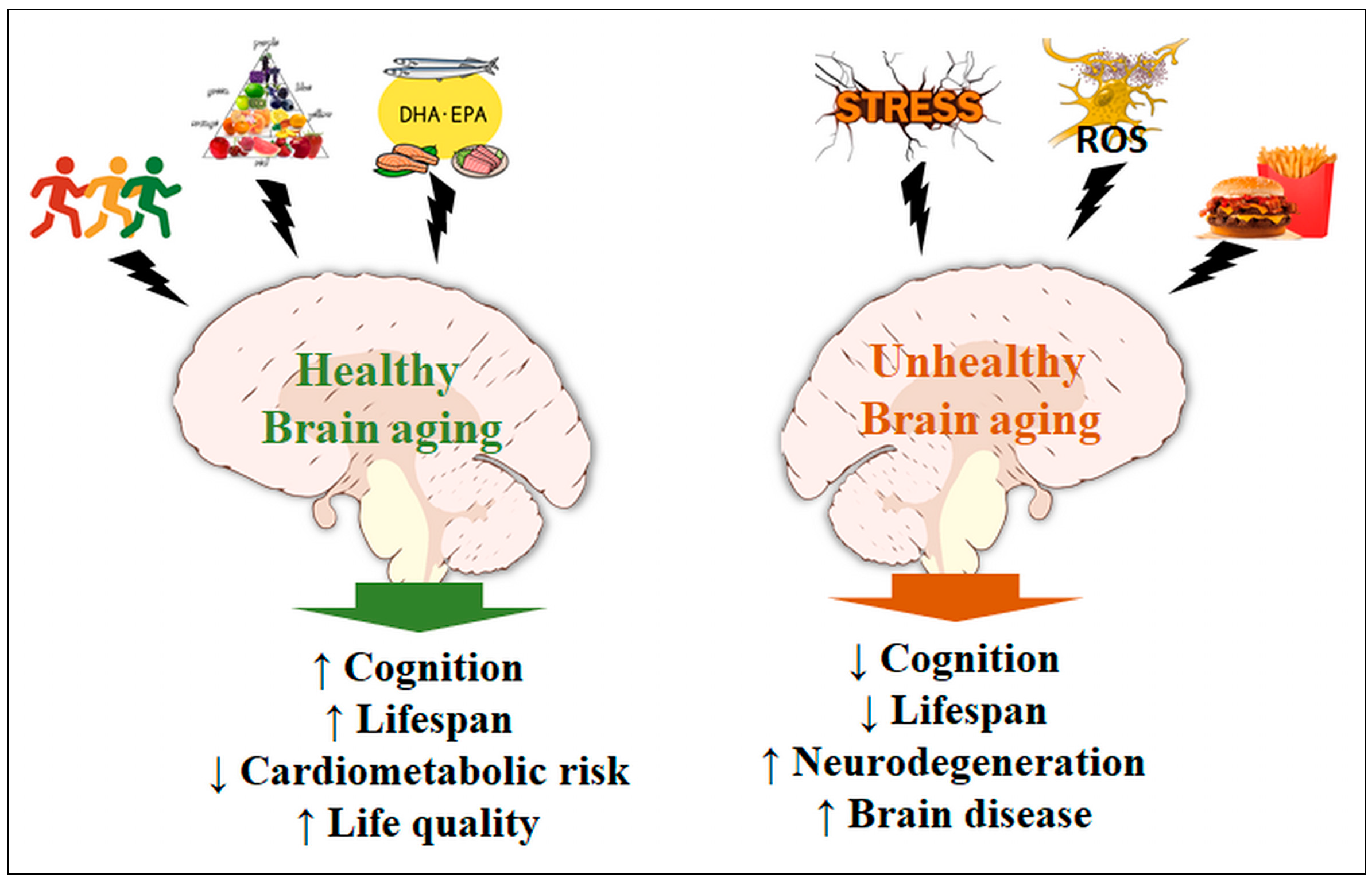
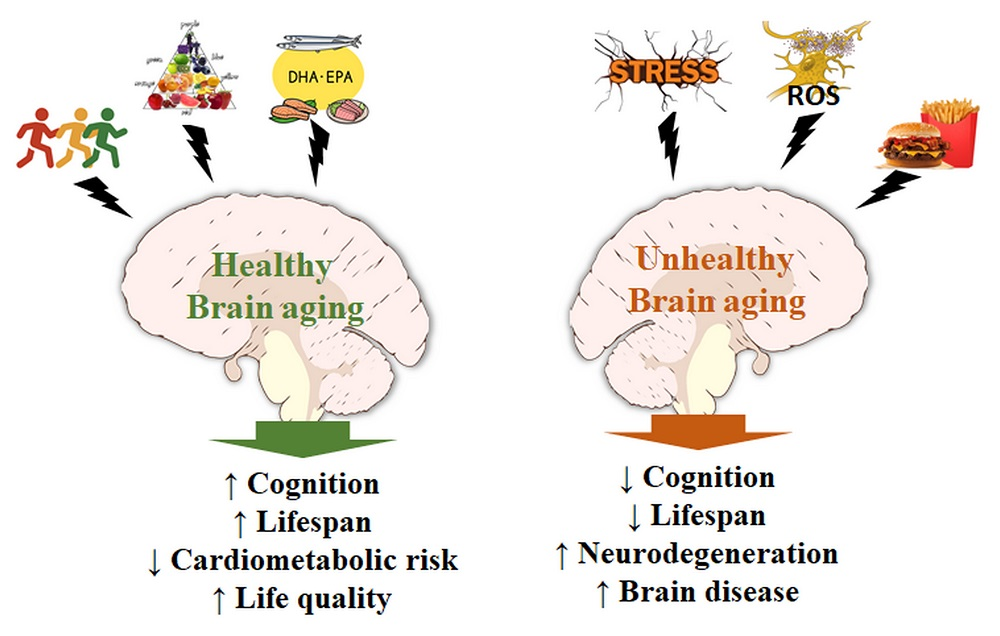{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Senescence of the Nervous System
3. Adiposity, Neuroinflammation and Senescence
4. Fatty Acids
5. Antioxidants
6. Physical Activity
6.1. Physical Activity and Disorders of Cerebral Blood Flow
6.2. Brain Volume and the Effect of Physical Activity on Brain Atrophy
6.3. Sexual Dimorphism and Physical Activity
6.4. Physical Activity and Brain Disorders of Metabolic Origin
6.5. Inflammation and a Role for Physical Activity
6.6. A Role for Physical Activity in Autophagy and Oxi-Inflammaging
6.7. Antineurodegenerative Effect of Muscle Metabolites
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khaw, K.T. Epidemiological aspects of ageing. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1997, 352, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.E. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: Roles of infection, inflammation, and nutrition. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. 1), 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Frangeskou, M.; Lopez-Valcarcel, B.; Serra-Majem, L. Dehydration in the Elderly: A Review Focused on Economic Burden. J. Nutr. Health Aging 2015, 19, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Milan, A.M.; D’Souza, R.F.; Pundir, S.; Pileggi, C.A.; Thorstensen, E.B.; Barnett, M.P.; Markworth, J.F.; Cameron-Smith, D.; Mitchell, C.J. Older adults have delayed amino acid absorption after a high protein mixed breakfast meal. J. Nutr. Health Aging 2015, 19, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Cumming, R.G.; Hilmer, S.N. A review of frailty in developing countries. J. Nutr. Health Aging 2015, 19, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Corona, L.P.; Andrade, F.C.D.; Duarte, Y.A.D.; Lebrao, M.L. The relationship between anemia, hemoglobin concentration and frailty in Brazilian older adults. J. Nutr. Health Aging 2015, 19, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Kearney, J. Food consumption trends and drivers. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2010, 365, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Callahan, D.M.; Toth, M.J. Skeletal muscle myofilament adaptations to aging, disease, and disuse and their effects on whole muscle performance in older adult humans. Front. Physiol. 2014, 5, 369. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; Pessin, J.E. Recent progress in metabolic signaling pathways regulating aging and life span. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S21–S27. [Google Scholar] [CrossRef] [PubMed]
- Popkin, B.M. The nutrition transition and obesity in the developing world. J. Nutr. 2001, 131, 871s–873s. [Google Scholar] [PubMed]
- Hossain, P.; Kawar, B.; Nahas, M.E. Obesity and diabetes in the developing world—A growing challenge. N. Engl. J. Med. 2007, 356, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Steptoe, A.; Deaton, A.; Stone, A.A. Subjective wellbeing, health, and ageing. Lancet 2015, 385, 640–648. [Google Scholar] [CrossRef]
- Goodman, E.; Whitaker, R.C. A prospective study of the role of depression in the development and persistence of adolescent obesity. Pediatrics 2002, 110, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Onyike, C.U.; Crum, R.M.; Lee, H.B.; Lyketsos, C.G.; Eaton, W.W. Is obesity associated with major depression? Results from the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 2003, 158, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- McCarty, C.A.; Kosterman, R.; Mason, W.A.; McCauley, E.; Hawkins, J.D.; Herrenkohl, T.I.; Lengua, L.J. Longitudinal associations among depression, obesity and alcohol use disorders in young adulthood. Gen. Hosp. Psychiatry 2009, 31, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Saunier, K.; Dore, J. Gastrointestinal tract and the elderly: Functional foods, gut microflora and healthy ageing. Dig. Liver Dis. 2002, 34 (Suppl. 2), S19–S24. [Google Scholar] [CrossRef]
- Lopez-Leon, M.; Outeiro, T.F.; Goya, R.G. Cell reprogramming: Therapeutic potential and the promise of rejuvenation for the aging brain. Ageing Res. Rev. 2017, 40, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Cararo, J.H.; Streck, E.L.; Schuck, P.F.; Ferreira Gda, C. Carnosine and related peptides: Therapeutic potential in age-related disorders. Aging Dis. 2015, 6, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.; Walter, M. Therapeutic targeting of telomerase. Genes 2016, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Q.; Sack, L.; Kang, C.; Elledge, S.J. A gain-of-function senescence bypass screen identifies the homeobox transcription factor DLX2 as a regulator of ATM-p53 signaling. Genes Dev. 2016, 30, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Poon, H.F.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: Insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid. Redox Signal. 2006, 8, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, E.; Tavernarakis, N. Oxidative stress and mitochondrial protein quality control in aging. J. Proteom. 2013, 92, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Samson, R.D.; Barnes, C.A. Impact of aging brain circuits on cognition. Eur. J. Neurosci. 2013, 37, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Camello, P.J.; Verkhratsky, A.; Toescu, E.C. Mitochondrial polarisation status and [Ca2+]i signalling in rat cerebellar granule neurones aged in vitro. Neurobiol. Aging 2004, 25, 349–359. [Google Scholar] [CrossRef]
- Murchison, D.; Zawieja, D.C.; Griffith, W.H. Reduced mitochondrial buffering of voltage-gated calcium influx in aged rat basal forebrain neurons. Cell Calcium 2004, 36, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Tatarkova, Z.; Kovalska, M.; Timkova, V.; Racay, P.; Lehotsky, J.; Kaplan, P. The effect of aging on mitochondrial complex I and the extent of oxidative stress in the rat brain cortex. Neurochem. Res. 2016, 41, 2160–2172. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J. An update on the mitochondrial-DNA mutation hypothesis of cell aging. Mutat. Res. 1992, 275, 209–216. [Google Scholar] [CrossRef]
- Bertoni-Freddari, C.; Fattoretti, P.; Casoli, T.; Spagna, C.; Meier-Ruge, W. Morphological alterations of synaptic mitochondria during aging. The effect of Hydergine treatment. Ann. N. Y. Acad. Sci. 1994, 717, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Millan, A.; Garcia de la Asuncion, J.; Pla, R.; Juan, G.; Pallardo, F.V.; O’Connor, E.; Martin, J.A.; Droy-Lefaix, M.T.; Vina, J. A Ginkgo biloba extract (EGb 761) prevents mitochondrial aging by protecting against oxidative stress. Free Radic. Biol. Med. 1998, 24, 298–304. [Google Scholar]
- Wakabayashi, T. Megamitochondria formation-physiology and pathology. J. Cell. Mol. Med. 2002, 6, 497–538. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Sik, A.; Penttonen, M.; Buzsaki, G. Interneurons in the hippocampal dentate gyrus: An in vivo intracellular study. Eur. J. Neurosci. 1997, 9, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Papageorgiou, I.E.; Draguhn, A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J. Cereb. Blood Flow Metab. 2014, 34, 1270–1282. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 1812, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Cornu, M.; Albert, V.; Hall, M.N. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 2013, 23, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bitarafan, S.; Harirchian, M.H.; Nafissi, S.; Sahraian, M.A.; Togha, M.; Siassi, F.; Saedisomeolia, A.; Alipour, E.; Mohammadpour, N.; Chamary, M.; et al. Dietary intake of nutrients and its correlation with fatigue in multiple sclerosis patients. Iran. J. Neurol. 2014, 13, 28–32. [Google Scholar] [PubMed]
- Mi, W.; van Wijk, N.; Cansev, M.; Sijben, J.W.; Kamphuis, P.J. Nutritional approaches in the risk reduction and management of Alzheimer’s disease. Nutrition 2013, 29, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Tomi, M.; Zhao, Y.; Thamotharan, S.; Shin, B.C.; Devaskar, S.U. Early life nutrient restriction impairs blood-brain metabolic profile and neurobehavior predisposing to Alzheimer’s disease with aging. Brain Res. 2013, 1495, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Abrass, C.K.; Hansen, K.M. Differentiating the influences of aging and adiposity on brain weights, levels of serum and brain cytokines, gastrointestinal hormones, and amyloid precursor protein. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, S.; Cai, D. Disruption of neurogenesis by hypothalamic inflammation in obesity or aging. Rev. Endocr. Metab. Disord. 2013, 14, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S. Digging deeper into obesity. J. Clin. Investig. 2011, 121, 2076–2079. [Google Scholar] [CrossRef] [PubMed]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Batsis, J.A.; Mackenzie, T.A.; Vasquez, E.; Germain, C.M.; Emeny, R.T.; Rippberger, P.; Lopez-Jimenez, F.; Bartels, S.J. Association of adiposity, telomere length and mortality: Data from the NHANES 1999–2002. Int. J. Obes. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Ezquerro, S.; Mendez-Gimenez, L.; Becerril, S.; Fruhbeck, G. Revisiting the adipocyte: A model for integration of cytokine signaling in the regulation of energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E691–E714. [Google Scholar] [CrossRef] [PubMed]
- Strissel, K.J.; DeFuria, J.; Shaul, M.E.; Bennett, G.; Greenberg, A.S.; Obin, M.S. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity 2010, 18, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, A.; Naffah de Souza, C.; Camara, N.O.; Moraes-Vieira, P.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2015, 6, 637. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A. Leptin decreases food intake induced by melanin-concentrating hormone (MCH), galanin (GAL) and neuropeptide Y (NPY) in the rat. Endocrinology 1998, 139, 4739–4742. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Osei, S.Y. Leptin signaling. Physiol. Behav. 2004, 81, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Bjorbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Horrillo, D.; Sierra, J.; Arribas, C.; Garcia-San Frutos, M.; Carrascosa, J.M.; Lauzurica, N.; Fernandez-Agullo, T.; Ros, M. Age-associated development of inflammation in Wistar rats: Effects of caloric restriction. Arch. Physiol. Biochem. 2011, 117, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.; Luheshi, G.N.; Wenz, T.; Gerstberger, R.; Roth, J.; Rummel, C. Leptin is involved in age-dependent changes in response to systemic inflammation in the rat. Brain Behav. Immun. 2014, 36, 128–138. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.J.; Ishii, M. Leptin Dysfunction and Alzheimer’s Disease: Evidence from Cellular, Animal, and Human Studies. Cell. Mol. Neurobiol. 2016, 36, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Ronan, L.; Alexander-Bloch, A.F.; Wagstyl, K.; Farooqi, S.; Brayne, C.; Tyler, L.K.; Fletcher, P.C. Obesity associated with increased brain age from midlife. Neurobiol. Aging 2016, 47, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Kosaka, H.; Okazawa, H.; Murata, T.; Wada, Y. Relationship between plasma leptin level and brain structure in elderly: A voxel-based morphometric study. Biol. Psychiatry 2009, 65, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Holden, K.F.; Lindquist, K.; Tylavsky, F.A.; Rosano, C.; Harris, T.B.; Yaffe, K. Serum leptin level and cognition in the elderly: Findings from the Health ABC Study. Neurobiol. Aging 2009, 30, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Lieb, W.; Beiser, A.S.; Vasan, R.S.; Tan, Z.S.; Au, R.; Harris, T.B.; Roubenoff, R.; Auerbach, S.; DeCarli, C.; Wolf, P.A.; et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. J. Am. Med. Assoc. 2009, 302, 2565–2572. [Google Scholar] [CrossRef] [PubMed]
- Gorska-Ciebiada, M.; Saryusz-Wolska, M.; Borkowska, A.; Ciebiada, M.; Loba, J. Adiponectin, leptin and IL-1 beta in elderly diabetic patients with mild cognitive impairment. Metab. Brain Dis. 2016, 31, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, P.; Toga, A.W.; Jack, C.R.; Weiner, M.W.; Thompson, P.M. Fat-mass-related hormone, plasma leptin, predicts brain volumes in the elderly. Neuroreport 2013, 24, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Lifelong brain health is a lifelong challenge: From evolutionary principles to empirical evidence. Ageing Res. Rev. 2015, 20, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C. Lifestyle modulators of neuroplasticity: How physical activity, mental engagement, and diet promote cognitive health during aging. Neural Plast. 2017, 2017, 3589271. [Google Scholar] [CrossRef] [PubMed]
- Jesko, H.; Wencel, P.; Strosznajder, R.P.; Strosznajder, J.B. Sirtuins and their roles in brain aging and neurodegenerative disorders. Neurochem. Res. 2017, 42, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.E.; Supinski, A.M.; Bonkowski, M.S.; Donmez, G.; Guarente, L.P. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes Dev. 2009, 23, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Fusco, S.; Ripoli, C.; Podda, M.V.; Ranieri, S.C.; Leone, L.; Toietta, G.; McBurney, M.W.; Schutz, G.; Riccio, A.; Grassi, C.; et al. A role for neuronal cAMP responsive-element binding (CREB)-1 in brain responses to calorie restriction. Proc. Natl. Acad. Sci. USA 2012, 109, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, N.; Zhao, H.H.; Zhang, X.; Kawano, H.; Liu, L.; Zhao, L.; Li, H.P. Interactions between Sirt1 and MAPKs regulate astrocyte activation induced by brain injury in vitro and in vivo. J. Neuroinflamm. 2017, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Freitas, H.R.; Isaac, A.R.; Malcher-Lopes, R.; Diaz, B.L.; Trevenzoli, I.H.; De Melo Reis, R.A. Polyunsaturated fatty acids and endocannabinoids in health and disease. Nutr. Neurosci. 2017, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. Essential fatty acids in health and chronic disease. Am. J. Clin. Nutr. 1999, 70, 560s–569s. [Google Scholar] [PubMed]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharm. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Denis, I.; Potier, B.; Vancassel, S.; Heberden, C.; Lavialle, M. Omega-3 fatty acids and brain resistance to ageing and stress: Body of evidence and possible mechanisms. Ageing Res. Rev. 2013, 12, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Denis, I.; Potier, B.; Heberden, C.; Vancassel, S. Omega-3 polyunsaturated fatty acids and brain aging. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Haast, R.A.; Kiliaan, A.J. Impact of fatty acids on brain circulation, structure and function. Prostaglandins Leukot. Essent. Fat. Acids 2015, 92, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Latour, A.; Grintal, B.; Champeil-Potokar, G.; Hennebelle, M.; Lavialle, M.; Dutar, P.; Potier, B.; Billard, J.M.; Vancassel, S.; Denis, I. Omega-3 fatty acids deficiency aggravates glutamatergic synapse and astroglial aging in the rat hippocampal CA1. Aging Cell 2013, 12, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Chouinard-Watkins, R.; Plourde, M. Fatty acid metabolism in carriers of apolipoprotein E epsilon 4 allele: Is it contributing to higher risk of cognitive decline and coronary heart disease? Nutrients 2014, 6, 4452–4471. [Google Scholar] [CrossRef] [PubMed]
- Salem, N., Jr.; Vandal, M.; Calon, F. The benefit of docosahexaenoic acid for the adult brain in aging and dementia. Prostaglandins Leukot. Essent. Fat. Acids 2015, 92, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, M.; Hara, T.; Kashino, A.; Akasaka, Y.; Ide, T.; Murakami, K. Identification of a functional peroxisome proliferator-activated receptor (PPAR) response element (PPRE) in the human apolipoprotein A-IV gene. Biochem. Pharmacol. 2009, 78, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Yang, Z.; Wang, X.; Shi, H. Omega-3 polyunsaturated fatty acids antagonize macrophage inflammation via activation of AMPK/SIRT1 pathway. PLoS ONE 2012, 7, e45990. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Minghetti, L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Landreth, G.E. PPARs in the brain. Biochim. Biophys. Acta 2007, 1771, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Culman, J.; Zhao, Y.; Gohlke, P.; Herdegen, T. PPAR-gamma: Therapeutic target for ischemic stroke. Trends Pharmacol. Sci. 2007, 28, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Afshordel, S.; Hagl, S.; Werner, D.; Rohner, N.; Kogel, D.; Bazan, N.G.; Eckert, G.P. Omega-3 polyunsaturated fatty acids improve mitochondrial dysfunction in brain aging—Impact of Bcl-2 and NPD-1 like metabolites. Prostaglandins Leukot. Essent. Fat. Acids 2015, 92, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Cutuli, D.; De Bartolo, P.; Caporali, P.; Laricchiuta, D.; Foti, F.; Ronci, M.; Rossi, C.; Neri, C.; Spalletta, G.; Caltagirone, C.; et al. n-3 polyunsaturated fatty acids supplementation enhances hippocampal functionality in aged mice. Front. Aging Neurosci. 2014, 6, 220. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.J.; Greenwood, C.E. Dietary saturated fatty acids and brain function. Neurochem. Res. 1998, 23, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Burke, L.E.; Dunbar-Jacob, J. Adherence to medication, diet, and activity recommendations: From assessment to maintenance. J. Cardiovasc. Nurs. 1995, 9, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Kanoski, S.E.; Davidson, T.L. Western diet consumption and cognitive impairment: Links to hippocampal dysfunction and obesity. Physiol. Behav. 2011, 103, 59–68. [Google Scholar] [CrossRef] [PubMed]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- El-Assaad, W.; Joly, E.; Barbeau, A.; Sladek, R.; Buteau, J.; Maestre, I.; Pepin, E.; Zhao, S.; Iglesias, J.; Roche, E.; et al. Glucolipotoxicity alters lipid partitioning and causes mitochondrial dysfunction, cholesterol, and ceramide deposition and reactive oxygen species production in INS832/13 ss-cells. Endocrinology 2010, 151, 3061–3073. [Google Scholar] [CrossRef] [PubMed]
- Fontes, G.; Zarrouki, B.; Hagman, D.K.; Latour, M.G.; Semache, M.; Roskens, V.; Moore, P.C.; Prentki, M.; Rhodes, C.J.; Jetton, T.L.; et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia 2010, 53, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontes, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; Diamant, M. Glucolipotoxicity and beta cells in type 2 diabetes mellitus: Target for durable therapy? Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S37–S46. [Google Scholar] [CrossRef]
- Liu, Z.; Stanojevic, V.; Brindamour, L.J.; Habener, J.F. GLP1-derived nonapeptide GLP1(28–36)amide protects pancreatic beta-cells from glucolipotoxicity. J. Endocrinol. 2012, 213, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; Molina, M.F.; Gordon, W.C. Docosahexaenoic acid signalolipidomics in nutrition: Significance in aging, neuroinflammation, macular degeneration, Alzheimer’s, and other neurodegenerative diseases. Ann. Rev. Nutr. 2011, 31, 321–351. [Google Scholar] [CrossRef] [PubMed]
- Head, E.; Rofina, J.; Zicker, S. Oxidative stress, aging, and central nervous system disease in the canine model of human brain aging. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci. USA 2000, 97, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo(-/-) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Hodges, R.E.; Hood, J.; Canham, J.E.; Sauberlich, H.E.; Baker, E.M. Clinical manifestations of ascorbic acid deficiency in man. Am. J. Clin. Nutr. 1971, 24, 432–443. [Google Scholar] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Gess, B.; Sevimli, S.; Strecker, J.K.; Young, P.; Schabitz, W.R. Sodium-dependent vitamin C transporter 2 (SVCT2) expression and activity in brain capillary endothelial cells after transient ischemia in mice. PLoS ONE 2011, 6, e17139. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Huang, Y.H.; Shen, Y.C.; Huang, H.C.; Liu, P.H. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. J. Cereb. Blood Flow Metab. 2010, 30, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010, 7, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.N.; Kumar, R.S.; Paval, J.; Nayak, S. Effect of ascorbic acid on the monosodium glutamate-induced neurobehavioral changes in periadolescent rats. Bratisl. Lek. Listy 2010, 111, 247–252. [Google Scholar] [PubMed]
- Moretti, M.; Colla, A.; de Oliveira Balen, G.; dos Santos, D.B.; Budni, J.; de Freitas, A.E.; Farina, M.; Severo Rodrigues, A.L. Ascorbic acid treatment, similarly to fluoxetine, reverses depressive-like behavior and brain oxidative damage induced by chronic unpredictable stress. J. Psychiatr. Res. 2012, 46, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Paleologos, M.; Cumming, R.G.; Lazarus, R. Cohort study of vitamin C intake and cognitive impairment. Am. J. Epidemiol. 1998, 148, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Guidi, I.; Galimberti, D.; Lonati, S.; Novembrino, C.; Bamonti, F.; Tiriticco, M.; Fenoglio, C.; Venturelli, E.; Baron, P.; Bresolin, N.; et al. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2006, 27, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Wilson, R.S.; Scherr, P.A. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. J. Am. Med. Assoc. 2002, 287, 3230–3237. [Google Scholar] [CrossRef]
- Joseph, J.A.; Shukitt-Hale, B.; Denisova, N.A.; Bielinski, D.; Martin, A.; McEwen, J.J.; Bickford, P.C. Reversals of age-related declines in neuronal signal transduction, cognitive, and motor behavioral deficits with blueberry, spinach, or strawberry dietary supplementation. J. Neurosci. 1999, 19, 8114–8121. [Google Scholar] [PubMed]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Kish, S.J.; Morito, C.; Hornykiewicz, O. Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci. Lett. 1985, 58, 343–346. [Google Scholar] [CrossRef]
- Volicer, L.; Crino, P.B. Involvement of free radicals in dementia of the Alzheimer type: A hypothesis. Neurobiol. Aging 1990, 11, 567–571. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione is a crucial guardian of protein integrity in the brain upon nitric oxide imbalance. Commun. Integr. Biol. 2011, 4, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Giorgi, A.; Schinina, M.E.; Coccia, R.; Cini, C.; Bellia, F.; Cambria, M.T.; Cornelius, C.; Butterfield, D.A.; et al. Redox proteomics in aging rat brain: Involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. J. Neurosci. Res. 2010, 88, 3498–3507. [Google Scholar] [CrossRef] [PubMed]
- Emir, U.E.; Raatz, S.; McPherson, S.; Hodges, J.S.; Torkelson, C.; Tawfik, P.; White, T.; Terpstra, M. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 2011, 24, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Tripathi, M.; Sugunan, S. Brain oxidative stress: Detection and mapping of anti-oxidant marker ‘Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 2012, 417, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Gawryluk, J.W.; Wang, J.F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
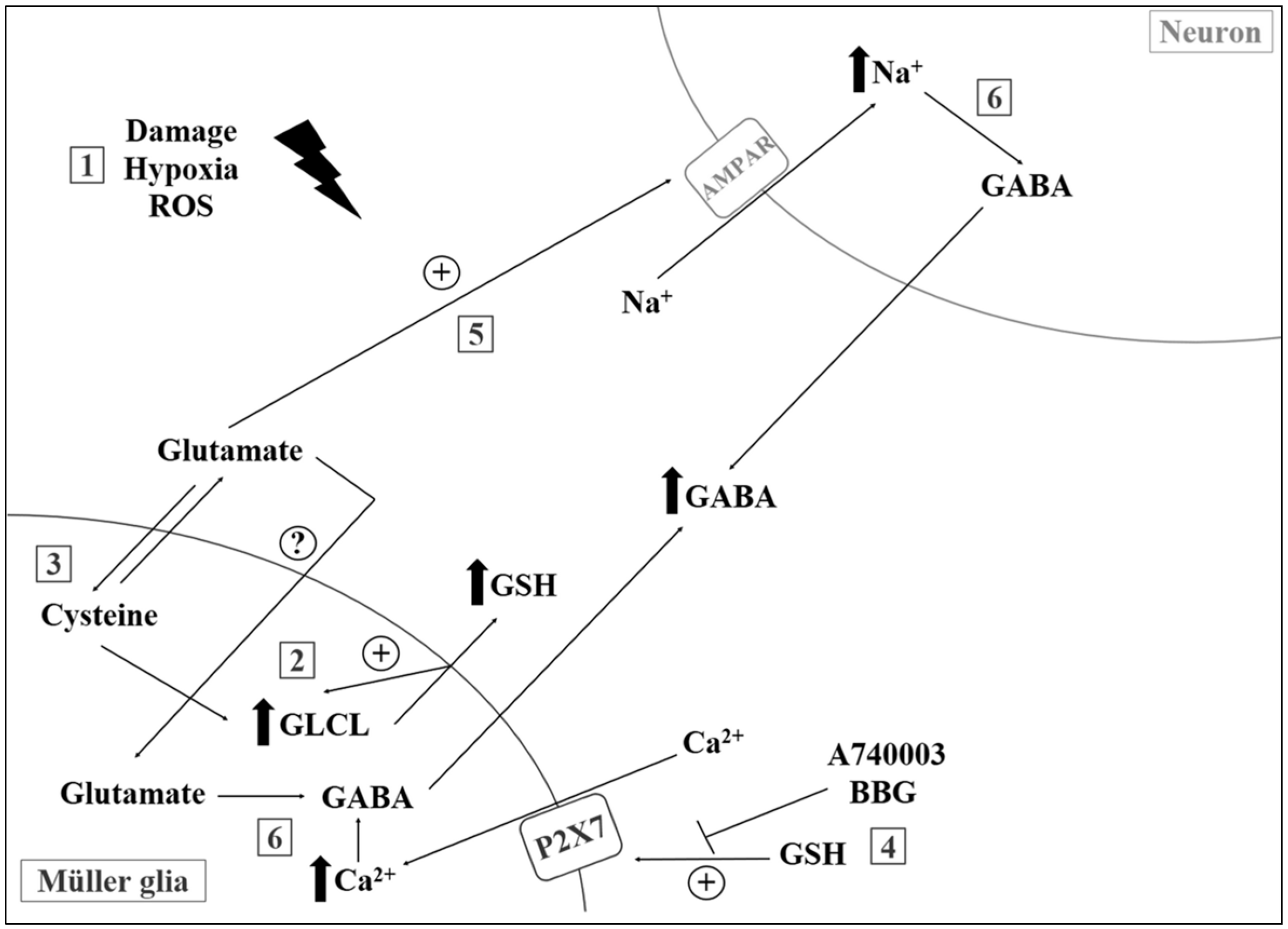
- Freitas, H.R.; Reis, R.A. Glutathione induces GABA release through P2X7R activation on Muller glia. Neurogenesis 2017, 4, e1283188. [Google Scholar] [CrossRef] [PubMed]
- Faria, R.X.; Freitas, H.R.; Reis, R.A.M. P2X7 receptor large pore signaling in avian Muller glial cells. J. Bioenerg. Biomembr. 2017, 49, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Blaha, M.J.; Hung, R.K.; Dardari, Z.; Feldman, D.I.; Whelton, S.P.; Nasir, K.; Blumenthal, R.S.; Brawner, C.A.; Ehrman, J.K.; Keteyian, S.J.; et al. Age-dependent prognostic value of exercise capacity and derivation of fitness-associated biologic age. Heart 2016, 102, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.W.; Nagamatsu, L.S.; Liu-Ambrose, T.; Kramer, A.F. Exercise, brain, and cognition across the life span. J. Appl. Physiol. 2011, 111, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; de Grey, A.D. Innovating aging: Promises and pitfalls on the road to life extension. Gerontology 2014, 60, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Justice, J.N.; LaRocca, T.J. Physiological geroscience: Targeting function to increase healthspan and achieve optimal longevity. J. Physiol. 2016, 594, 2001–2024. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, S.; Muller, L.; Wenger, E.; Duzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.A.; Pialoux, V.; Corbett, D.; Drogos, L.; Erickson, K.I.; Eskes, G.A.; Poulin, M.J. Promoting brain health through exercise and diet in older adults: A physiological perspective. J. Physiol. 2016, 594, 4485–4498. [Google Scholar] [CrossRef] [PubMed]
- Guure, C.B.; Ibrahim, N.A.; Adam, M.B.; Said, S.M. Impact of physical activity on cognitive decline, dementia, and its subtypes: meta-analysis of prospective studies. BioMed Res. Int. 2017, 2017, 9016924. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, V.; Thorin-Trescases, N.; Thorin, E. Endothelium-dependent control of cerebrovascular functions through age: Exercise for healthy cerebrovascular aging. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H620–H633. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; Corella, D.; Castaner, O.; Martinez-Gonzalez, M.A.; Salas-Salvador, J.; Vila, J.; Estruch, R.; Sorli, J.V.; Aros, F.; Fiol, M.; et al. Protective effect of homovanillyl alcohol on cardiovascular disease and total mortality: Virgin olive oil, wine, and catechol-methylathion. Am. J. Clin. Nutr. 2017, 105, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Ainslie, P.N.; Cotter, J.D.; George, K.P.; Lucas, S.; Murrell, C.; Shave, R.; Thomas, K.N.; Williams, M.J.; Atkinson, G. Elevation in cerebral blood flow velocity with aerobic fitness throughout healthy human ageing. J. Physiol. 2008, 586, 4005–4010. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.Y.; Tarumi, T.; Meijers, R.L.; Turner, M.; Repshas, J.; Xiong, L.; Ding, K.; Vongpatanasin, W.; Yuan, L.J.; Zhang, R. Arterial pressure, heart rate, and cerebral hemodynamics across the adult life span. Hypertension 2017, 69, 712–720. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Cardiovascular risk factors promote brain hypoperfusion leading to cognitive decline and dementia. Cardiovasc. Psychiatry Neurol. 2012, 2012, 367516. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.N. Exercise, cognitive function, and aging. Adv. Physiol. Educ. 2015, 39, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Viboolvorakul, S.; Patumraj, S. Exercise training could improve age-related changes in cerebral blood flow and capillary vascularity through the upregulation of VEGF and eNOS. BioMed Res. Int. 2014, 2014, 230791. [Google Scholar] [CrossRef] [PubMed]
- Gertz, K.; Priller, J.; Kronenberg, G.; Fink, K.B.; Winter, B.; Schrock, H.; Ji, S.; Milosevic, M.; Harms, C.; Bohm, M.; et al. Physical activity improves long-term stroke outcome via endothelial nitric oxide synthase-dependent augmentation of neovascularization and cerebral blood flow. Circ. Res. 2006, 99, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Lee, K.H.; Lee, J. Effect of exercise-induced neurogenesis on cognitive function deficit in a rat model of vascular dementia. Mol. Med. Rep. 2016, 13, 2981–2990. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Huddleston, D.E.; Brickman, A.M.; Sosunov, A.A.; Hen, R.; McKhann, G.M.; Sloan, R.; Gage, F.H.; Brown, T.R.; Small, S.A. An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. USA 2007, 104, 5638–5643. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Leckie, R.L.; Weinstein, A.M. Physical activity, fitness, and gray matter volume. Neurobiol. Aging 2014, 35 (Suppl. 2), S20–S28. [Google Scholar] [CrossRef] [PubMed]
- Maass, A.; Duzel, S.; Goerke, M.; Becke, A.; Sobieray, U.; Neumann, K.; Lovden, M.; Lindenberger, U.; Backman, L.; Braun-Dullaeus, R.; et al. Vascular hippocampal plasticity after aerobic exercise in older adults. Mol. Psychiatry 2015, 20, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Hanning, U.; Roesler, A.; Peters, A.; Berger, K.; Baune, B.T. Structural brain changes and all-cause mortality in the elderly population-the mediating role of inflammation. Age 2016, 38, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Brooks, S.J.; Kullberg, J.; Nordenskjold, R.; Burgos, J.; Le Greves, M.; Kilander, L.; Larsson, E.M.; Johansson, L.; Ahlstrom, H.; et al. Association between physical activity and brain health in older adults. Neurobiol. Aging 2013, 34, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Prakash, R.S.; Voss, M.W.; Chaddock, L.; Hu, L.; Morris, K.S.; White, S.M.; Wojcicki, T.R.; McAuley, E.; Kramer, A.F. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus 2009, 19, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Vidoni, E.D.; Johnson, D.K.; Van Sciver, A.; Mahnken, J.D.; Honea, R.A.; Wilkins, H.M.; Brooks, W.M.; Billinger, S.A.; Swerdlow, R.H.; et al. Aerobic exercise for Alzheimer’s disease: A randomized controlled pilot trial. PLoS ONE 2017, 12, e0170547. [Google Scholar] [CrossRef] [PubMed]
- Chaddock, L.; Erickson, K.I.; Prakash, R.S.; Kim, J.S.; Voss, M.W.; Vanpatter, M.; Pontifex, M.B.; Raine, L.B.; Konkel, A.; Hillman, C.H.; et al. A neuroimaging investigation of the association between aerobic fitness, hippocampal volume, and memory performance in preadolescent children. Brain Res. 2010, 1358, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, J.; Aberg, M.A.; Schioler, L.; Nilsson, M.; Wallin, A.; Toren, K.; Kuhn, H.G. Cardiovascular and cognitive fitness at age 18 and risk of early-onset dementia. Brain 2014, 137, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L. Why sex matters for neuroscience. Nat. Rev. Neurosci. 2006, 7, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Taaffe, D.R.; Harris, T.B.; Ferrucci, L.; Rowe, J.; Seeman, T.E. Cross-sectional and prospective relationships of interleukin-6 and C-reactive protein with physical performance in elderly persons: MacArthur studies of successful aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2000, 55, M709–M715. [Google Scholar] [CrossRef]
- De la Fuente, M.; Cruces, J.; Hernandez, O.; Ortega, E. Strategies to improve the functions and redox state of the immune system in aged subjects. Curr. Pharm. Des. 2011, 17, 3966–3993. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, M.; Cha, R.H.; Aakre, J.A.; Therneau, T.M.; Geda, Y.E.; Mielke, M.M.; Knopman, D.S.; Petersen, R.C.; Roberts, R.O. Mortality in mild cognitive impairment varies by subtype, sex, and lifestyle factors: The Mayo Clinic Study of Aging. J. Alzheimer Dis. 2015, 45, 1237–1245. [Google Scholar] [CrossRef]
- Xing, C.Y.; Tarumi, T.; Liu, J.; Zhang, Y.; Turner, M.; Riley, J.; Tinajero, C.D.; Yuan, L.J.; Zhang, R. Distribution of cardiac output to the brain across the adult lifespan. J. Cereb. Blood Flow Metab. 2017, 37, 2848–2856. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, N.C.; Cribbs, D.H.; Coleman, P.D.; Rogers, J.; Head, E.; Kim, R.; Beach, T.; Miller, C.; Troncoso, J.; Trojanowski, J.Q.; et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA 2008, 105, 15605–15610. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.M.; Peiffer, J.J.; Martins, R.N. Multiple effects of physical activity on molecular and cognitive signs of brain aging: Can exercise slow neurodegeneration and delay Alzheimer’s disease? Mol. Psychiatry 2013, 18, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.R.; Chuang, Y.F.; Harris, G.C.; Tan, E.J.; Carlson, M.C. Low-intensity daily walking activity is associated with hippocampal volume in older adults. Hippocampus 2015, 25, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Pentikainen, H.; Ngandu, T.; Liu, Y.; Savonen, K.; Komulainen, P.; Hallikainen, M.; Kivipelto, M.; Rauramaa, R.; Soininen, H. Cardiorespiratory fitness and brain volumes in men and women in the FINGER study. Age Ageing 2017, 46, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K. Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs. Neurodegener. Dis. Manag. 2014, 4, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Akintola, A.A.; van Heemst, D. Insulin, aging, and the brain: Mechanisms and implications. Front. Endocrinol. 2015, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Gambardella, A.; Ammendola, S.; D’Amore, A.; Balbi, V.; Varricchio, M.; D’Onofrio, F. Glucose tolerance and insulin action in healty centenarians. Am. J. Physiol. 1996, 270, E890–E894. [Google Scholar] [PubMed]
- Bertram, S.; Brixius, K.; Brinkmann, C. Exercise for the diabetic brain: How physical training may help prevent dementia and Alzheimer’s disease in T2DM patients. Endocrine 2016, 53, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Hoene, M.; Lehmann, R.; Hennige, A.M.; Pohl, A.K.; Haring, H.U.; Schleicher, E.D.; Weigert, C. Acute regulation of metabolic genes and insulin receptor substrates in the liver of mice by one single bout of treadmill exercise. J. Physiol. 2009, 587, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.G.; Venutolo, C.; Yau, P.L.; Convit, A. Fitness, insulin sensitivity, and frontal lobe integrity in adults with overweight and obesity. Obesity 2016, 24, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.B.; Cho, J.Y. Effects of treadmill exercise on brain insulin signaling and beta-amyloid in intracerebroventricular streptozotocin induced-memory impairment in rats. J. Exerc. Nutr. Biochem. 2014, 18, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.P.; Gnoatto, J.; Moreira, J.D.; Zimmer, E.R.; Haas, C.B.; Lulhier, F.; Perry, M.L.; Souza, D.O.; Torres-Aleman, I.; Portela, L.V. Exercise increases insulin signaling in the hippocampus: Physiological effects and pharmacological impact of intracerebroventricular insulin administration in mice. Hippocampus 2011, 21, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, C. Adipose “talks” to distant organs to regulate insulin sensitivity and vascular function. Obesity 2010, 18, 2071–2076. [Google Scholar] [CrossRef] [PubMed]
- Ringseis, R.; Eder, K.; Mooren, F.C.; Kruger, K. Metabolic signals and innate immune activation in obesity and exercise. Exerc. Immunol. Rev. 2015, 21, 58–68. [Google Scholar] [PubMed]
- Khoo, J.; Dhamodaran, S.; Chen, D.D.; Yap, S.Y.; Chen, R.Y.; Tian, R.H. Exercise-induced weight loss is more effective than dieting for improving adipokine profile, insulin resistance, and inflammation in obese men. Int. J. Sport Nutr. Exerc. Metab. 2015, 25, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Lowndes, J.; Zoeller, R.F.; Kyriazis, G.E.; Miles, M.P.; Seip, R.L.; Moyna, N.M.; Visich, P.; Pescatello, L.; Gordon, P.; Thompson, P.D.; et al. Hyperleptinemia is associated with CRP, but not apolipoprotein E, and is reduced by exercise training. Int. J. Sport Nutr. Exerc. Metab. 2014, 24, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Markofski, M.M.; Carrillo, A.E.; Timmerman, K.L.; Jennings, K.; Coen, P.M.; Pence, B.D.; Flynn, M.G. Exercise training modifies ghrelin and adiponectin concentrations and is related to inflammation in older adults. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Yau, S.Y.; Li, A.; Hoo, R.L.; Ching, Y.P.; Christie, B.R.; Lee, T.M.; Xu, A.; So, K.F. Physical exercise-induced hippocampal neurogenesis and antidepressant effects are mediated by the adipocyte hormone adiponectin. Proc. Natl. Acad. Sci. USA 2014, 111, 15810–15815. [Google Scholar] [CrossRef] [PubMed]
- Vella, C.A.; Allison, M.A.; Cushman, M.; Jenny, N.S.; Miles, M.P.; Larsen, B.; Lakoski, S.G.; Michos, E.D.; Blaha, M.J. Physical Activity and Adiposity-related Inflammation: The MESA. Med. Sci. Sports Exerc. 2017, 49, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Sanada, K.; Machida, S.; Okutsu, M.; Suzuki, K. Resistance exercise training-induced muscle hypertrophy was associated with reduction of inflammatory markers in elderly women. Mediat. Inflamm. 2010, 2010, 171023. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.V.; Viana, V.A.; Boscolo, R.A.; Marques, V.G.; Santana, M.G.; Lira, F.S.; Tufik, S.; de Mello, M.T. Moderate exercise training modulates cytokine profile and sleep in elderly people. Cytokine 2012, 60, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Miguelez, P.; Fernandez-Gonzalo, R.; Almar, M.; Mejias, Y.; Rivas, A.; de Paz, J.A.; Cuevas, M.J.; Gonzalez-Gallego, J. Role of Toll-like receptor 2 and 4 signaling pathways on the inflammatory response to resistance training in elderly subjects. Age 2014, 36, 9734. [Google Scholar] [CrossRef] [PubMed]
- Chupel, M.U.; Direito, F.; Furtado, G.E.; Minuzzi, L.G.; Pedrosa, F.M.; Colado, J.C.; Ferreira, J.P.; Filaire, E.; Teixeira, A.M. Strength training decreases inflammation and increases cognition and physical fitness in older women with cognitive impairment. Front. Physiol. 2017, 8, 377. [Google Scholar] [CrossRef] [PubMed]
- Braskie, M.N.; Boyle, C.P.; Rajagopalan, P.; Gutman, B.A.; Toga, A.W.; Raji, C.A.; Tracy, R.P.; Kuller, L.H.; Becker, J.T.; Lopez, O.L.; et al. Physical activity, inflammation, and volume of the aging brain. Neuroscience 2014, 273, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Chennaoui, M.; Gomez-Merino, D.; Drogou, C.; Geoffroy, H.; Dispersyn, G.; Langrume, C.; Ciret, S.; Gallopin, T.; Sauvet, F. Effects of exercise on brain and peripheral inflammatory biomarkers induced by total sleep deprivation in rats. J. Inflamm. 2015, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C.; Davis, J.M.; Brown, V.A.; Henson, D.A.; Dumke, C.L.; Utter, A.C.; Vinci, D.M.; Downs, M.F.; Smith, J.C.; Carson, J.; et al. Influence of carbohydrate ingestion on immune changes after 2 h of intensive resistance training. J. Appl. Physiol. 2004, 96, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Della Gatta, P.A.; Garnham, A.P.; Peake, J.M.; Cameron-Smith, D. Effect of exercise training on skeletal muscle cytokine expression in the elderly. Brain Behav. Immun. 2014, 39, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Piao, C.S.; Stoica, B.A.; Wu, J.; Sabirzhanov, B.; Zhao, Z.; Cabatbat, R.; Loane, D.J.; Faden, A.I. Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol. Dis. 2013, 54, 252–263. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Are major dementias triggered by poor blood flow to the brain? Theoretical considerations. J. Alzheimer Dis. 2017, 57, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed]
- Mejias-Pena, Y.; Estebanez, B.; Rodriguez-Miguelez, P.; Fernandez-Gonzalo, R.; Almar, M.; de Paz, J.A.; Gonzalez-Gallego, J.; Cuevas, M.J. Impact of resistance training on the autophagy-inflammation-apoptosis crosstalk in elderly subjects. Aging 2017, 9, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Mejias-Pena, Y.; Rodriguez-Miguelez, P.; Fernandez-Gonzalo, R.; Martinez-Florez, S.; Almar, M.; de Paz, J.A.; Cuevas, M.J.; Gonzalez-Gallego, J. Effects of aerobic training on markers of autophagy in the elderly. Age 2016, 38, 33. [Google Scholar] [CrossRef] [PubMed]
- Marosi, K.; Bori, Z.; Hart, N.; Sarga, L.; Koltai, E.; Radak, Z.; Nyakas, C. Long-term exercise treatment reduces oxidative stress in the hippocampus of aging rats. Neuroscience 2012, 226, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mesa, Y.; Colie, S.; Corpas, R.; Cristofol, R.; Comellas, F.; Nebreda, A.R.; Gimenez-Llort, L.; Sanfeliu, C. Oxidative stress is a central target for physical exercise neuroprotection against pathological brain aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Raefsky, S.M.; Mattson, M.P. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 2017, 102, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Marques-Aleixo, I.; Santos-Alves, E.; Balca, M.M.; Rizo-Roca, D.; Moreira, P.I.; Oliveira, P.J.; Magalhaes, J.; Ascensao, A. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 2015, 301, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Panati, K.; Suneetha, Y.; Narala, V.R. Irisin/FNDC5—An updated review. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 689–697. [Google Scholar] [PubMed]
- Raschke, S.; Eckel, J. Adipo-myokines: Two sides of the same coin—Mediators of inflammation and mediators of exercise. Mediat. Inflamm. 2013, 2013, 320724. [Google Scholar] [CrossRef] [PubMed]
- Dinoff, A.; Herrmann, N.; Swardfager, W.; Liu, C.S.; Sherman, C.; Chan, S.; Lanctot, K.L. The effect of exercise training on resting concentrations of peripheral brain-derived neurotrophic factor (BDNF): A Meta-Analysis. PLoS ONE 2016, 11, e0163037. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Miller, D.L.; Roecklein, K.A. The aging hippocampus: Interactions between exercise, depression, and BDNF. Neuroscientist 2012, 18, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D. FNDC5/irisin—Their role in the nervous system and as a mediator for beneficial effects of exercise on the brain. Brain Plast. 2015, 1, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.S.; Ghaedi, K.; Salamian, A.; Karbalaie, K.; Emadi-Baygi, M.; Tanhaei, S.; Nasr-Esfahani, M.H.; Baharvand, H. Fndc5 knockdown significantly decreased neural differentiation rate of mouse embryonic stem cells. Neuroscience 2013, 231, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.S.; Sheng, H.; Batinic-Haberle, I. Oxidants, antioxidants and the ischemic brain. J. Exp. Biol. 2004, 207, 3221–3231. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Atamna, H.; Kuratsune, H.; Ames, B.N. Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann. N. Y. Acad. Sci. 2002, 959, 133–166. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Slemmer, J.E.; Shacka, J.J.; Sweeney, M.I.; Weber, J.T. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr. Med. Chem. 2008, 15, 404–414. [Google Scholar] [PubMed]
- De Domenico, S.; Giudetti, A.M. Nutraceutical intervention in ageing brain. J. Gerontol. Geriatr. 2017, 65, 79–92. [Google Scholar]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitas, H.R.; Ferreira, G.D.C.; Trevenzoli, I.H.; Oliveira, K.D.J.; De Melo Reis, R.A. Fatty Acids, Antioxidants and Physical Activity in Brain Aging. Nutrients 2017, 9, 1263. https://doi.org/10.3390/nu9111263
Freitas HR, Ferreira GDC, Trevenzoli IH, Oliveira KDJ, De Melo Reis RA. Fatty Acids, Antioxidants and Physical Activity in Brain Aging. Nutrients. 2017; 9(11):1263. https://doi.org/10.3390/nu9111263
Chicago/Turabian StyleFreitas, Hércules Rezende, Gustavo Da Costa Ferreira, Isis Hara Trevenzoli, Karen De Jesus Oliveira, and Ricardo Augusto De Melo Reis. 2017. "Fatty Acids, Antioxidants and Physical Activity in Brain Aging" Nutrients 9, no. 11: 1263. https://doi.org/10.3390/nu9111263
APA StyleFreitas, H. R., Ferreira, G. D. C., Trevenzoli, I. H., Oliveira, K. D. J., & De Melo Reis, R. A. (2017). Fatty Acids, Antioxidants and Physical Activity in Brain Aging. Nutrients, 9(11), 1263. https://doi.org/10.3390/nu9111263