Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper



Abstract
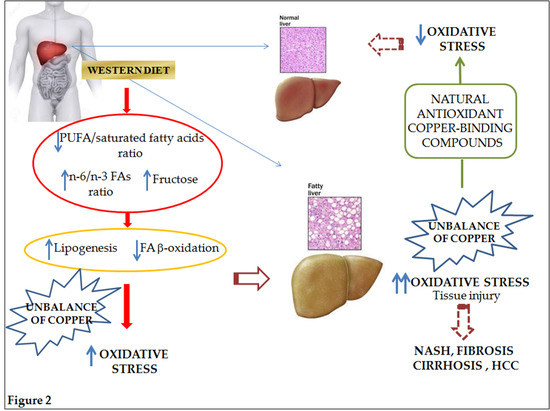
:
1. Introduction
2. Pathophysiology of NAFLD and Nutritional Implications
2.1. Dietary Fats
2.2. Dietary Carbohydrates
3. NAFLD-Related Oxidative Stress
3.1. Special Focus on Copper
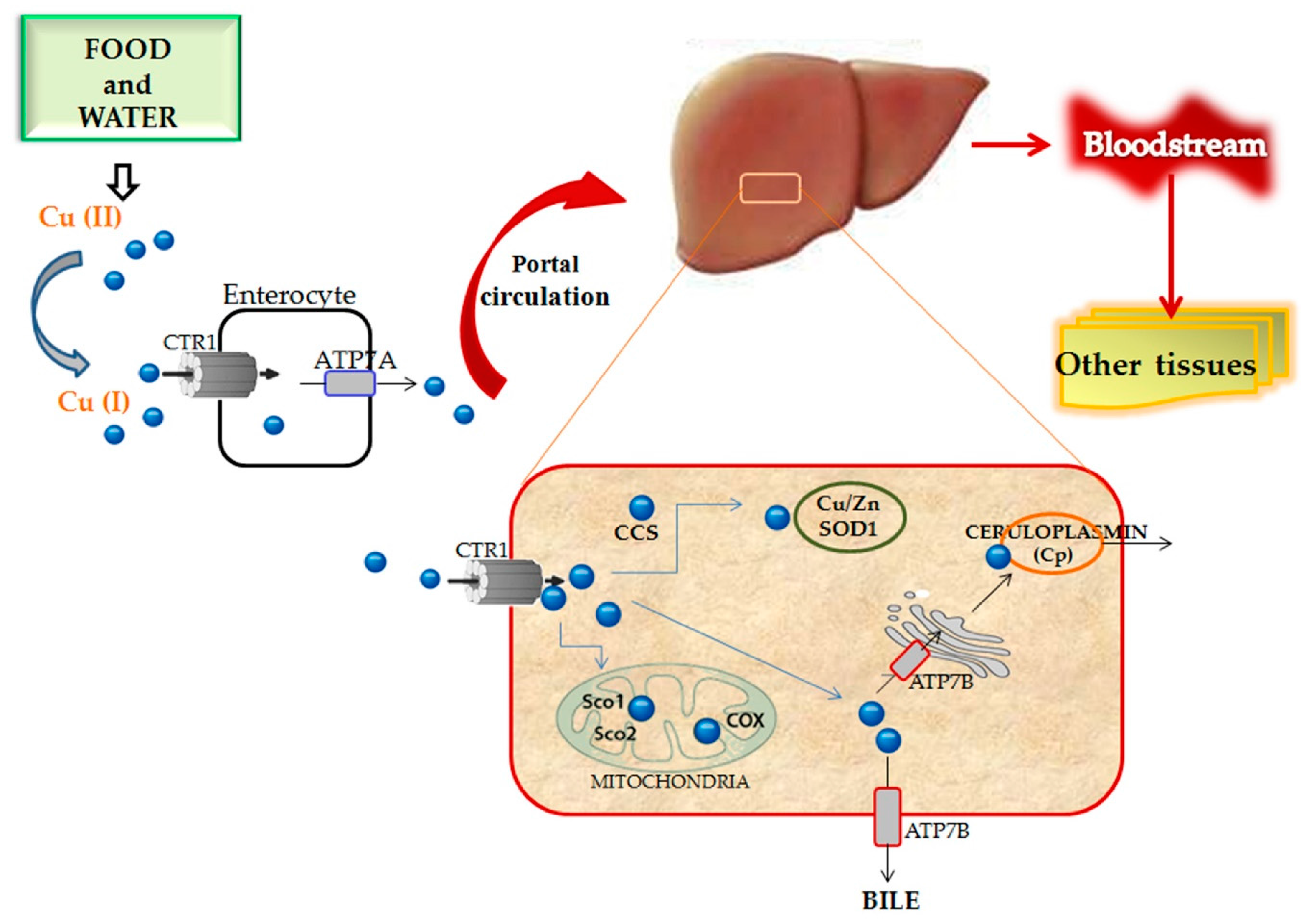
3.1.1. Copper Homeostasis and Metabolism
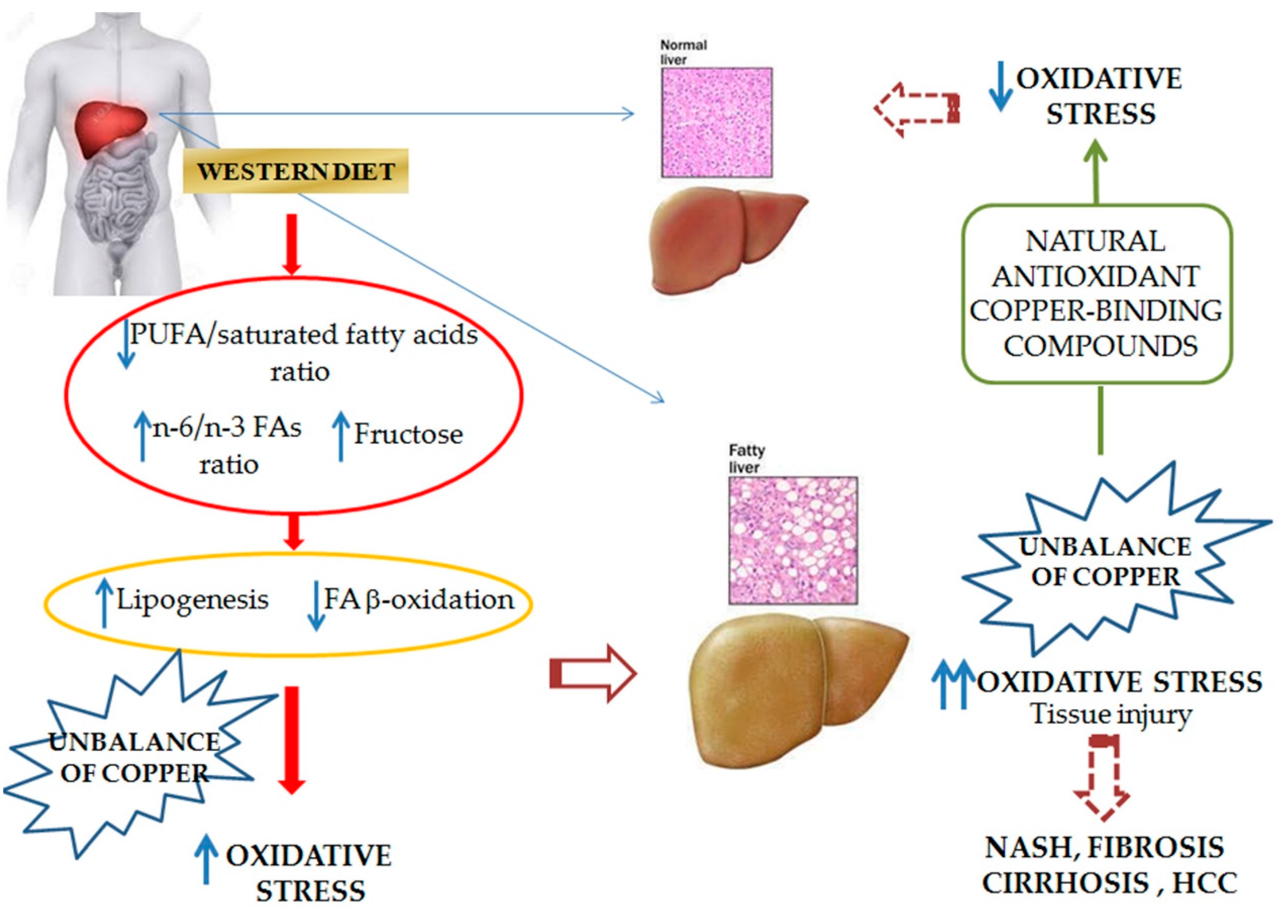
3.1.2. Copper and Its Role in NAFLD Onset and Progression
3.1.3. Copper and Nutrients in NAFLD
Curcumin
Epigallocatechin-3-Gallate (EGCG)
Luteolin and Luteolin-7-Glucoside
Caffeic Acid and Caffeine
Oleuropein
Quercetin and Rutin (Quercetin-3-O-Rutinoside)
Resveratrol (3,5,4′-Trihydroxy-Trans-Stilbene)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Systematic review with meta-analysis: Risk factors for non-alcoholic fatty liver disease suggest a shared altered metabolic and cardiovascular profile between lean and obese patients. Aliment. Pharmacol. Ther. 2017, 46, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Tallino, S.; Duffy, M.; Ralle, M.; Cortés, M.P.; Latorre, M.; Burkhead, J.L. Nutrigenomics analysis reveals that copper deficiency and dietary sucrose upregulate inflammation, fibrosis and lipogenic pathways in a mature rat model of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2015, 26, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, K.; Matsuura, B.; Ohkubo, I.; Niiya, T.; Furukawa, S.; Hiasa, Y.; Kawamura, M.; Ebihara, K.; Onji, M. Dietary habits and nutrient intake in non-alcoholic steatohepatitis. Nutrition 2007, 23, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Ravasco, P.; Jesus, L.; Marques-Vidal, P.; Oliveira, C.R.; Proença, T.; Baldeiras, I.; Camilo, M.E.; Cortez-Pinto, H. Blood oxidative stress markers in non-alcoholic steatohepatitis and how it correlates with diet. Scand. J. Gastroenterol. 2008, 43, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Jornayvaz, F.R. Diets and nonalcoholic fatty liver disease: The good and the bad. Clin. Nutr. 2014, 33, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Capanni, M.; Calella, F.; Biagini, M.R.; Genise, S.; Raimondi, L.; Bedogni, G.; Svegliati-Baroni, G.; Sofi, F.; Milani, S.; Abbate, R.; et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: A pilot study. Aliment. Pharmacol. Ther. 2006, 23, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Zúiga, J.; Cancino, M.; Medina, F.; Varela, P.; Vargas, R.; Tapia, G.; Videla, L.A.; Fernández, V. N-3 PUFA supplementation triggers PPAR-α activation and PPAR-α/NF-κB interaction: Anti-inflammatory implications in liver ischemia-reperfusion injury. PLoS ONE 2011, 6, e28502. [Google Scholar]
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V., Jr.; Zechner, R.; Newgard, C.B.; Walther, T.C. The Problem of Establishing Relationships between Hepatic Steatosis and Hepatic Insulin Resistance. Cell Metab. 2012, 15, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Després, J.-P.; Hu, F.B. Sugar Sweetend Beverages, Obesity, Type 2 Diabetes and Cardiovascular Disease risk. Circulation 2010, 121, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg. Nutr. 2015, 4, 109–116. [Google Scholar] [PubMed]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Gambino, R.; Bugianesi, E.; Rosso, C.; Mezzabotta, L.; Pinach, S.; Alemanno, N.; Saba, F.; Cassader, M. Different serum free fatty acid profiles in NAFLD subjects and healthy controls after oral fat load. Int. J. Mol. Sci. 2016, 17, 479. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carreras, M.; del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Perlegas, P.; Zhao, Y.; Angdisen, J.; Borgerink, H.; Shadoan, M.K.; Wagner, J.D.; Matern, D.; Rinaldo, P.; Cline, J.M. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 2005, 128, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Opazo, C.M.; Greenough, M.A.; Bush, A.I. Copper: From neurotransmission to neuroproteostasis. Front. Aging Neurosci. 2014, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Dorts, J.; Falisse, E.; Schoofs, E.; Flamion, E.; Kestemont, P.; Silvestre, F. DNA methyltransferases and stress-related genes expression in zebrafish larvae after exposure to heat and copper during reprogramming of DNA methylation. Sci. Rep. 2016, 6, 34254. [Google Scholar] [CrossRef] [PubMed]
- Hordyjewska, A.; Popiołek, Ł.; Kocot, J. The many ‘faces’ of copper in medicine and treatment. BioMetals 2014, 27, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Heffern, M.C.; Park, H.M.; Au-Yeung, H.Y.; Van de Bittner, G.C.; Ackerman, C.M.; Stahl, A.; Chang, C.J. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2016, 113, 14219–14224. [Google Scholar] [CrossRef] [PubMed]
- Morrell, A.; Tallino, S.; Yu, L.; Burkhead, J.L. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life 2017, 69, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H.; Maryon, E.B. How Mammalian Cells Acquire Copper: An Essential but Potentially Toxic Metal. Biophys. J. 2016, 110, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.; Mar, D.; Ishida, M.; Vargas, R.; Gaite, M.; Montgomery, A.; Linder, M.C. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS ONE 2016, 9, 815–826. [Google Scholar]
- Nose, Y.; Kim, B.E.; Thiele, D.J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Bissig, K.D.; Honer, M.; Zimmermann, K.; Summer, K.H.; Solioz, M. Whole animal copper flux assessed by positron emission tomography in the Long-Evans cinnamon rat—A feasibility study. BioMetals 2005, 18, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Lanti, C.; Riso, P.; Valenti, L. Nutritional therapy for nonalcoholic fatty liver disease. J. Nutr. Biochem. 2016, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tadini-Buoninsegni, F.; Smeazzetto, S. Mechanisms of charge transfer in human copper ATPases ATP7A and ATP7B. IUBMB Life 2017, 69, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.F.; Knutson, M.D. Metabolic crossroads of iron and copper. Nutr. Rev. 2010, 68, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Peng, J.; Wu, Q.; Ren, Z.; Pan, L.; Tang, Z.; Jiang, Z.; Wang, G.; Liu, L. Imbalanced cholesterol metabolism in Alzheimer’s disease. Clin. Chim. Acta 2016, 456, 107–114. [Google Scholar]
- Burkhead, J.L.; Lutsenko, S. The Role of Copper as a Modifier of Lipid Metabolism. In Lipid Metabolism; Valenzuela Baez, R., Ed.; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Aigner, E.; Strasser, M.; Haufe, H.; Sonnweber, T.; Hohla, F.; Stadlmayr, A.; Solioz, M.; Tilg, H.; Patsch, W.; et al. A role for low hepatic copper concentrations in nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2010, 105, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Tosco, A.; Fontanella, B.; Danise, R.; Cicatiello, L.; Grober, OM.; Ravo, M.; Weisz, A.; Marzullo, L. Molecular bases of copper and iron deficiency-associated dyslipidemia: A microarray analysis of the rat intestinal transcriptome. Genes Nutr. 2010, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Siotto, M.; Bedogni, G.; Ravà, L.; Pietrobattista, A.; Panera, N.; Alisi, A.; Squitti, R. Levels of serum ceruloplasmin associate with pediatric nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Porcu, C.; Antonucci, L.; Barbaro, B.; Illi, B.; Nasi, S.; Licata, A.; Miele, L.; Grieco, A.; Balsano, C. The copper/MYC interplay: A dangerous relationship promoting hepatocellular carcinoma. Oncotarget 2017. Submitted. [Google Scholar]
- Xu, R.; Hajdu, C.H. Wilson disease and hepatocellular carcinoma. Gastroenterol. Hepatol. 2008, 4, 438–439. [Google Scholar]
- Iwadate, H.; Ohira, H.; Suzuki, T.; Abe, K.; Yokokawa, J.; Takiguchi, J.; Rai, T.; Orikasa, H.; Irisawa, A.; et al. Hepatocellular Carcinoma Associated with Wilson’s Disease. Intern. Med. 2004, 43, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; The National Academies Press: Washington, DC, USA, 2001. [Google Scholar]
- World Health Organization. Copper in Drinking-Water; 2004 WHO/SDE/WSH/03.04/88; WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Noguchi, N.; Watanabe, A.; Shi, H. Diverse functions of antioxidants. Free Radic. Res. 2000, 33, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Polimeni, L.; Baratta, F.; Pastori, D.; Angelico, F. The role of nutraceuticals for the treatment of non-alcoholic fatty liver disease. Br. J. Clin. Pharmacol. 2017, 83, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Godos, J.; Federico, A.; Dallio, M.; Scazzina, F. Mediterranean diet and nonalcoholic fatty liver disease: Molecular mechanisms of protection. Int. J. Food Sci. Nutr. 2017, 68, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Godos, J.; Zelber-Sagi, S. Natural antioxidants for non-alcoholic fatty liver disease: Molecular targets and clinical perspectives. Liver Int. 2016, 36, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, H.; Bruck, R. Therapeutic potential of curcumin in non-alcoholic steatohepatitis. Nutr. Res. Rev. 2005, 18, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Inzaugarat, M.E.; De Matteo, E.; Baz, P.; Lucero, D.; García, C.C.; Gonzalez Ballerga, E.; Daruich, J.; Sorda, J.A.; Wald, M.R.; Cherñavsky, A.C. New evidence for the therapeutic potential of curcumin to treat nonalcoholic fatty liver disease in humans. PLoS ONE 2017, 12, e0172900. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Jiang, T.; Wang, L.; Yang, H.; Zhang, S.; Zhou, P. Interaction of curcumin with Zn (II) and Cu (II) ions based on experiment and theoretical calculation. J. Mol. Struct. 2010, 984, 316–325. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, C.; Shi, H.; Yang, M.; Liu, Y.; Ji, P.; Chen, H.; Tan, R.X.; Li, E. Curcumin is a biologically active copper chelator with antitumor activity. Phytomedicine 2016, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Santamarina, A.B.; Carvalho-Silva, M.; Gomes, L.M.; Okuda, M.H.; Santana, A.A.; Streck, E.L.; Seelaender, M.; do Nascimento, C.M.; Ribeiro, E.B.; Lira, F.S.; et al. Decaffeinated green tea extract rich in epigallocatechin-3-gallate prevents fatty liver disease by increased activities of mitochondrial respiratory chain complexes in diet-induced obesity mice. J. Nutr. Biochem. 2015, 26, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Wing, V.; Hung, S.; Bressan, P.; Seo, K.; Kerman, K. Electroanalysis of Natural Compounds as Copper Chelating Agents for Alzheimer’s Disease Therapy. Electroanalysis 2015, 27, 2670–2678. [Google Scholar]
- Yin, Y.; Gao, L.; Lin, H.; Wu, Y.; Han, X.; Zhu, Y.; Li, J. Luteolin improves non-alcoholic fatty liver disease in db/db mice by inhibition of liver X receptor activation to downregulate expression of sterol regulatory element binding protein 1c. Biochem. Biophys. Res. Commun. 2017, 482, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Sá, C.; Oliveira, A.R.; Machado, C.; Azevedo, M.; Pereira-wilson, C. Effects on Liver Lipid Metabolism of the Naturally Occurring Dietary Flavone Luteolin-7-glucoside. Evid. Based Complement. Altern. Med. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.E.; Khodr, H.; Hider, R.C.; Rice-Evans, C.A. Structural dependence of flavonoid interactions with Cu2+ ions: Implications for their antioxidant properties. Biochem. J. 1998, 330, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Yesil, A.; Yilmaz, Y. Review article: Coffee consumption, the metabolic syndrome and non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013, 38, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Lipka, S.; Shahzad, G.; Kumar, A.; Mustacchia, P. Association between caffeine consumption and nonalcoholic fatty liver disease: A systemic review and meta-analysis. Am. J. Gastroenterol. 2016, 9, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.C.; Ou, T.T.; Huang, H.P.; Wang, C.J. The inhibition of oleic acid induced hepatic lipogenesis and the promotion of lipolysis by caffeic acid via upregulation of AMP-activated kinase. J. Sci. Food Agric. 2014, 94, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Nkhili, E.; Loonis, M.; Mihai, S.; el Hajji, H.; Dangles, O. Reactivity of food phenols with iron and copper ions: Binding, dioxygen activation and oxidation mechanisms. Food Funct. 2014, 5, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, B.; Toietta, G.; Maggio, R.; Arciello, M.; Tarocchi, M.; Galli, A.; Balsano, C. Effects of the olive-derived polyphenol oleuropein on human health. Int. J. Mol. Sci. 2014, 15, 18508–18524. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Hur, W.; Li, T.Z.; Lee, Y.K.; Choi, J.E.; Hong, S.W.; Lyoo, K.S.; You, C.R.; Jung, E.S.; Jung, C.K.; Park, T.; et al. Oleuropein prevents the progression of steatohepatitis to hepatic fibrosis induced by a high-fat diet in mice. Exp. Mol. Med. 2014, 46, e92. [Google Scholar] [CrossRef] [PubMed]
- Bendini, A.; Cerretani, L.; Vecchi, S.; Carrasco-Pancorbo, A.; Lercker, G. Protective effects of extra virgin olive oil phenolics on oxidative stability in the presence or absence of copper ions. J. Agric. Food Chem. 2006, 54, 4880–4887. [Google Scholar] [CrossRef] [PubMed]
- Porras, D.; Nistal, E.; Martinez-Flores, S.; Pisonero-Vaquero, S.; Olcoz, J.L.; Jover, R.; González-Gallego, J.; García-Mediavilla, M.V.; Sánchez-Campos, S. Protective effect of quercetin on high-fat diet-induced non-alcoholic fatty liver disease in mice is mediated by modulating intestinal microbiota imbalance and related gut-liver axis activation. Free Radic. Biol. Med. 2017, 102, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Panchal, S.K.; Poudyal, H.; Arumugam, T.V.; Brown, L. Rutin Attenuates Metabolic Changes, Nonalcoholic Steatohepatitis, and Cardiovascular Remodeling in High-Carbohydrate, High-Fat Diet-Fed Rats. J. Nutr. 2011, 141, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.B.; Memon, S.; Mahroof-Tahir, M.; Bhanger, M.I. Synthesis, characterization and antioxidant activity copper-quercetin complex. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2009, 71, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.; Portillo, M.P.; Hijona, E.; Bujanda, L. Effects of resveratrol and other polyphenols in hepatic steatosis. World J. Gastroenterol. 2014, 20, 7366–7380. [Google Scholar] [CrossRef] [PubMed]
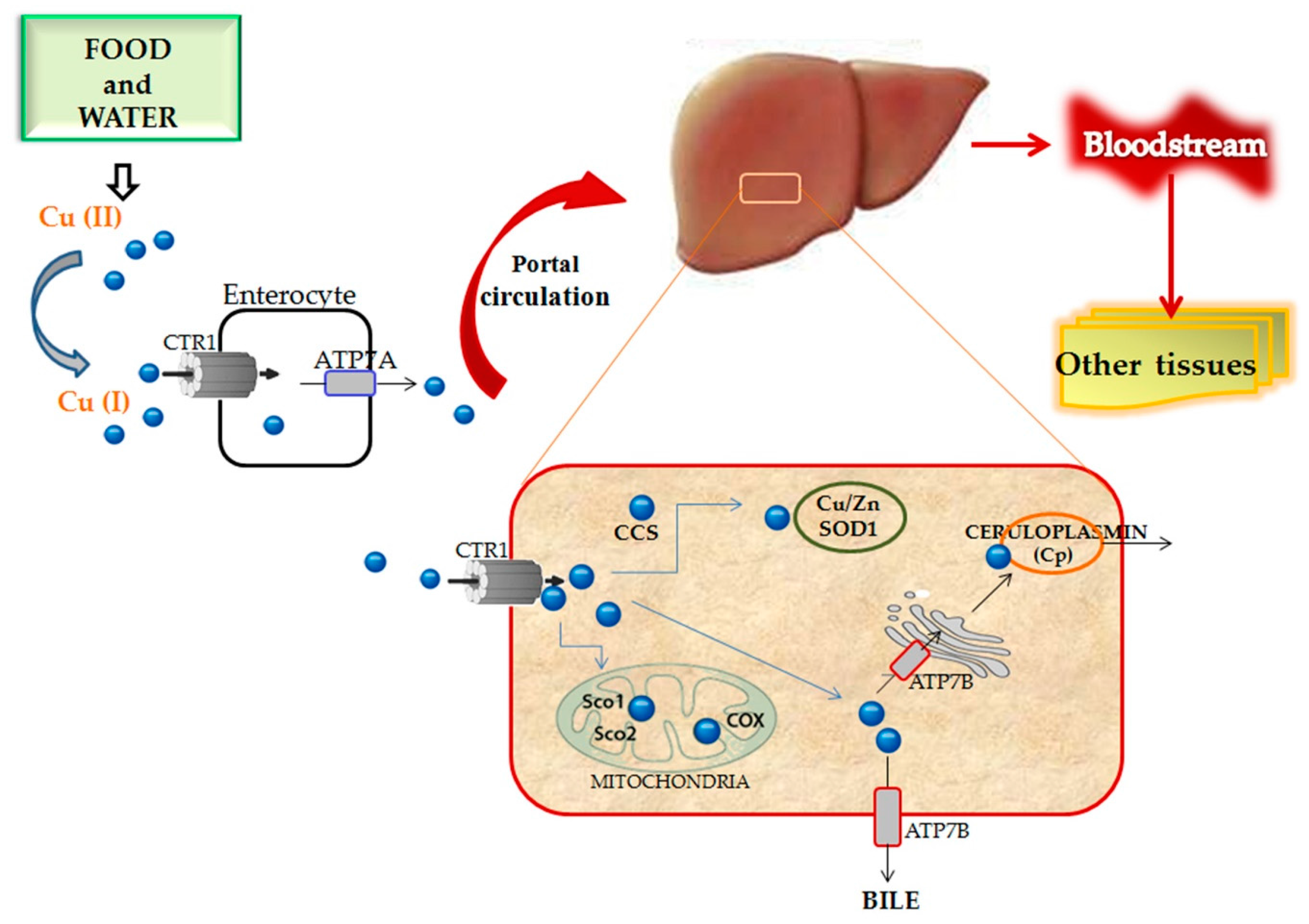
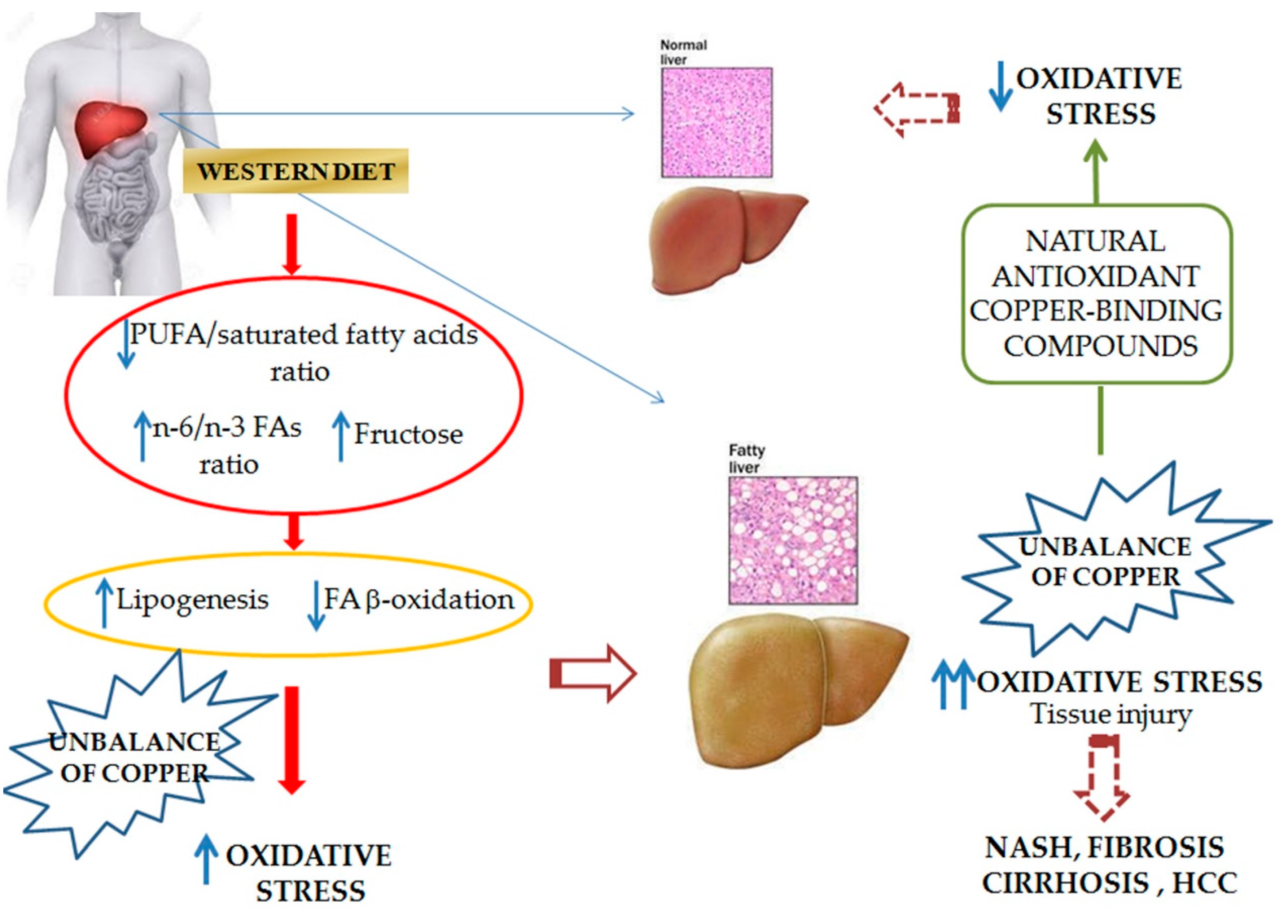

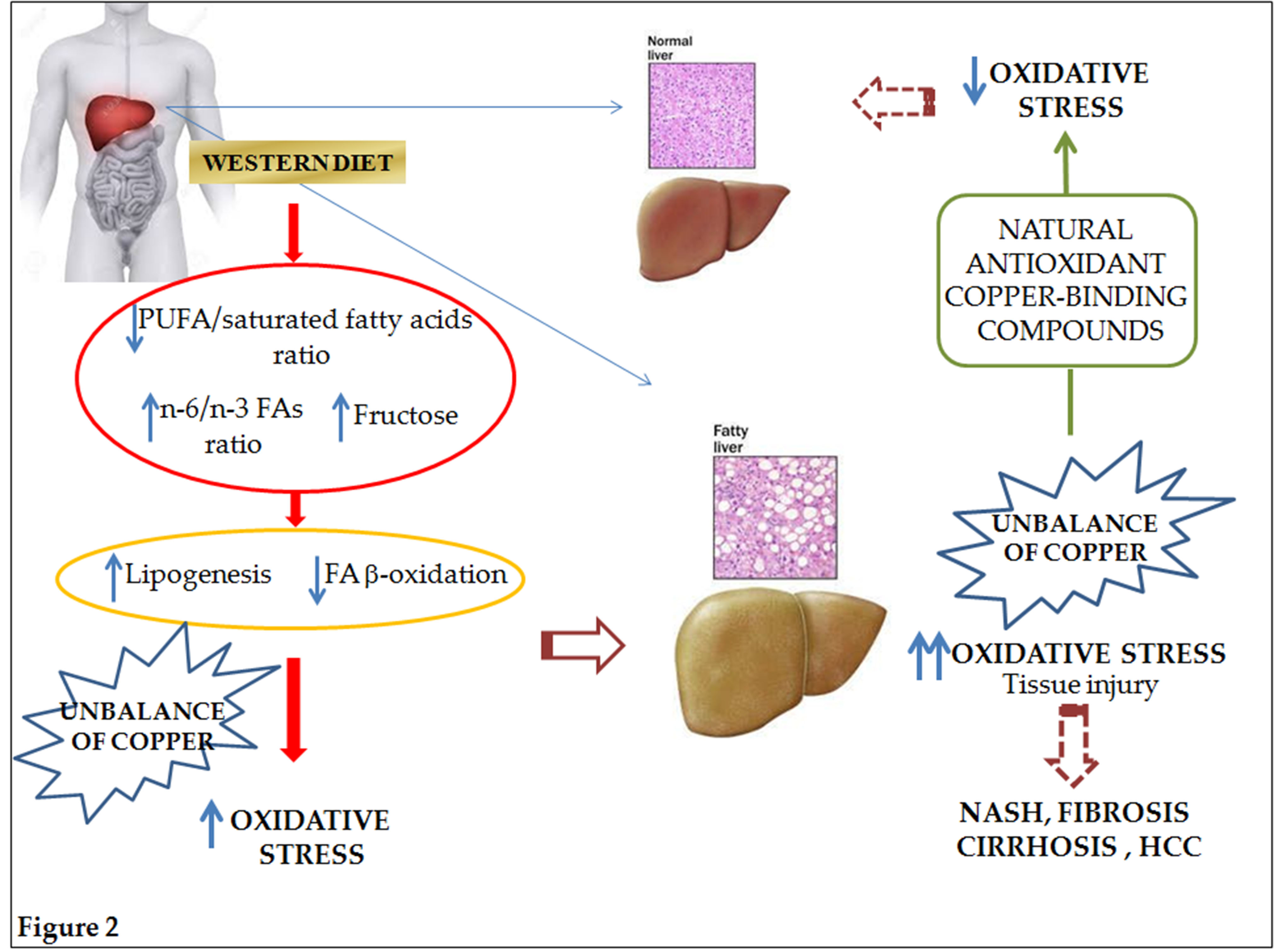{kind=link}
{kind=link}
{kind=link}
| Natural Antioxidant | Food Source | Ability to Counteract NAFLD and Its Progression | Copper Binding Ability |
|---|---|---|---|
| Curcumin | Rhizomes of Curcuma longa | Salomone et al., 2016 [51] Shapiro and Bruck, 2005 [52] Inzaugarat et al., 2017 [53] | Zhao et al., 2016 [54] Zhang et al., 2010 [55] |
| Epigallocatechin-3-Gallate (EGCG) | Green tea | Aline B. Santamarina et al., 2015 [56] | Wing et al., 2015 [57] |
| Luteolin and Luteolin-7-Glucoside | Aromatic plants | Yin et al., 2017 [58] Sá et al., 2015 [59] | Brown et al., 1998 [60] |
| Caffeic Acid and Caffeine | Coffee | Yesil et al., 2013 [61] Shen et al., 2014 [62] Liao et al., 2014 [63] | Nkhili et al., 2014 [64] |
| Oleuropein | Olive and olive leaves | Barbaro et al., 2014 [65] Kim et al., 2014 [66] | Bendini et al., 2006 [67] |
| Quercetin and Rutin | Vegetables, mostly capers and radish, citrus fruit | Porras et al., 2016 [68] Panchal et al., 2011 [69] | Brown et al., 1998 [60] Bukhari et al., 2009 [70] |
| Resveratrol | Grapes | Aguirre Let al., 2014 [71] | Wing et al., 2015 [57] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonucci, L.; Porcu, C.; Iannucci, G.; Balsano, C.; Barbaro, B. Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper. Nutrients 2017, 9, 1137. https://doi.org/10.3390/nu9101137
Antonucci L, Porcu C, Iannucci G, Balsano C, Barbaro B. Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper. Nutrients. 2017; 9(10):1137. https://doi.org/10.3390/nu9101137
Chicago/Turabian StyleAntonucci, Laura, Cristiana Porcu, Gino Iannucci, Clara Balsano, and Barbara Barbaro. 2017. "Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper" Nutrients 9, no. 10: 1137. https://doi.org/10.3390/nu9101137
APA StyleAntonucci, L., Porcu, C., Iannucci, G., Balsano, C., & Barbaro, B. (2017). Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper. Nutrients, 9(10), 1137. https://doi.org/10.3390/nu9101137