Premenopausal Obesity and Breast Cancer Growth Rates in a Rodent Model



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Immunohistochemical Evaluation of Proliferation (Ki67)
2.3. Histological Evaluation of Apoptosis
2.4. Lysate Preparation
2.5. Western Blot-Based Detection/CE-Based Protein Expression
2.6. Immunoprecipitation
2.7. Statistical Methods
2.7.1. Apoptotic/Mitotic Indices
2.7.2. Western Blot/CE-Based Evaluation of Protein Expression
2.7.3. Multivariate Statistical Modeling
3. Results
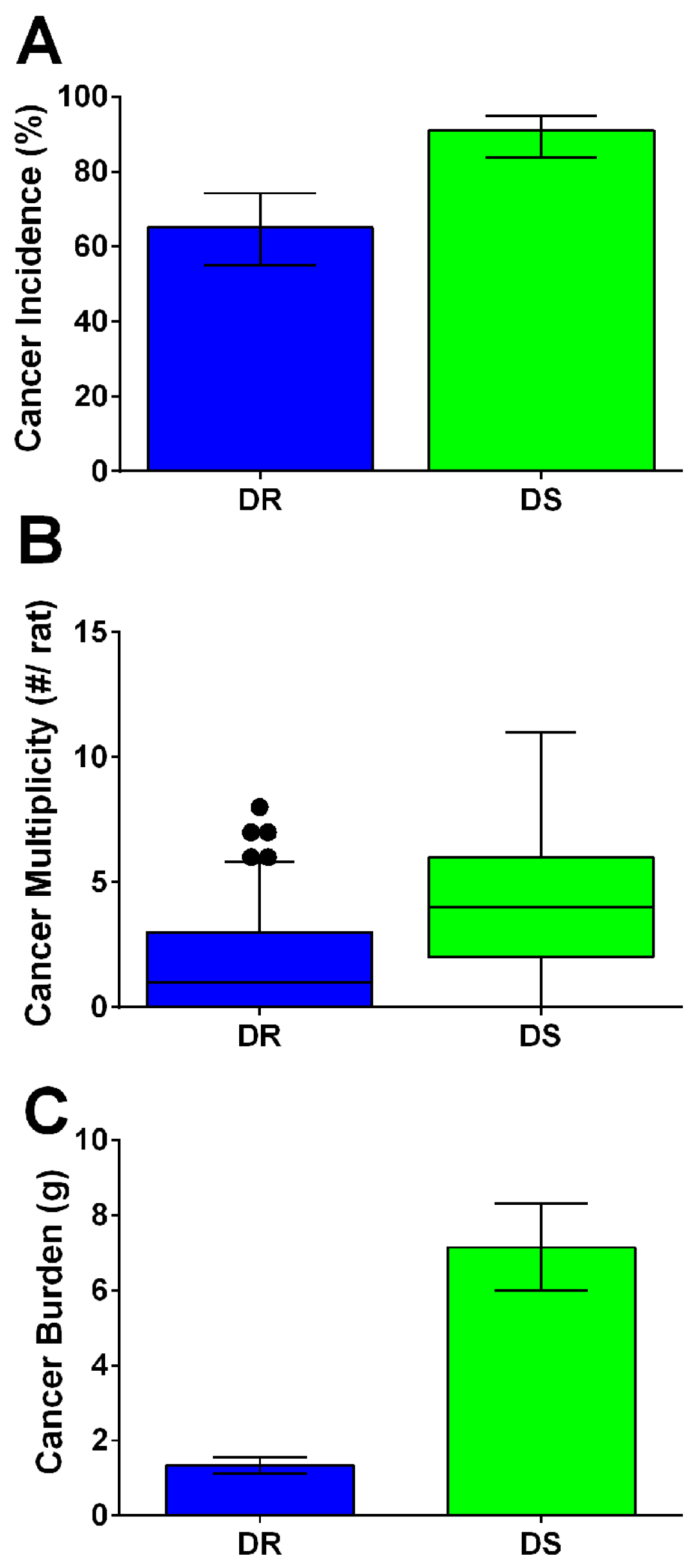
3.1. Chemically-Induced Mammary Carcinogenesis Is Accelerated in DS Rats
3.2. Tumor Burden, Body Weight, and Adiposity
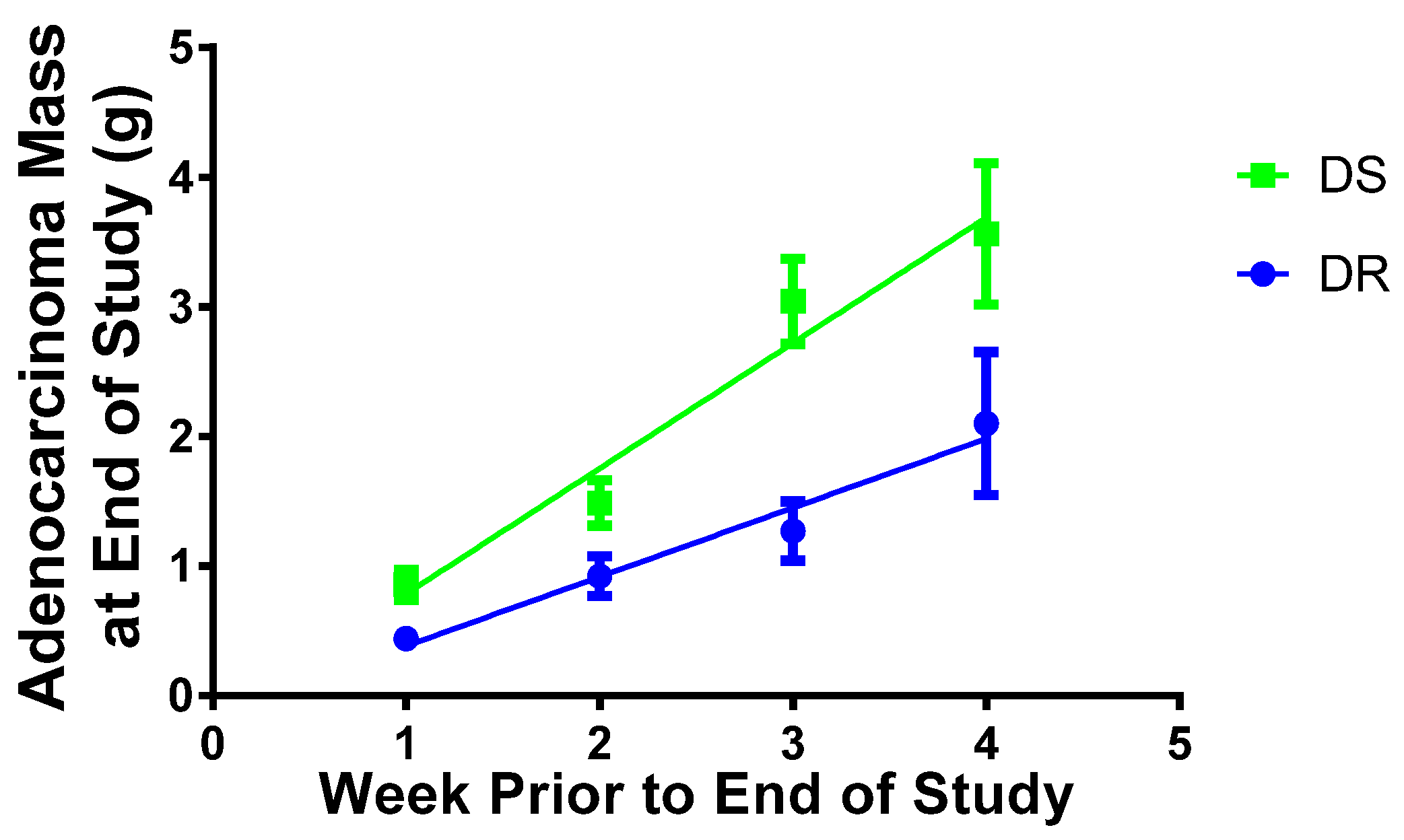
3.3. Change in Tumor Mass
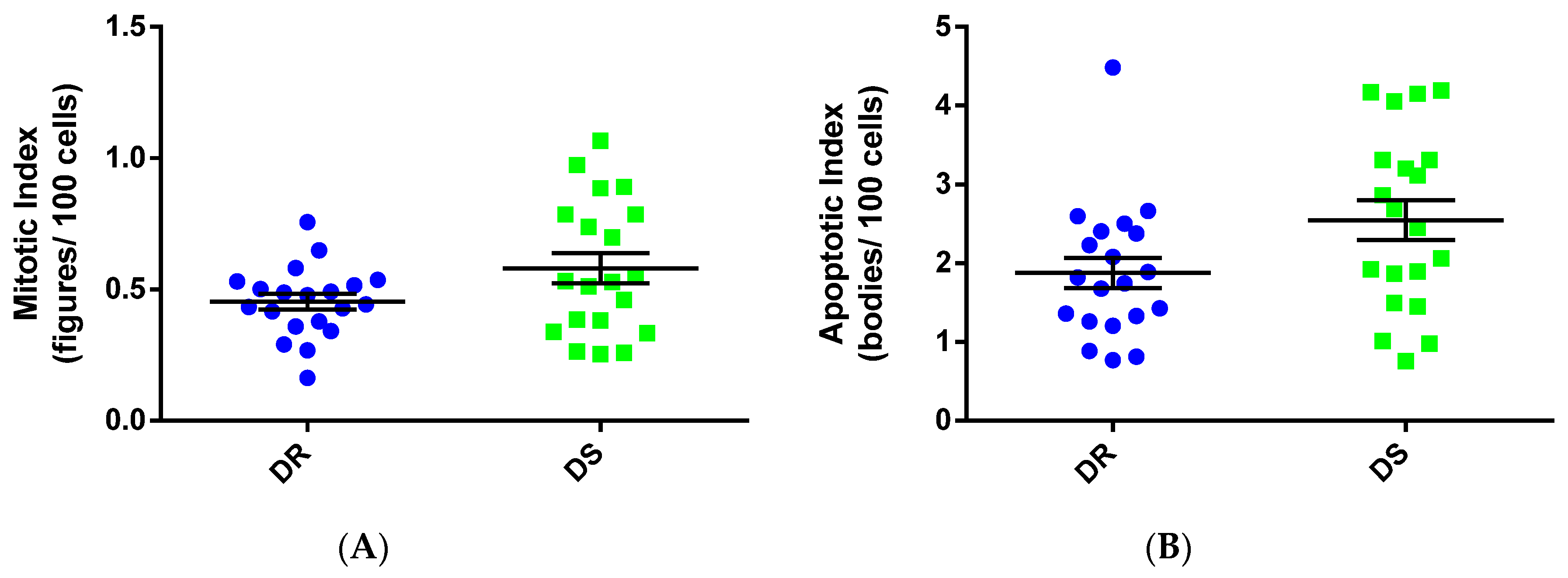
3.4. Cellular Processes Associated with Tumor Growth
3.5. Cell Cycle Length and Duration of Apoptosis
3.5.1. Cell Cycle Length
3.5.2. Apoptotic Duration
3.5.3. Multivariate Analyses of Cell Proliferation and Apoptosis Data
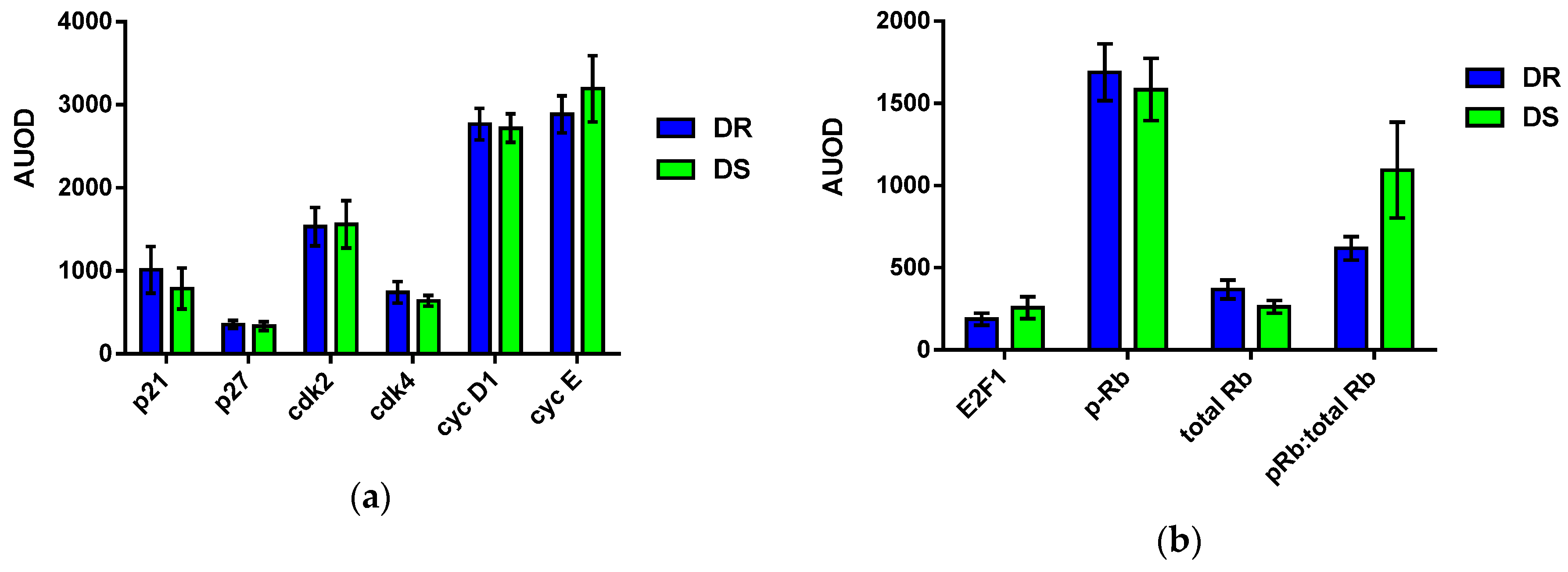
3.6. Effects on Cellular Machinery
3.6.1. Cell Cycle
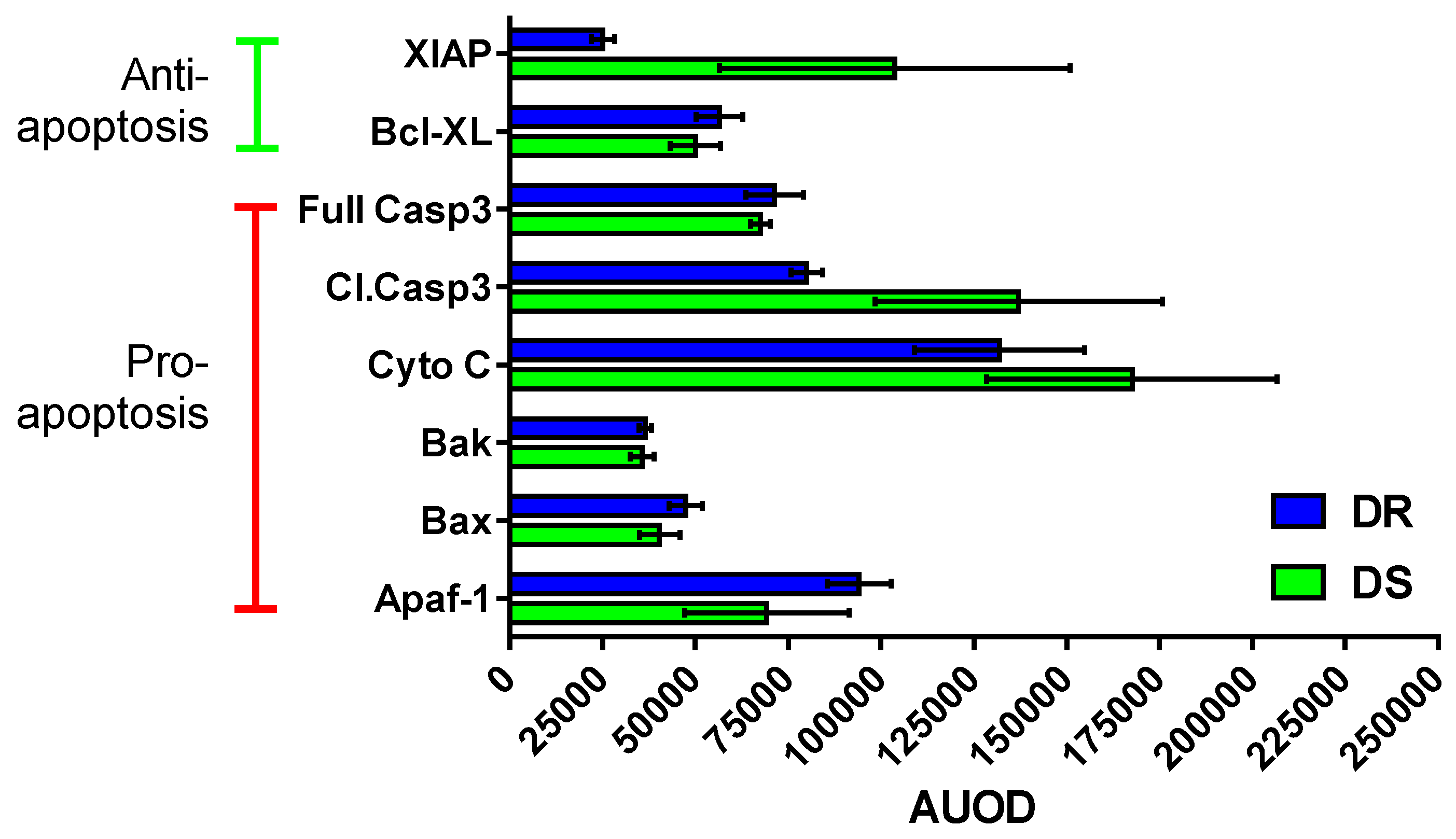
3.6.2. Effects on Apoptotic Machinery
3.6.3. Multivariate Analysis of Tumor Growth Characteristics
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AUOD | arbitrary units of opical density |
| DR | dietary obesity resistant |
| DS | dietary obesity sensitive |
| FFPE | formalin fixed paraffin embedded |
| MNU | 1-methyl-1-nitrosourea |
| APAF | apoptotic protease activating factor 1 |
| BAX | BCL2-Associated X |
| BCL2 | B-cell lymphoma 2 |
| Cdc6 | cell division cycle 6 |
| Cdk2 | cyclin-dependent kinase 2 |
| Cdk4 | cyclin-dependent kinase 4 |
| DAB | 3,3′-diaminobenzidine |
| E2F1 transcription factor | Retinoblastoma-Associated Protein1 |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| H & E | hematoxylin and eosin |
| IRS | insulin receptor substrate |
| Ki67 | nuclear protein that is associated with and may be necessary for cellular proliferation |
| P21 | cyclin-dependent kinase inhibitor 1 |
| P27 | cyclin-dependent kinase inhibitor 1B |
| PBS-T | phosphate buffered saline + 0.05% tween 20 |
| XIAP | X-linked inhibitor of apoptosis |
References
- Bouchard, C.; Tremblay, A.; Despres, J.P.; Nadeau, A.; Lupien, P.J.; Theriault, G.; Dussault, J.; Moorjani, S.; Pinault, S.; Fournier, G. The response to long-term overfeeding in identical twins. N. Engl. J. Med. 1990, 322, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Wiseman, M. The second World Cancer Research Fund/American Institute for Cancer Research expert report. Food, nutrition, physical activity, and the prevention of cancer: A global perspective. Proc. Nutr. Soc. 2008, 67, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Berstad, P.; Coates, R.J.; Bernstein, L.; Folger, S.G.; Malone, K.E.; Marchbanks, P.A.; Weiss, L.K.; Liff, J.M.; McDonald, J.A.; Strom, B.L.; et al. A case-control study of body mass index and breast cancer risk in white and African-American women. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Cheraghi, Z.; Poorolajal, J.; Hashem, T.; Esmailnasab, N.; Irani, A.D. Effect of body mass index on breast cancer during premenopausal and postmenopausal periods: A meta-analysis. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.L.; Neuhouser, M.L. Obesity and the risk for premenopausal and postmenopausal breast cancer. Cancer Prev. Res. 2012, 5, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, R.S.; Costantino, J.P.; Cauley, J.A.; Cronin, W.M.; Wickerham, D.L.; Land, S.R.; Weissfeld, J.L.; Wolmark, N. Body mass index and the risk for developing invasive breast cancer among high-risk women in NSABP P-1 and STAR breast cancer prevention trials. Cancer Prev. Res. 2012, 5, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Gail, M.H.; Brinton, L.A.; Byar, D.P.; Corle, D.K.; Green, S.B.; Schairer, C.; Mulvihill, J.J. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J. Natl. Cancer Inst. 1989, 81, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.B.; Zhu, Z.; Jiang, W.; McGinley, J.N.; Neil, E.S.; Thompson, H.J. Excess weight gain accelerates 1-methyl-1-nitrosourea-induced mammary carcinogenesis in a rat model of premenopausal breast cancer. Cancer Prev. Res. 2014, 7, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Playdon, M.C.; Matthews, S.B.; Thompson, H.J. Weight change patterns and breast cancer risk: A brief review and analysis. Crit. Rev. Eukaryot. Gene Expr. 2013, 23, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Haakinson, D.J.; Leeds, S.G.; Dueck, A.C.; Gray, R.J.; Wasif, N.; Stucky, C.C.; Northfelt, D.W.; Apsey, H.A.; Pockaj, B. The impact of obesity on breast cancer: A retrospective review. Ann. Surg. Oncol. 2012, 19, 3012–3018. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P. Impact of obesity on development and progression of mammary tumors in preclinical models of breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Biglia, N.; Peano, E.; Sgandurra, P.; Moggio, G.; Pecchio, S.; Maggiorotto, F.; Sismondi, P. Body mass index (BMI) and breast cancer: Impact on tumor histopathologic features, cancer subtypes and recurrence rate in pre and postmenopausal women. Gynecol. Endocrinol. 2013, 29, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Obesity and inflammation: New insights into breast cancer development and progression. Am. Soc. Clin. Oncol. Educ. Book 2013. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Dannenberg, A.J. Obesity and breast cancer prognosis: Weight of the evidence. J. Clin. Oncol. 2011, 29, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Vargo-Gogola, T.; Rosen, J.M. Modelling breast cancer: One size does not fit all. Nat. Rev. Cancer 2007, 7, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Poiley, S.M. A systematic method of breeder rotation for non-inbred laboratory animal colonies. Proc. Anim. Care Panel 1960, 10, 159–166. [Google Scholar]
- Thompson, H.J.; McGinley, J.N.; Rothhammer, K.; Singh, M. Rapid induction of mammary intraductal proliferations, ductal carcinoma in situ and carcinomas by the injection of sexually immature female rats with 1-methyl-1-nitrosourea. Carcinogenesis 1995, 16, 2407–2411. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Adlakha, H. Dose-responsive induction of mammary gland carcinomas by the intraperitoneal injection of 1-methyl-1-nitrosourea. Cancer Res. 1991, 51, 3411–3415. [Google Scholar] [PubMed]
- Thompson, H.J.; McGinley, J.N.; Wolfe, P.; Singh, M.; Steele, V.E.; Kelloff, G.J. Temporal sequence of mammary intraductal proliferations, ductal carcinomas in situ and adenocarcinomas induced by 1-methyl-1-nitrosourea in rats. Carcinogenesis 1998, 19, 2181–2185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, Z.; Jiang, W.; Thompson, H.J. Effect of energy restriction on the expression of cyclin D1 and p27 during premalignant and malignant stages of chemically induced mammary carcinogenesis. Mol. Carcinog. 1999, 24, 241–245. [Google Scholar] [CrossRef]
- Regan, M.M.; Viale, G.; Mastropasqua, M.G.; Maiorano, E.; Golouh, R.; Carbone, A.; Brown, B.; Suurkula, M.; Langman, G.; Mazzucchelli, L.; et al. Re-evaluating adjuvant breast cancer trials: Assessing hormone receptor status by immunohistochemical versus extraction assays. J. Natl. Cancer Inst. 2006, 98, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, V.J.; Ruotoistenmaki, S.; Viitanen, A.; Jumppanen, M.; Isola, J. ImmunoRatio: A publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res. 2010, 12, R56. [Google Scholar] [CrossRef] [PubMed]
- SAS Institute Inc. Example 38.14 Generalized Poisson mixed model for overdispersed count data. In SAS/STAT(R) 9.2 User’s Guide; SAS Institute Inc.: Cary, NC, USA, 2015. [Google Scholar]
- Morrison, D.F. Multivariate Statistical Methods; McGraw-Hill Publishing Co.: New York, NY, USA, 1990. [Google Scholar]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemom. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Wiklund, S.; Johansson, E.; Sjostrom, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 2008, 80, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, S. Multivariate data analysis and modeling in “omics”. In Multivariate Analysis for Omics, Proceedings of the Umetrics, Umea, Sweden, 2 September 2008.
- Thompson, H.J.; Strange, R.; Schedin, P.J. Apoptosis in the genesis and prevention of cancer. Cancer Epidemiol. Biomark. Prev. 1992, 1, 597–602. [Google Scholar]
- Gerdes, J.; Li, L.; Schlueter, C.; Duchrow, M.; Wohlenberg, C.; Gerlach, C.; Stahmer, I.; Kloth, S.; Brandt, E.; Flad, H.D. Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am. J. Pathol. 1991, 138, 867–873. [Google Scholar] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. An overview of the cell cycle. In Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Steel, G.G. Experimental techniques for cell kinetic studies. In Growth Kinetics of Tumours: Cell Population Kinetics in Relation to the Growth and Treatment of Cancer; Oxford University Press: Oxford, UK, 1977; pp. 86–119. [Google Scholar]
- Steel, G.G. Growth rate of tumours. In Growth Kinetics of Tumours: Cell Population Kinetics in Relation to the Growth and Treatment of Cancer; Oxford University Press: Oxford, UK, 1977; pp. 5–55. [Google Scholar]
- Zetterberg, A.; Larsson, O. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. USA 1985, 82, 5365–5369. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Tsujimoto, A.; Ikeda, M.; Nakamura, M. Regulation of cell growth-dependent expression of mammalian CDC6 gene by the cell cycle transcription factor E2F. Oncogene 1998, 17, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, N.; Zhang, H.; Durden, D.L.; Feng, Y.; Zhou, M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 2009, 15, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Apovian, C.; Hess, D.; Carmine, B.; Saha, A.; Ruderman, N. Improved insulin sensitivity 3 months after RYGB surgery is associated with increased subcutaneous adipose tissue AMPK activity and decreased oxidative stress. Diabetes 2015, 64, 3155–3159. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Pories, W.J.; Dohm, L.G.; Ruderman, N.B. What distinguishes adipose tissue of severely obese humans who are insulin sensitive and resistant? Curr. Opin. Lipidol. 2013, 24, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Rideout, D.A.; Rakita, S.S.; Gower, W.R., Jr.; You, M.; Murr, M.M. Does LKB1 mediate activation of hepatic AMP-protein kinase (AMPK) and sirtuin1 (SIRT1) after Roux-en-Y gastric bypass in obese rats? J. Gastrointest. Surg. 2010, 14, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Rappou, E.; Jukarainen, S.; Rinnankoski-Tuikka, R.; Kaye, S.; Heinonen, S.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Saunavaara, V.; Rissanen, A.; et al. Weight loss is associated with increased NAD+/SIRT1 expression but reduced PARP activity in white adipose tissue. J. Clin. Endocrinol. Metab. 2016. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Wieser, V.; Gerner, R.R.; Bichler, A.; Enrich, B.; Moser, P.; Ebenbichler, C.F.; Kaser, S.; Tilg, H. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J. Hepatol. 2013, 59, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Leyvraz, C.; Verdumo, C.; Suter, M.; Paroz, A.; Calmes, J.M.; Marques-Vidal, P.M.; Giusti, V. Changes in Gene Expression Profile in Human Subcutaneous Adipose Tissue during Significant Weight Loss. Obes. Facts 2012, 5, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, S.P.M.; Camps, S.G.J.A.; Bouwman, F.G.; Mariman, E.C.M.; Westerterp, K.R. Genetic predisposition, dietary restraint and disinhibition in relation to short and long-term weight loss. Physiol. Behav. 2014, 128, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Jennissen, K.; Siegel, F.; Liebig-Gonglach, M.; Hermann, M.R.; Kipschull, S.; van Sander, D.; Kunz, W.S.; Fassler, R.; Pfeifer, A. A VASP-Rac-soluble guanylyl cyclase pathway controls cGMP production in adipocytes. Sci. Signal. 2012. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Bolanos, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Stewart, I.; Tiyyagura, S.R.; Lin, J.E.; Kazerounian, S.; Pitari, G.M.; Schulz, S.; Martin, E.; Murad, F.; Waldman, S.A. Guanylyl cyclase is an ATP sensor coupling nitric oxide signaling to cell metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat Strain 1 | Perirenal 2 (mg/mm) | Retroperitoneal (mg/mm) | Parametrial (mg/mm) | Mammary Gland 3 Adipocytes (µm2/Field) |
|---|---|---|---|---|
| DR | 6 ± 1 | 23 ± 4 | 37 ± 5 | 1128 ± 44 |
| DS | 26 ± 3 | 65 ± 8 | 192 ± 37 | 1561 ± 51 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthews, S.B.; McGinley, J.N.; Neil, E.S.; Thompson, H.J. Premenopausal Obesity and Breast Cancer Growth Rates in a Rodent Model. Nutrients 2016, 8, 214. https://doi.org/10.3390/nu8040214
Matthews SB, McGinley JN, Neil ES, Thompson HJ. Premenopausal Obesity and Breast Cancer Growth Rates in a Rodent Model. Nutrients. 2016; 8(4):214. https://doi.org/10.3390/nu8040214
Chicago/Turabian StyleMatthews, Shawna B., John N. McGinley, Elizabeth S. Neil, and Henry J. Thompson. 2016. "Premenopausal Obesity and Breast Cancer Growth Rates in a Rodent Model" Nutrients 8, no. 4: 214. https://doi.org/10.3390/nu8040214
APA StyleMatthews, S. B., McGinley, J. N., Neil, E. S., & Thompson, H. J. (2016). Premenopausal Obesity and Breast Cancer Growth Rates in a Rodent Model. Nutrients, 8(4), 214. https://doi.org/10.3390/nu8040214