Glutamine Modulates Macrophage Lipotoxicity

Abstract
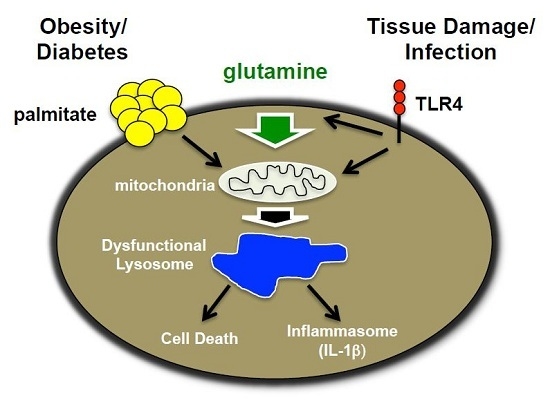
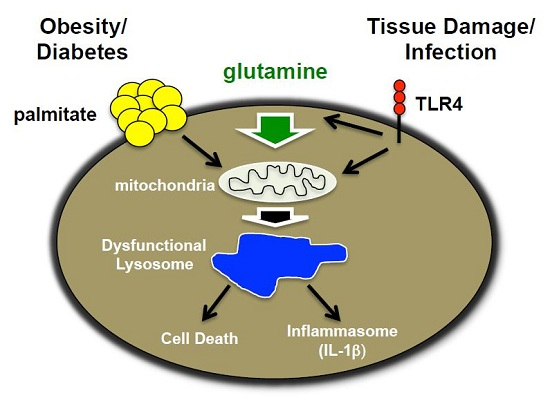
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Reagents
2.2. Cell Culture
2.3. Mice
2.4. RNA Isolation and Quantitative RT-PCR
2.5. Western Blotting
2.6. Lysosome Imaging
2.7. Metabolism Assays
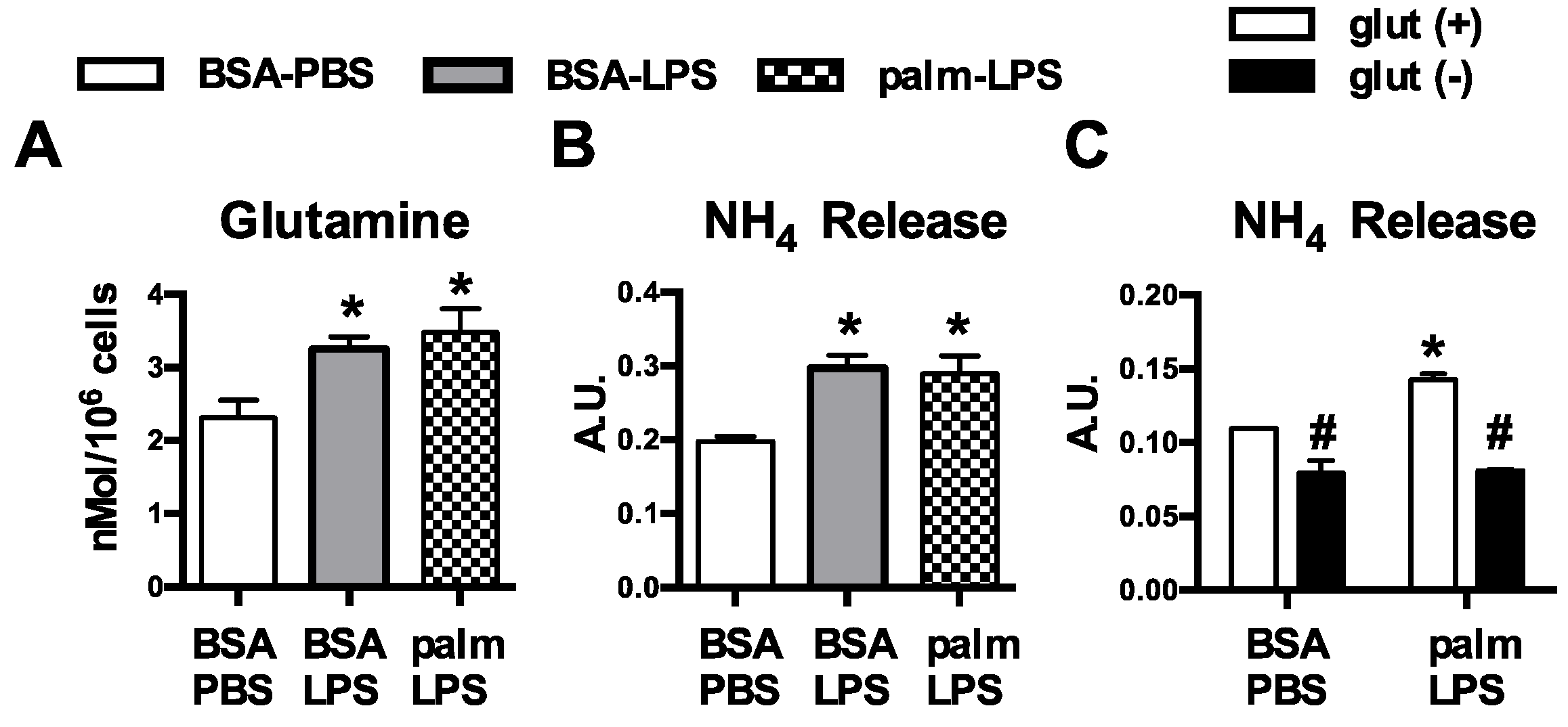
2.8. Ammonia Quantification
2.9. Intracellular Glutamine Quantification
3. Results
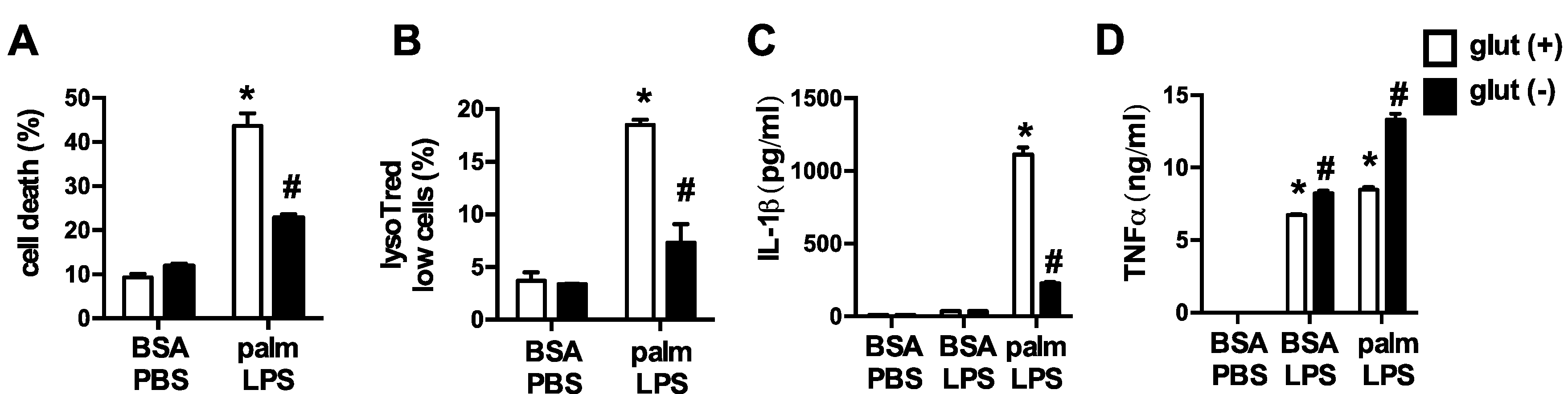
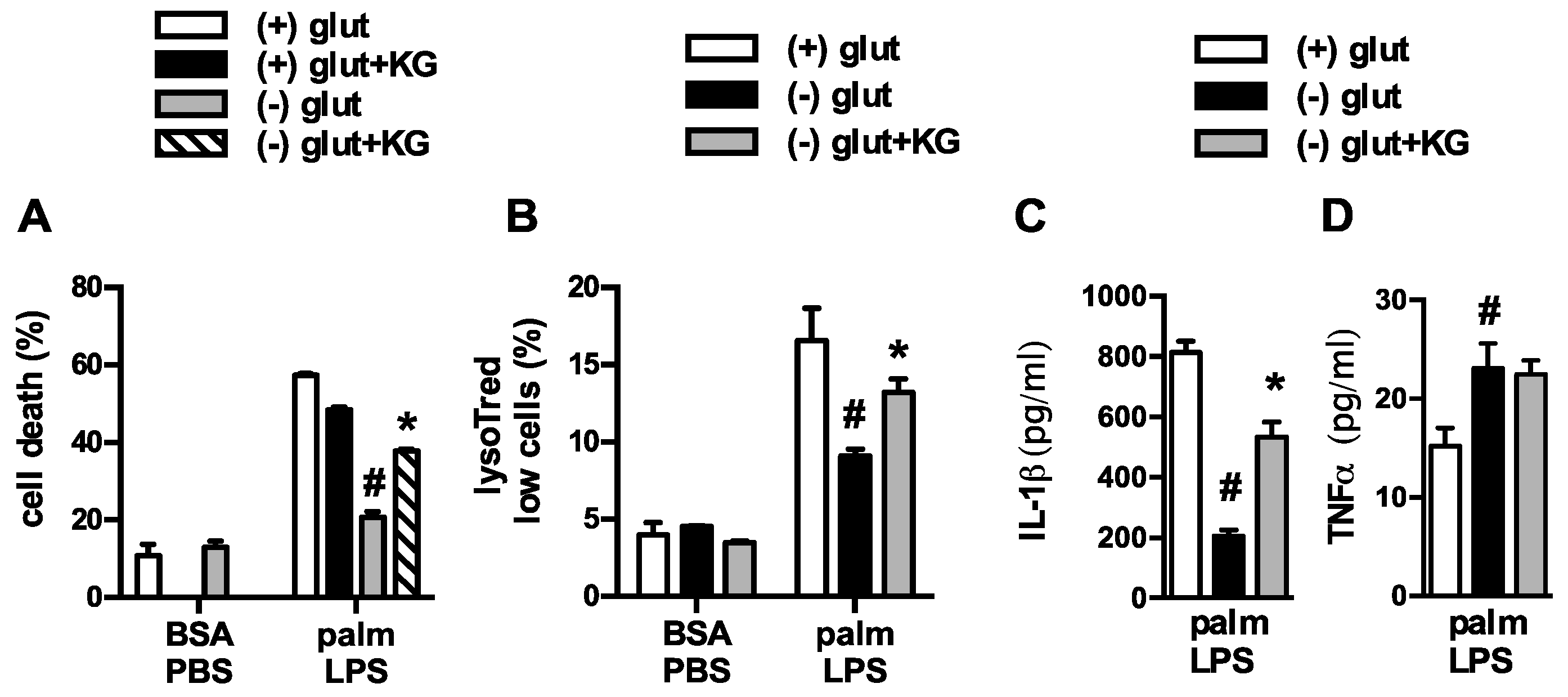
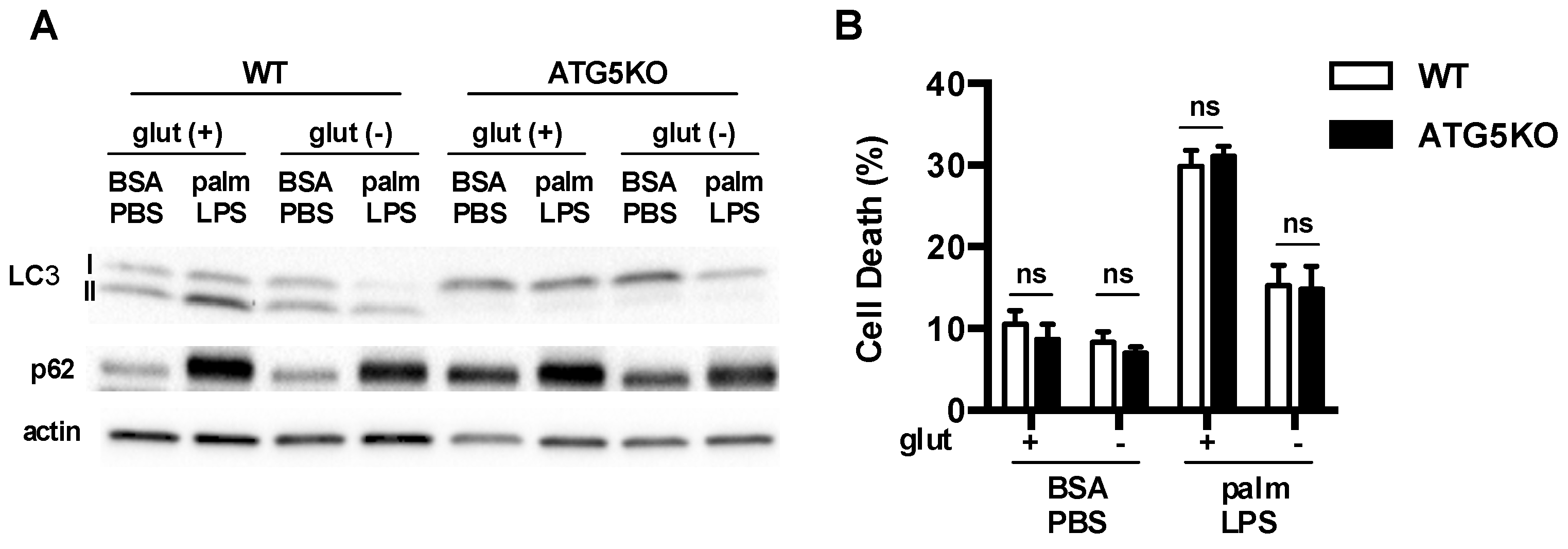
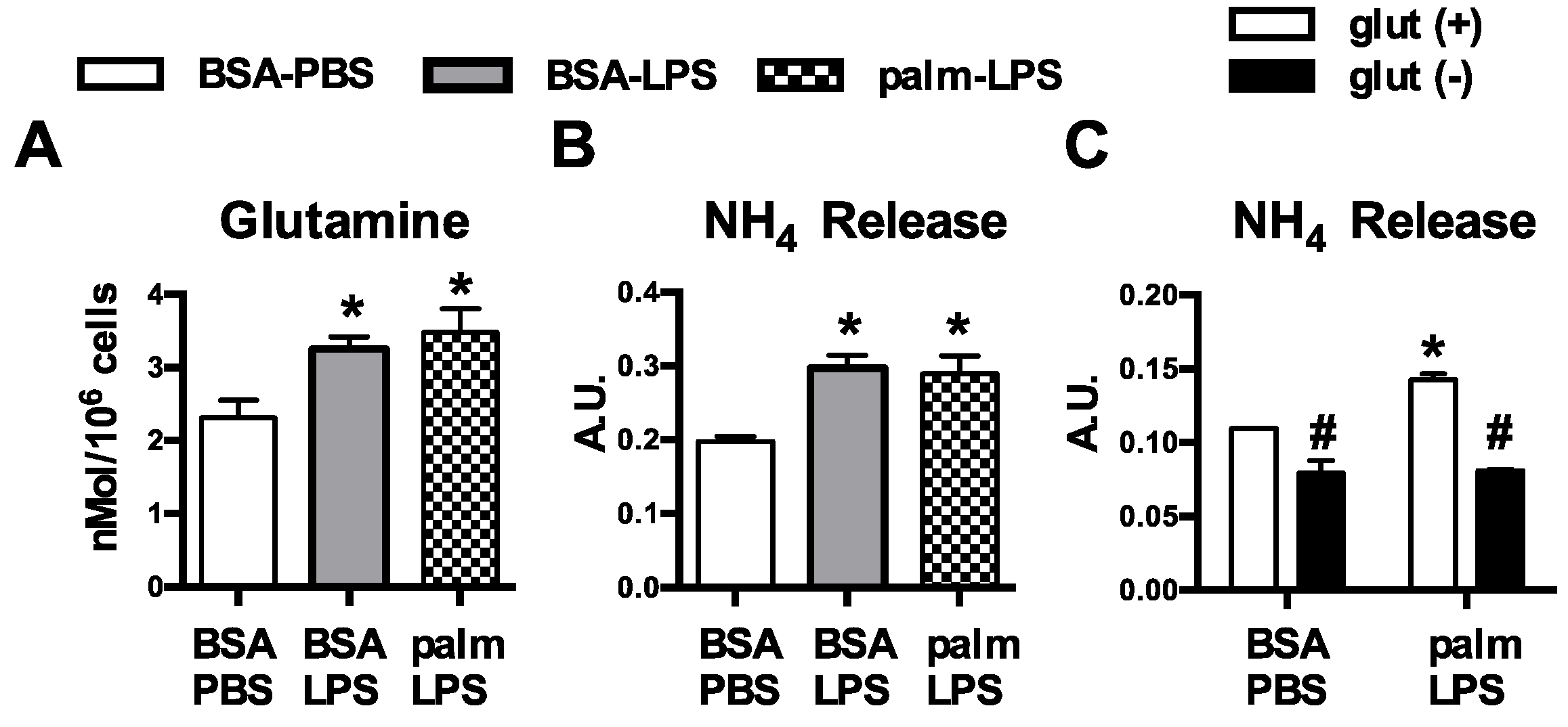
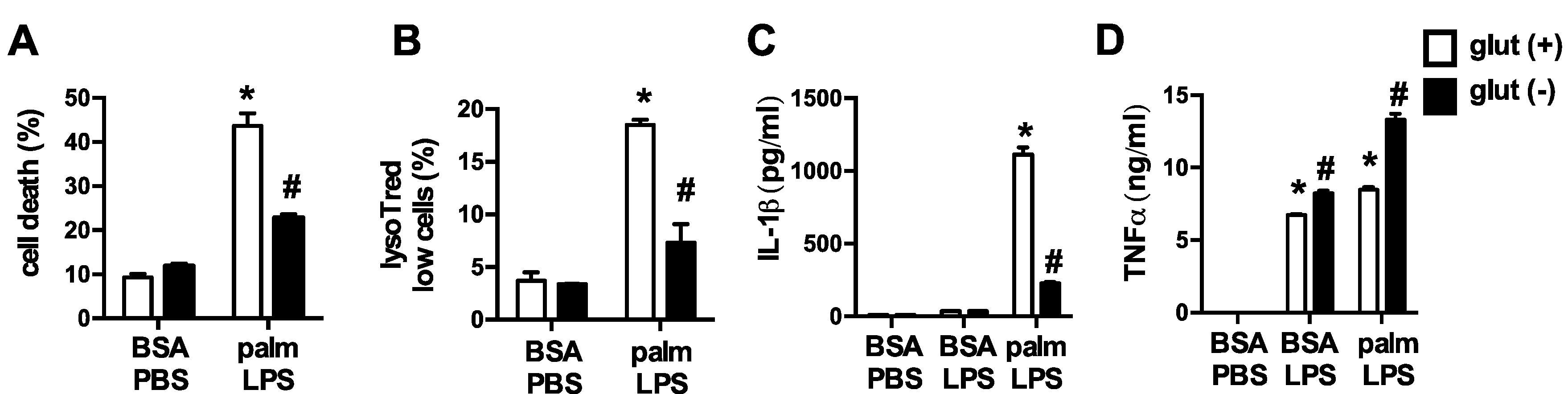
3.1. Glutamine Deficiency Attenuates Macrophage Lipotoxic Responses
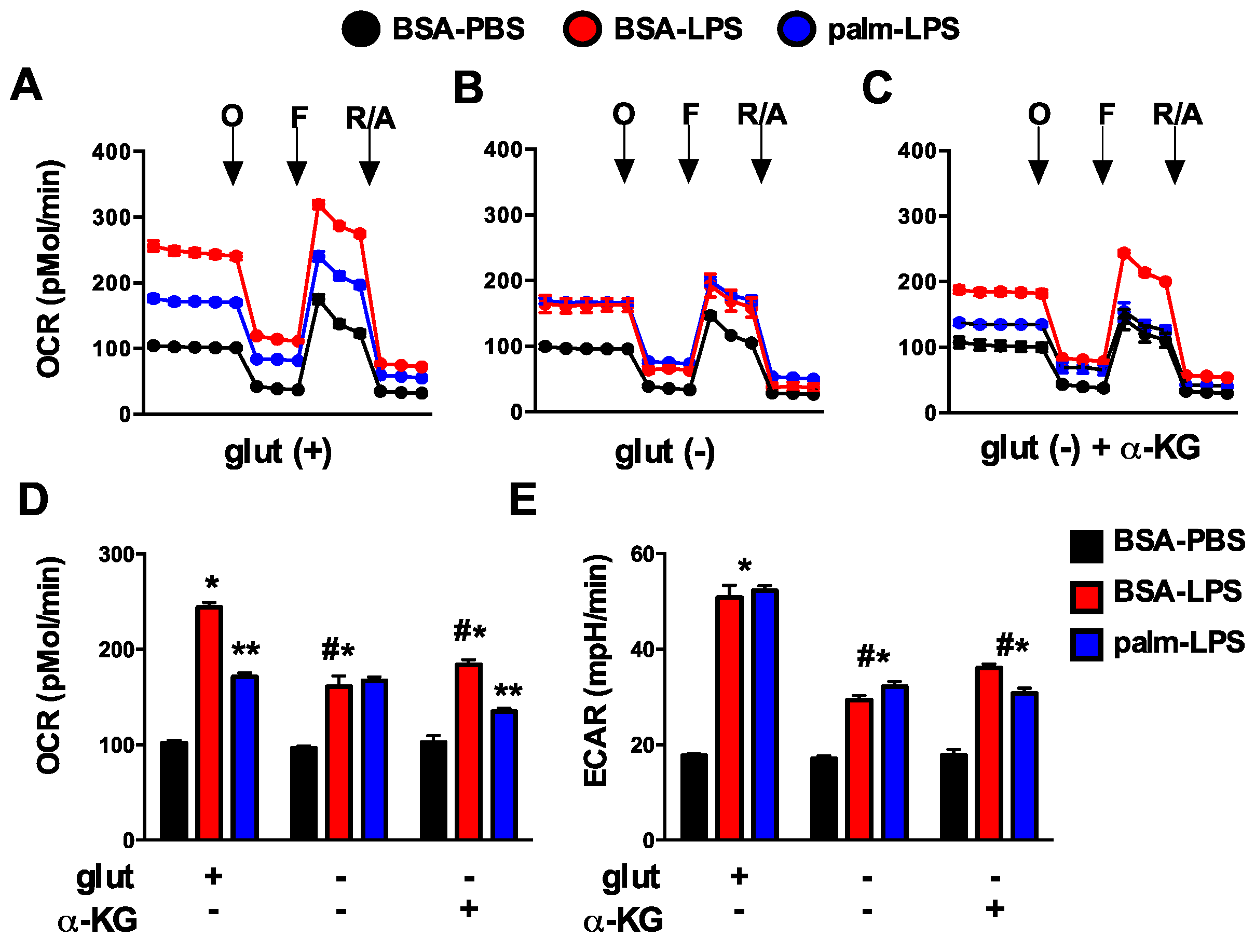
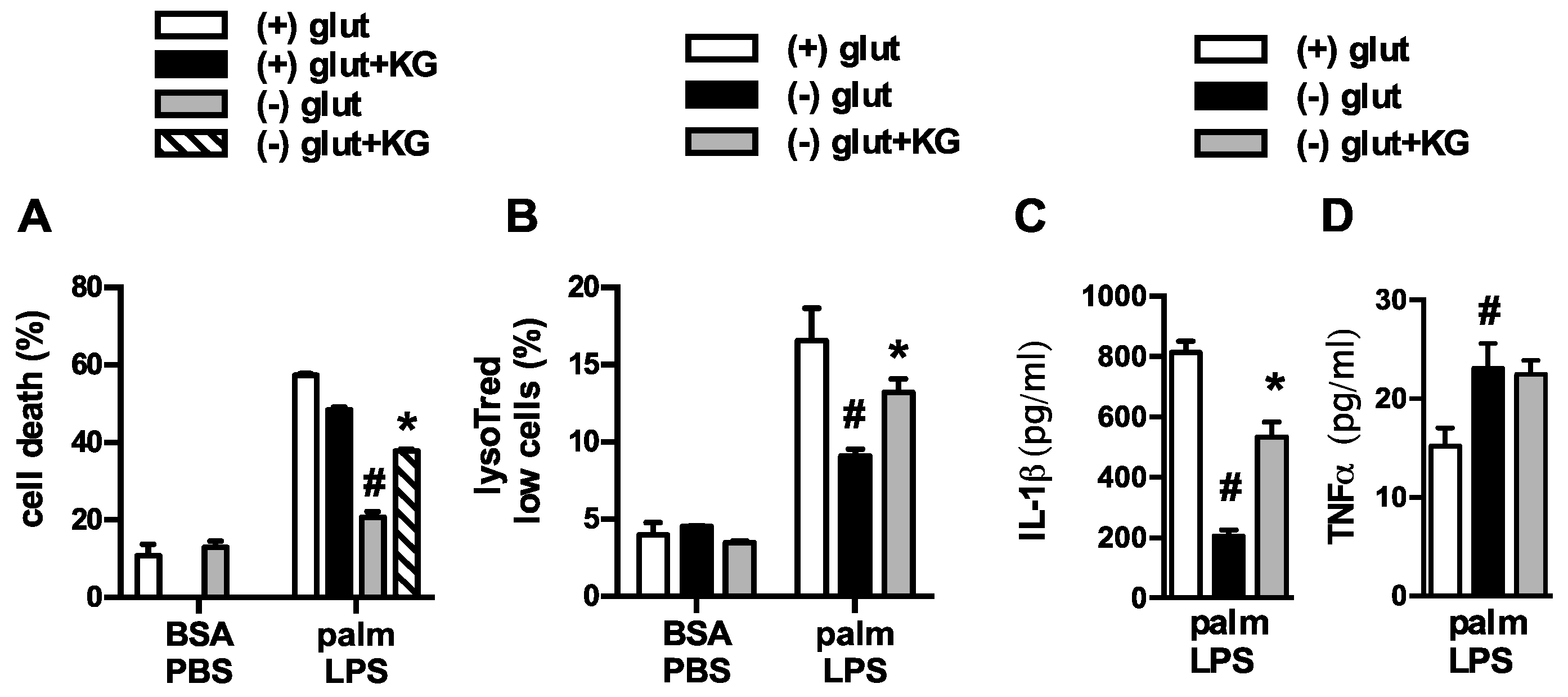
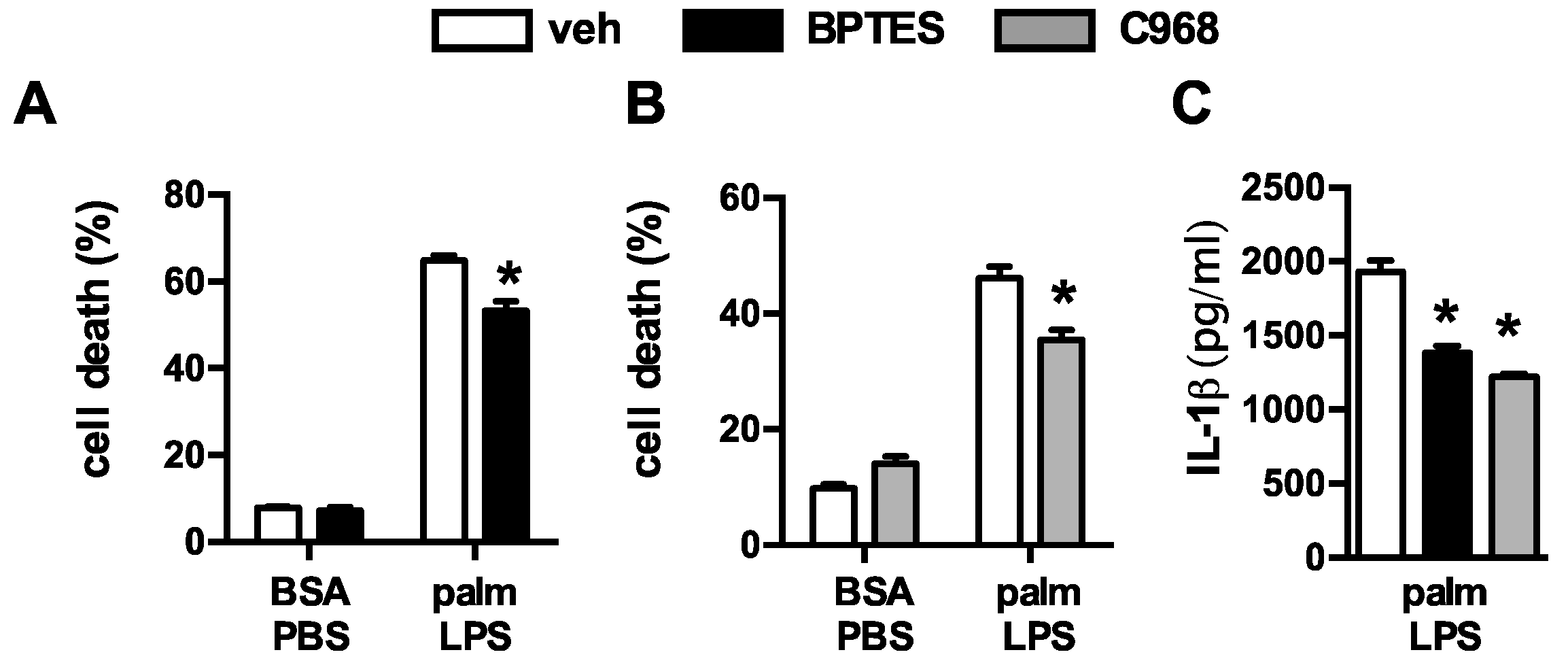
3.2. Oxidative Glutamine Metabolism Is Partially Responsible for the Protection from Lipid Toxicity
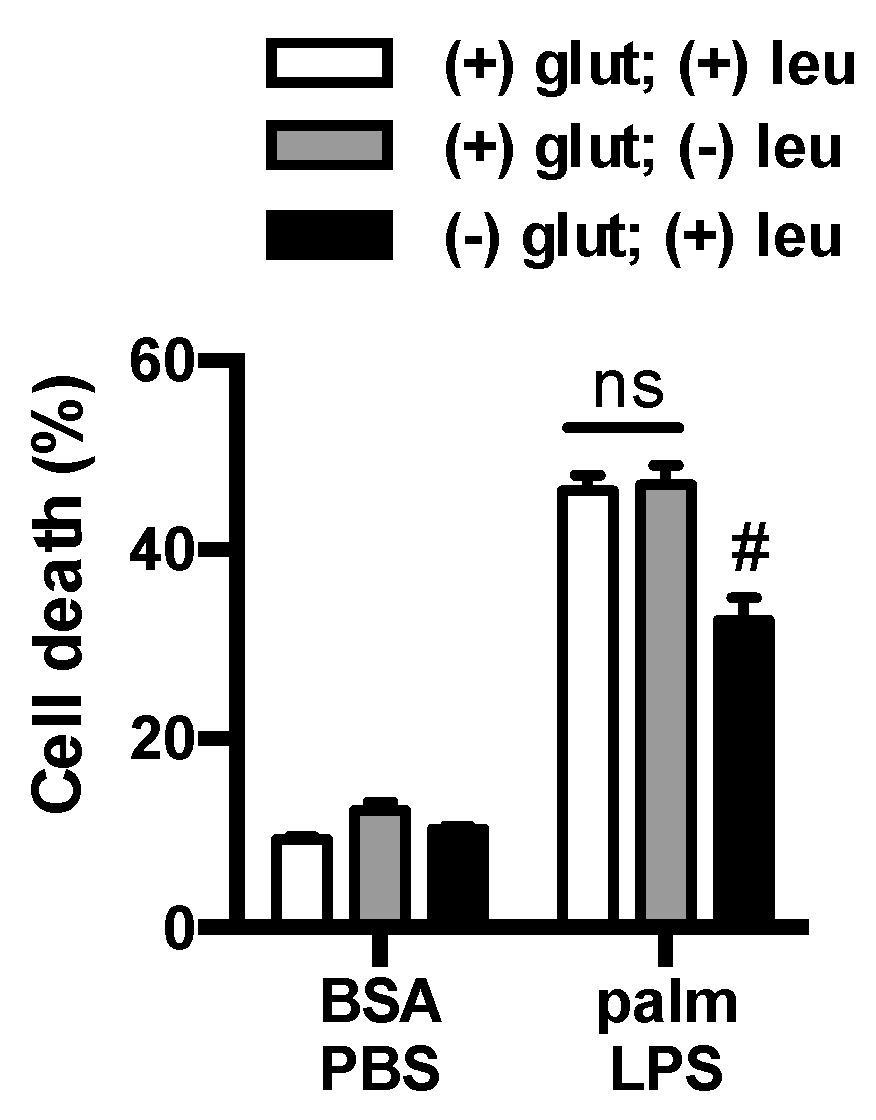
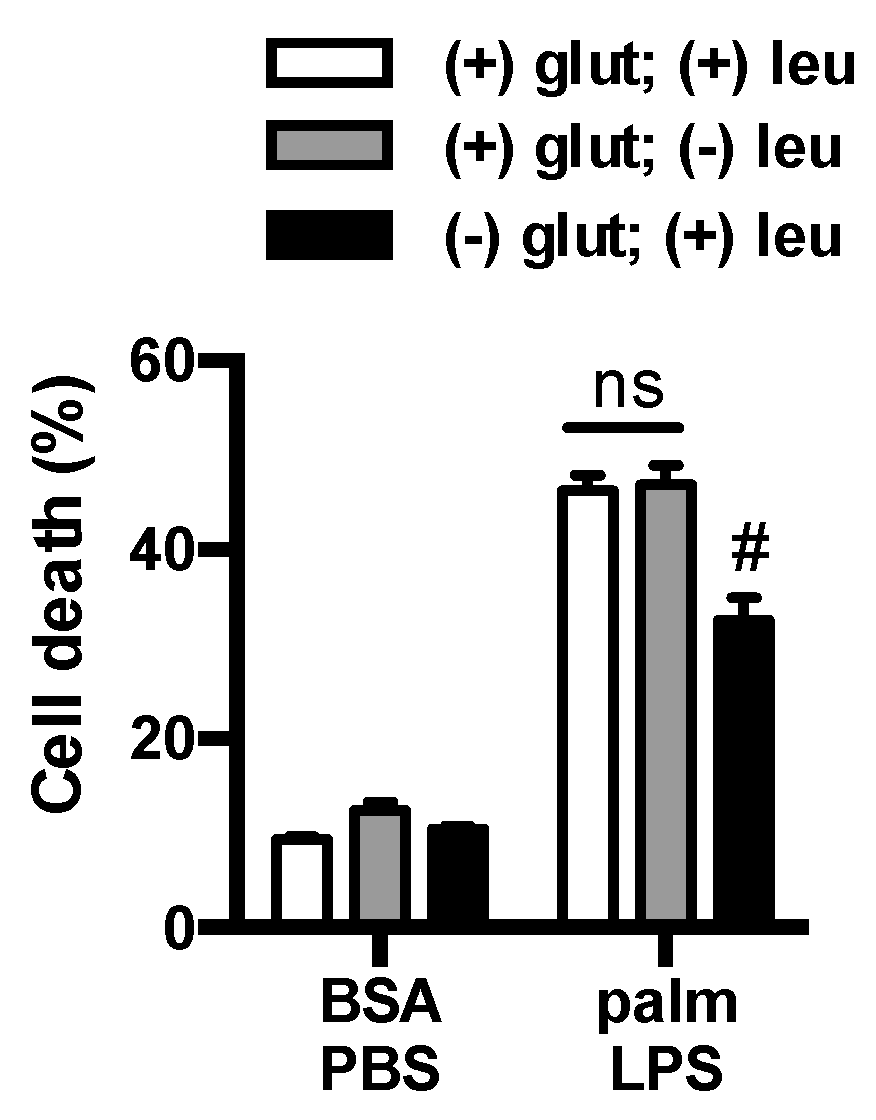
3.3. Glutamine Deficiency Is Protective Independent of Leucine
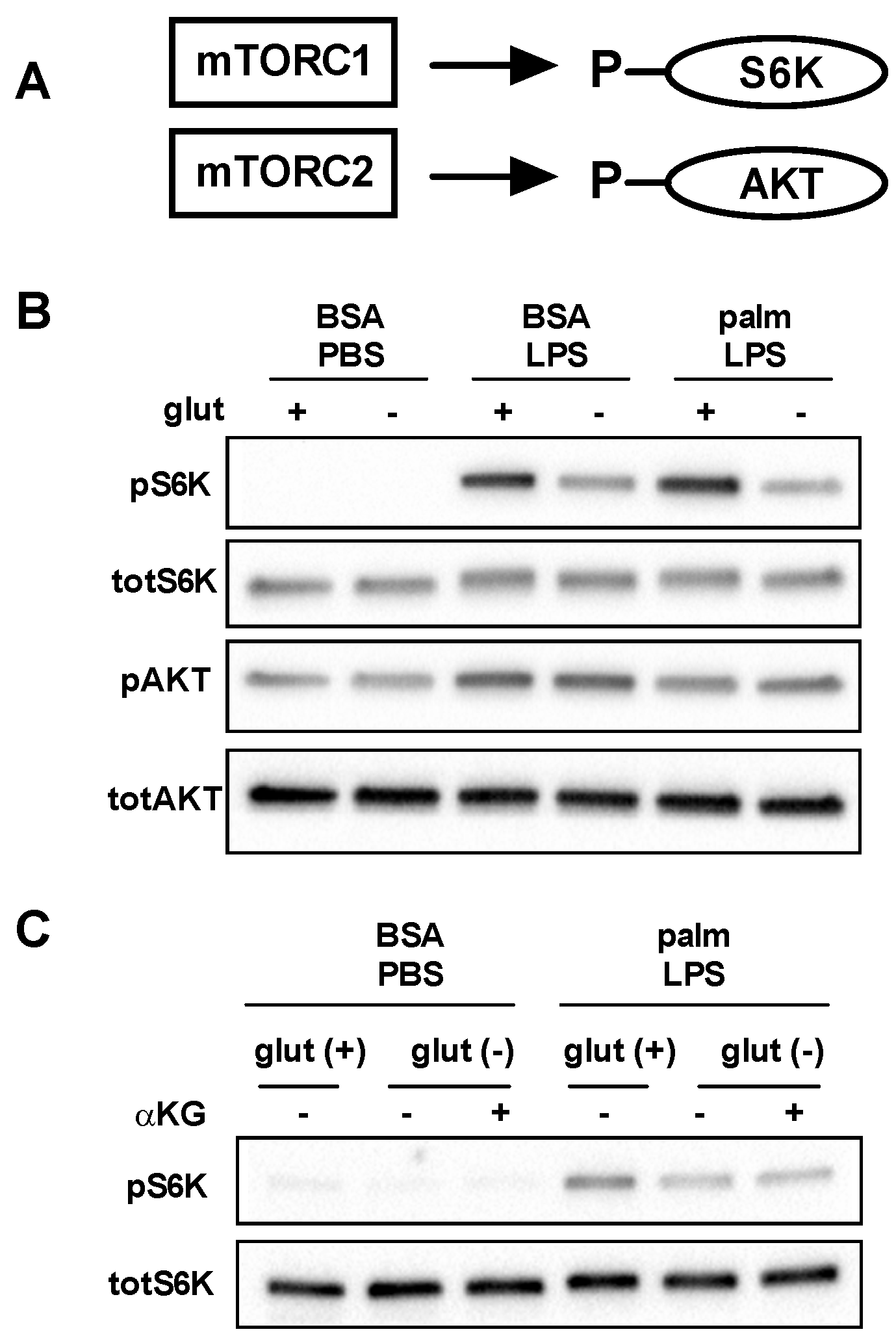
3.4. mTOR Signaling Is Reduced in the Absence of Glutamine
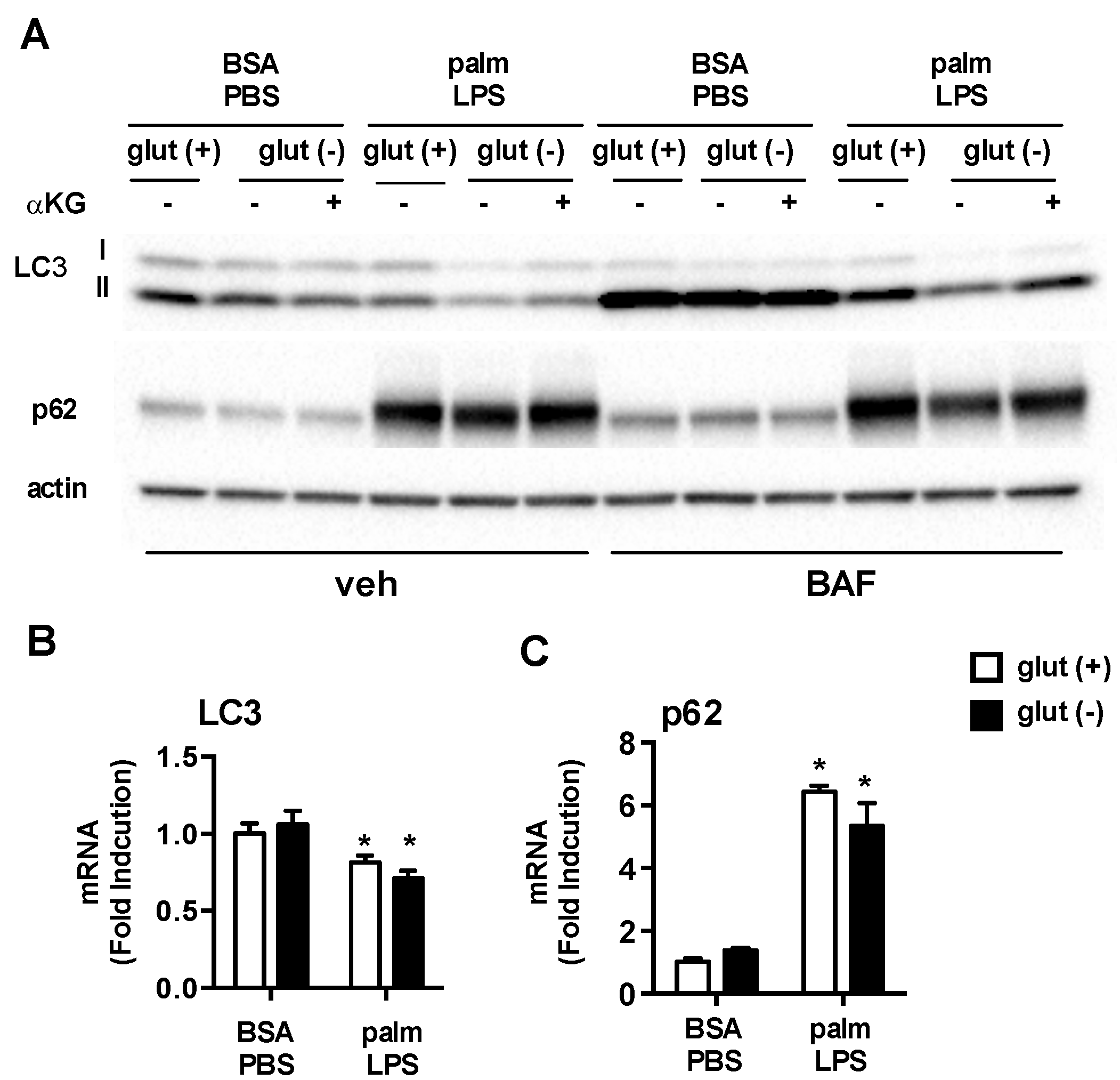
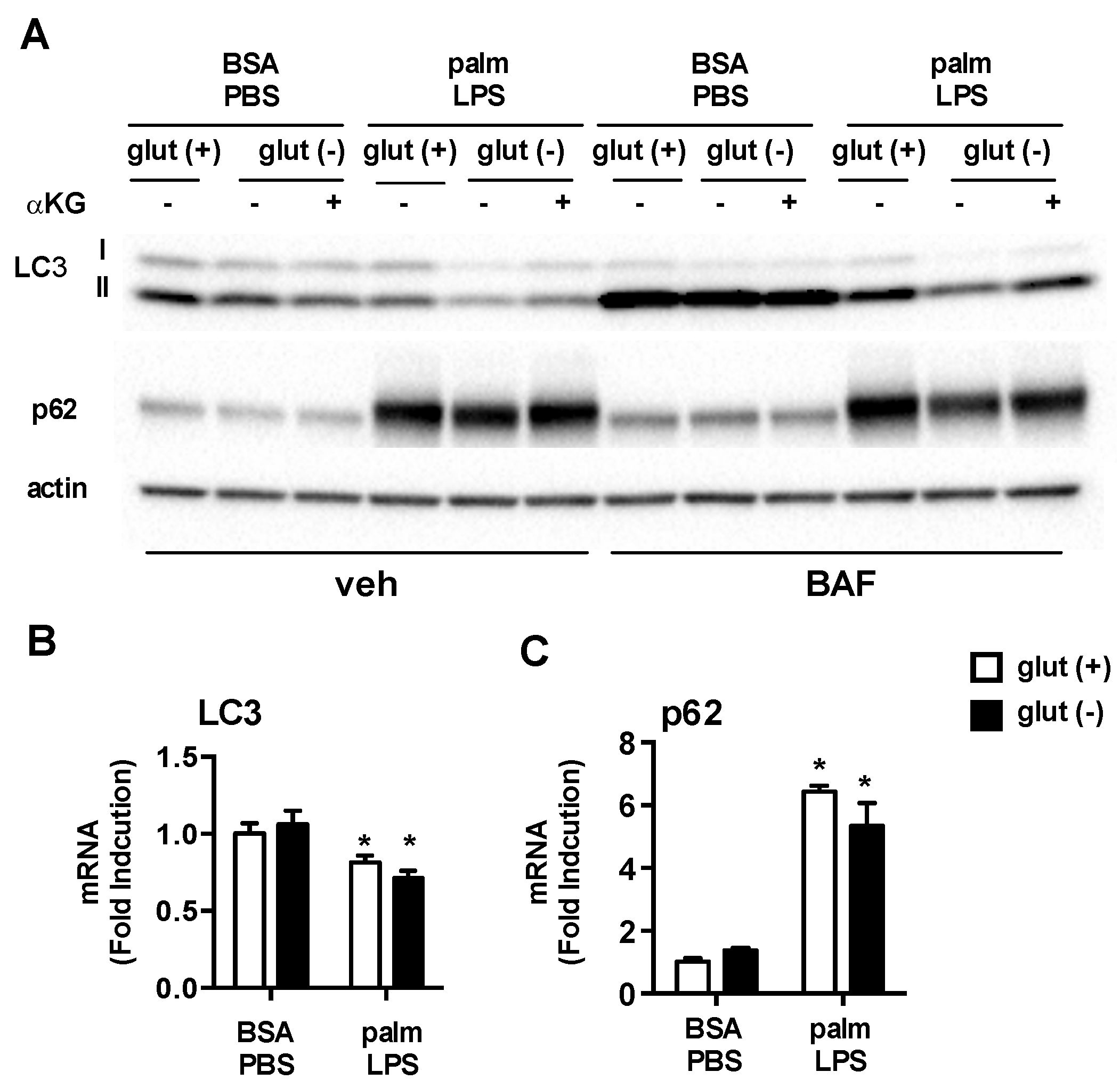
3.5. Glutamine Deficiency Modulates Macrophage Autophagy
3.6. Glutamine Deficiency Prevents the Suppression of Mitochondrial Function by Palmitate
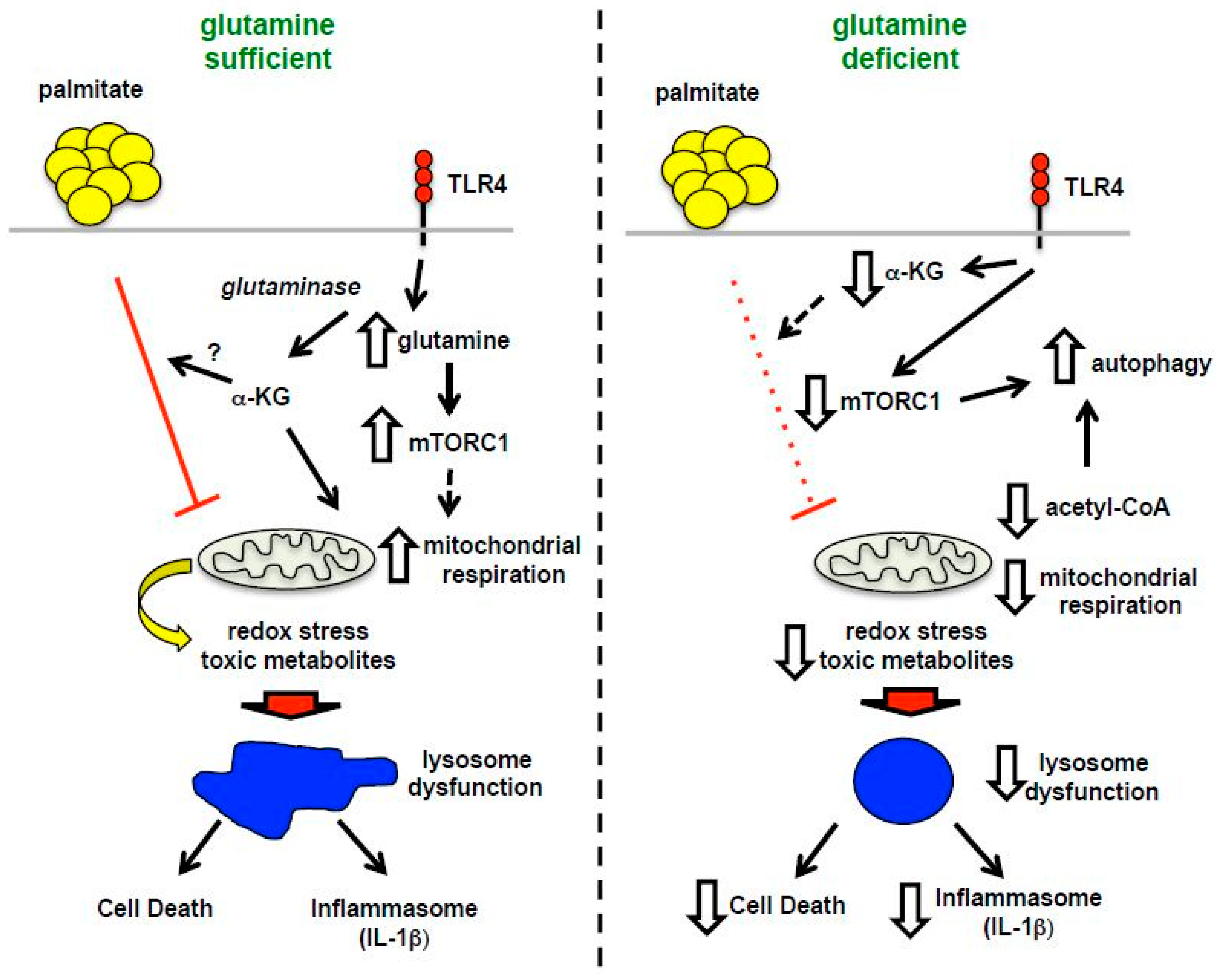
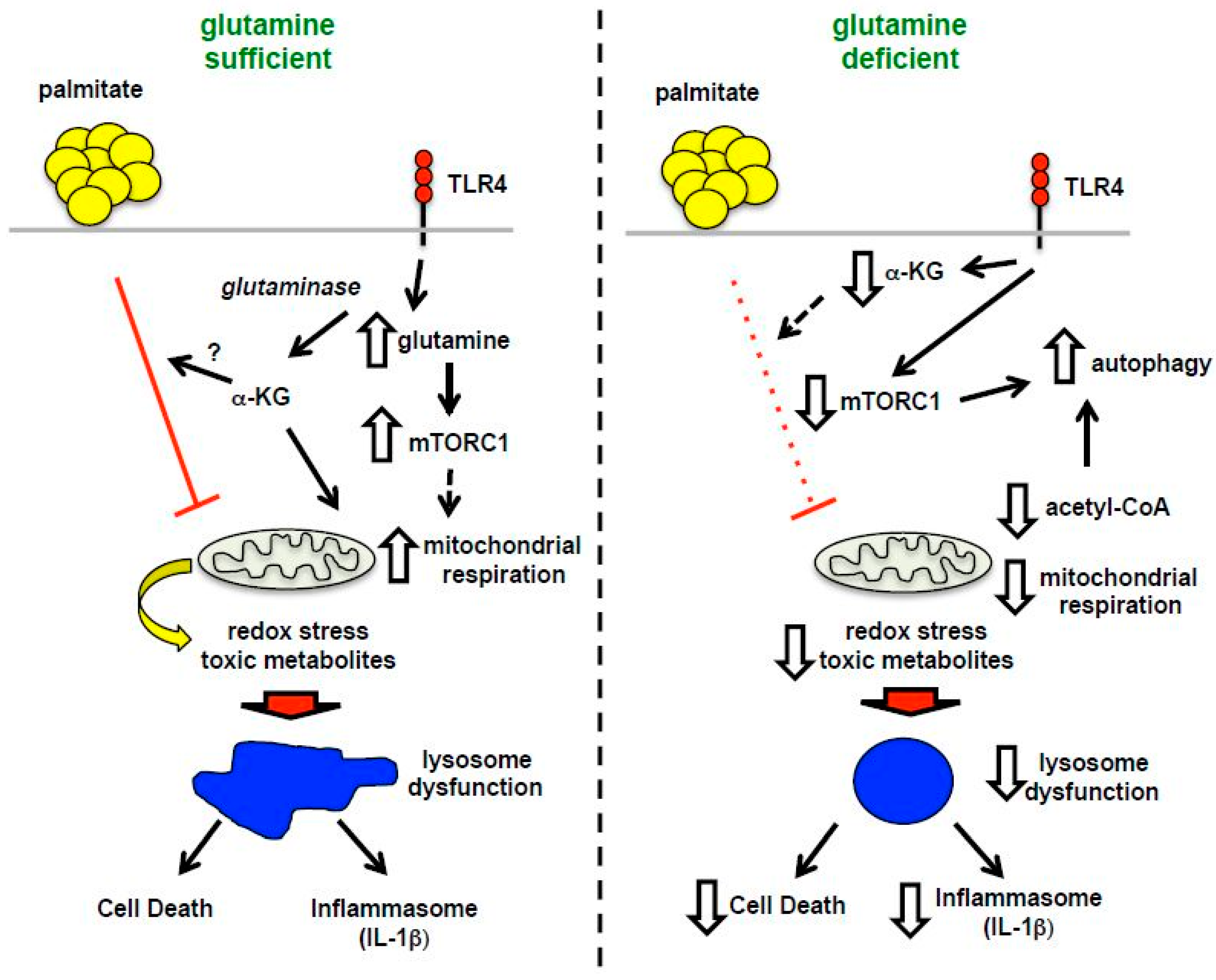
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mirza, R.; Koh, T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011, 56, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.; Rayfield, E.J.; Chesebro, J.H. Mechanisms determining course and outcome of diabetic patients who have had acute myocardial infarction. Ann. Intern. Med. 1997, 126, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Biswas, S.; Shang, Y.; Collard, E.; Azad, A.; Kauh, C.; Bhasker, V.; Gordillo, G.M.; Sen, C.K.; Roy, S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE 2010, 5, e9539. [Google Scholar] [CrossRef] [PubMed]
- Wetzler, C.; Kampfer, H.; Stallmeyer, B.; Pfeilschifter, J.; Frank, S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: Prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Investig. Dermatol. 2000, 115, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, B.; Stenhagen, S.; Svanborg, A.; Svennerholm, L. Gas chromatographic analysis of the fatty acid composition of the plasma lipids in normal and diabetic subjects. J. Clin. Investig. 1960, 39, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, D.J.; Tsung, A.; Billiar, T.R. Innate immune mechanisms in ischemia/reperfusion. Front. Biosci. (Elite Ed.) 2009, 1, 91–98. [Google Scholar] [PubMed]
- Takeda, K.; Akira, S. Tlr signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.D.; Machkovech, H.M.; He, L.; Diwan, A.; Schaffer, J.E. Tlr4 activation under lipotoxic conditions leads to synergistic macrophage cell death through a trif-dependent pathway. J. Immunol. 2013, 190, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.D.; Machkovech, H.M.; He, L.; Sidhu, R.; Fujiwara, H.; Weber, K.; Ory, D.S.; Schaffer, J.E. Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J. Biol. Chem. 2013, 288, 2923–2932. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.J.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory crosstalk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular metabolism turns immune regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. Nad+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Prados, J.C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragones, J.; Martin-Sanz, P.; Cascante, M.; Bosca, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Curi, R.; Pithon Curi, T.C.; Murphy, C.J.; Garcia, C.; Pires de Melo, M. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: Its importance in health and disease. J. Nutr. Biochem. 1999, 10, 316–324. [Google Scholar] [CrossRef]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mtorc1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Ristic, Z.; Klauser, S.; Verrey, F. Activation of system l heterodimeric amino acid exchangers by intracellular substrates. EMBO J. 2002, 21, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Lorin, S.; Blommaart, E.F.; Codogno, P. Regulation of autophagy by amino acids and mtor-dependent signal transduction. Amino Acids 2015, 47, 2037–2063. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mtor and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Nakai, M.; Iwamaru, Y.; Sugama, S.; Tsukimoto, M.; Fujita, M.; Wei, J.; Sekigawa, A.; Sato, M.; Kojima, S.; et al. The activation of p2x7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J. Immunol. 2009, 182, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Stenmark, K.R. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin. Immunol. 2015, 27, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Huffman, K.M.; Shah, S.H.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.; Slentz, C.A.; Tanner, C.J.; Kuchibhatla, M.; Houmard, J.A.; Newgard, C.B.; et al. Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care 2009, 32, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Granger, C.B.; Craig, D.; Haynes, C.; Bain, J.; Stevens, R.D.; Hauser, E.R.; Newgard, C.B.; Kraus, W.E.; Newby, L.K.; et al. Validation of the association between a branched chain amino acid metabolite profile and extremes of coronary artery disease in patients referred for cardiac catheterization. Atherosclerosis 2014, 232, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.; Newsholme, P. Importance of glutamine metabolism in murine macrophages and human monocytes to l-arginine biosynthesis and rates of nitrite or urea production. Clin. Sci. 1998, 95, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Newsholme, E.A. Rates of utilization of glucose, glutamine and oleate and formation of end-products by mouse peritoneal macrophages in culture. Biochem. J. 1989, 261, 211–218. [Google Scholar] [CrossRef] [PubMed]
- van der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the pi(3)k-pkb-foxo network regulates autophagy. Nat. Cell Biol. 2012, 14, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Kuttner, V.; Bhukel, A.; Marino, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Karinch, A.M.; Pan, M.; Lin, C.M.; Strange, R.; Souba, W.W. Glutamine metabolism in sepsis and infection. J. Nutr. 2001, 131, 2535S–2538S. [Google Scholar] [PubMed]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated nlrp3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. Nlrp3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.E.; Fang, M.M.; Ennis, W.J.; Koh, T.J. Blocking il-1beta induces a healing-associated wound macrophage phenotype and improves healing in type-2 diabetes. Diabetes 2013, 62, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Erbay, E. Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 2008, 8, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. Ccr2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Grijalva, A.; Skowronski, A.; van Eijk, M.; Serlie, M.J.; Ferrante, A.W., Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Thakker, G.D.; Frangogiannis, N.G.; Bujak, M.; Zymek, P.; Gaubatz, J.W.; Reddy, A.K.; Taffet, G.; Michael, L.H.; Entman, M.L.; Ballantyne, C.M. Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2504–H2514. [Google Scholar] [CrossRef] [PubMed]
- Greer, J.J.; Ware, D.P.; Lefer, D.J. Myocardial infarction and heart failure in the db/db diabetic mouse. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H146–H153. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.E.; Fang, M.M.; Novak, M.L.; Urao, N.; Sui, A.; Ennis, W.J.; Koh, T.J. Macrophage ppargamma and impaired wound healing in type 2 diabetes. J. Pathol. 2015, 236, 433–444. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Weber, K.J.; Schilling, J.D. Glutamine Modulates Macrophage Lipotoxicity. Nutrients 2016, 8, 215. https://doi.org/10.3390/nu8040215
He L, Weber KJ, Schilling JD. Glutamine Modulates Macrophage Lipotoxicity. Nutrients. 2016; 8(4):215. https://doi.org/10.3390/nu8040215
Chicago/Turabian StyleHe, Li, Kassandra J. Weber, and Joel D. Schilling. 2016. "Glutamine Modulates Macrophage Lipotoxicity" Nutrients 8, no. 4: 215. https://doi.org/10.3390/nu8040215
APA StyleHe, L., Weber, K. J., & Schilling, J. D. (2016). Glutamine Modulates Macrophage Lipotoxicity. Nutrients, 8(4), 215. https://doi.org/10.3390/nu8040215