Docosahexaenoic Acid Ameliorates Fructose-Induced Hepatic Steatosis Involving ER Stress Response in Primary Mouse Hepatocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Primary Mouse Hepatocytes Culture
2.3. Oil Red O Staining
2.4. RNA Extraction and Real-Time PCR Assays
2.5. Western Blot
2.6. Statistical Analysis
3. Results
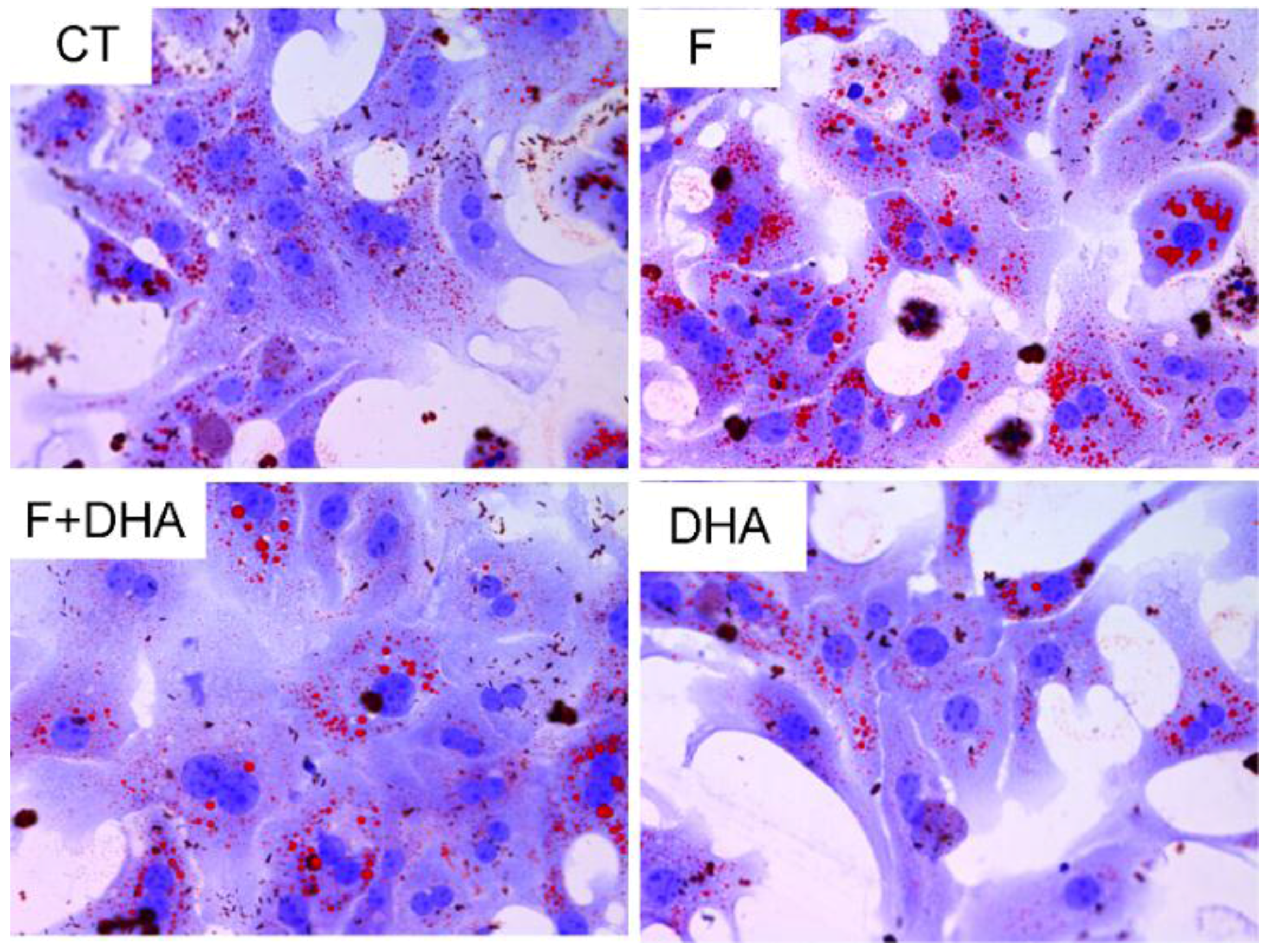
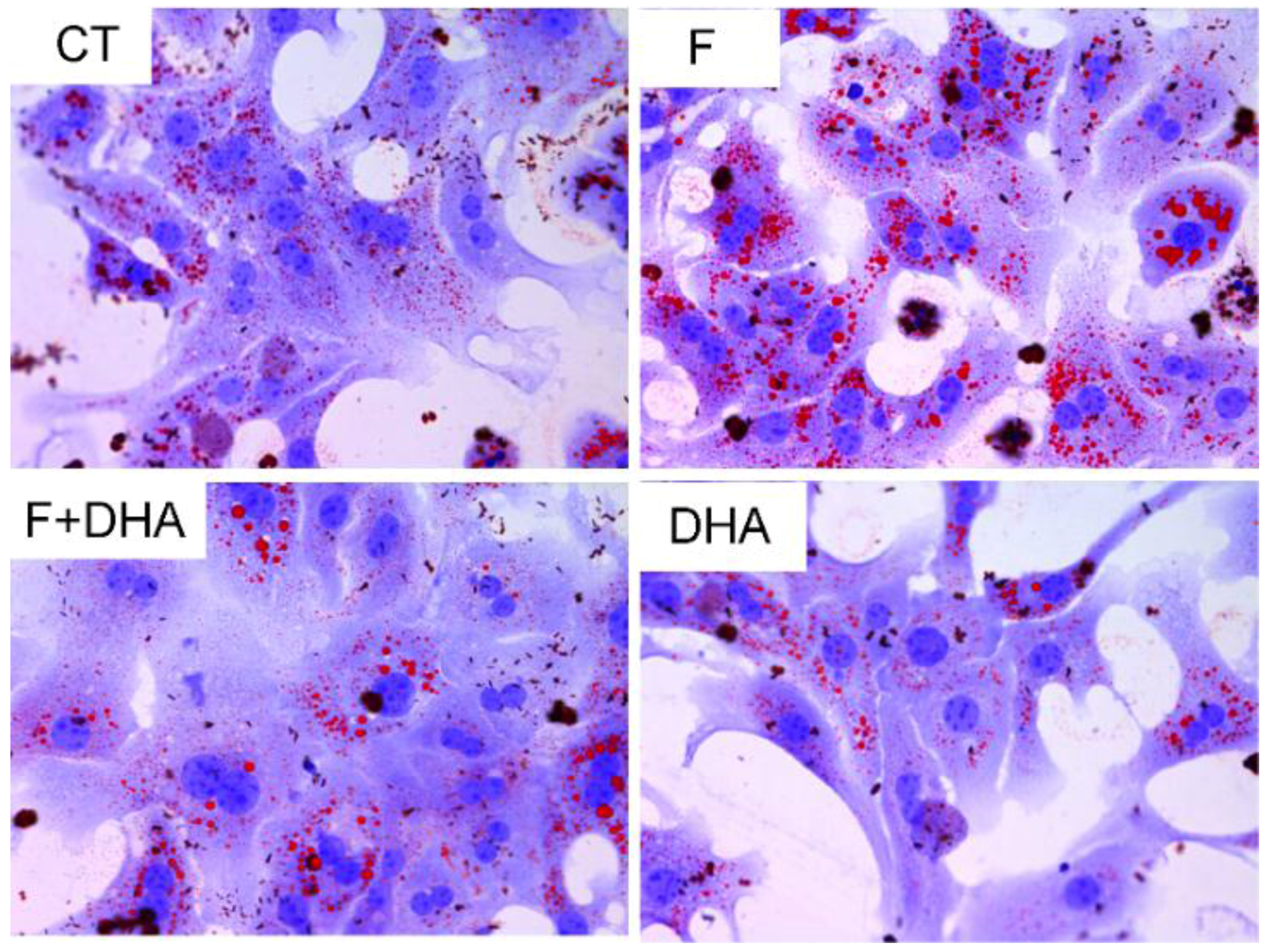
3.1. DHA Prevents Fructose-Induced Lipid Accumulation in Primary Mouse Hepatocytes
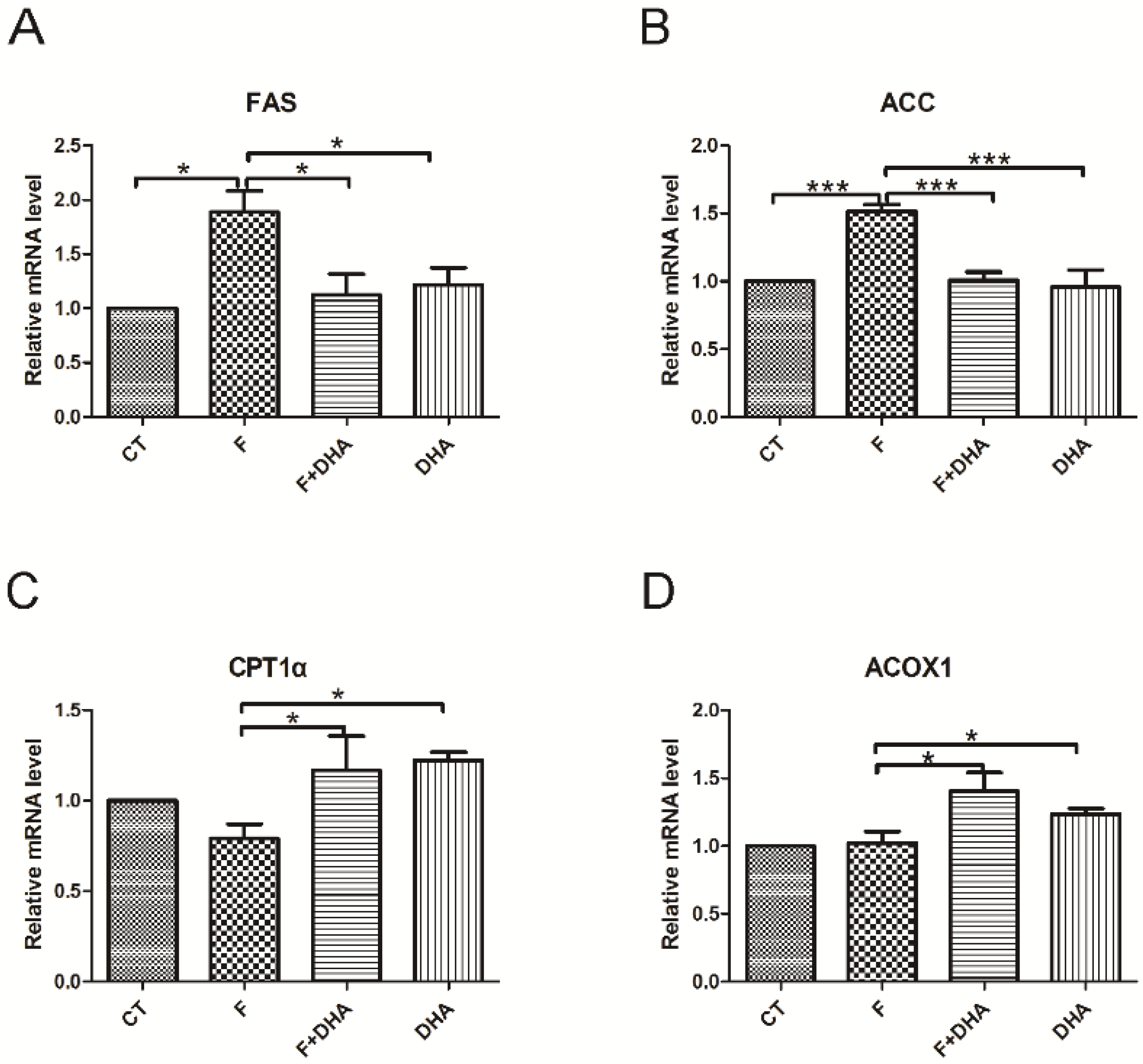
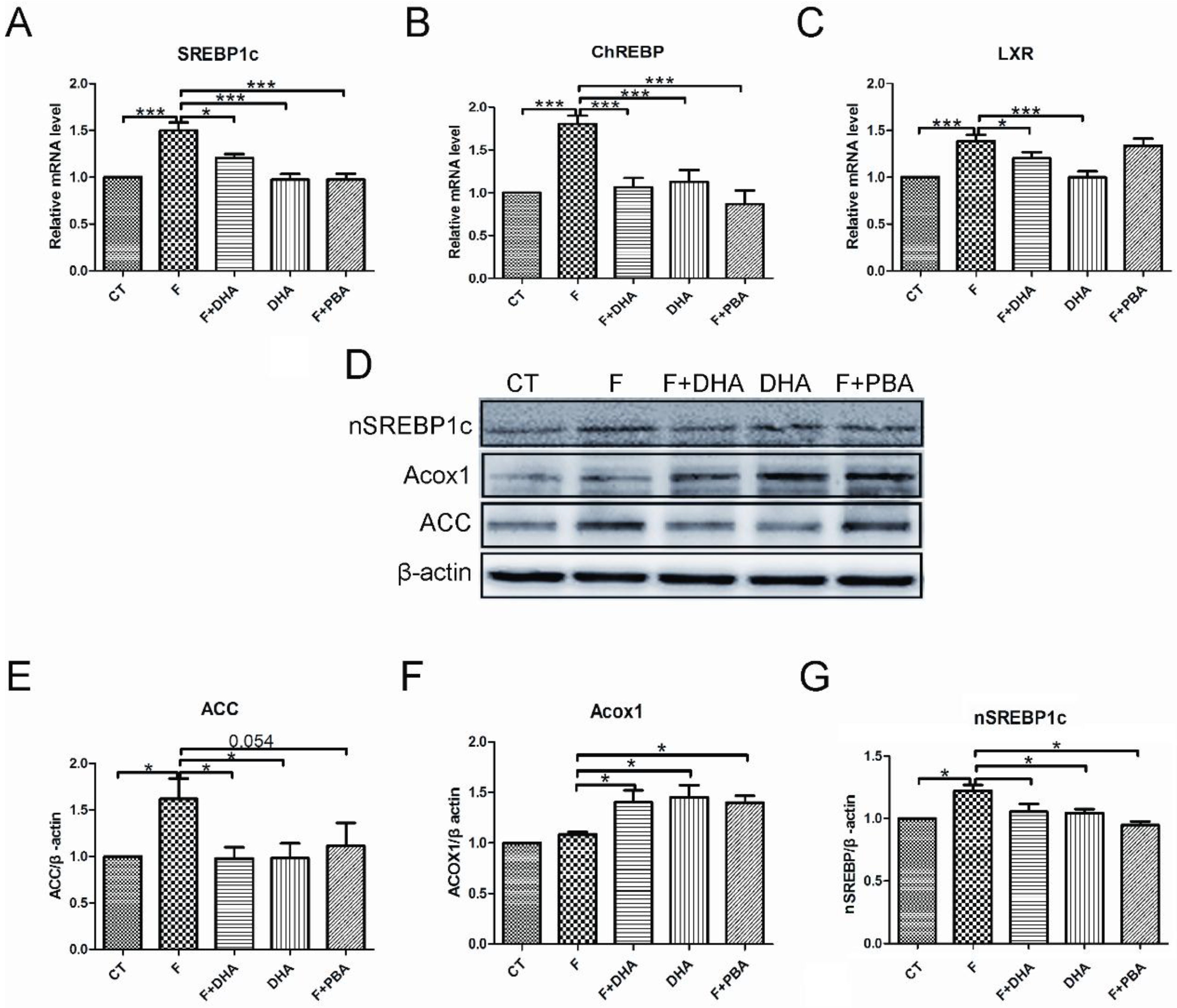
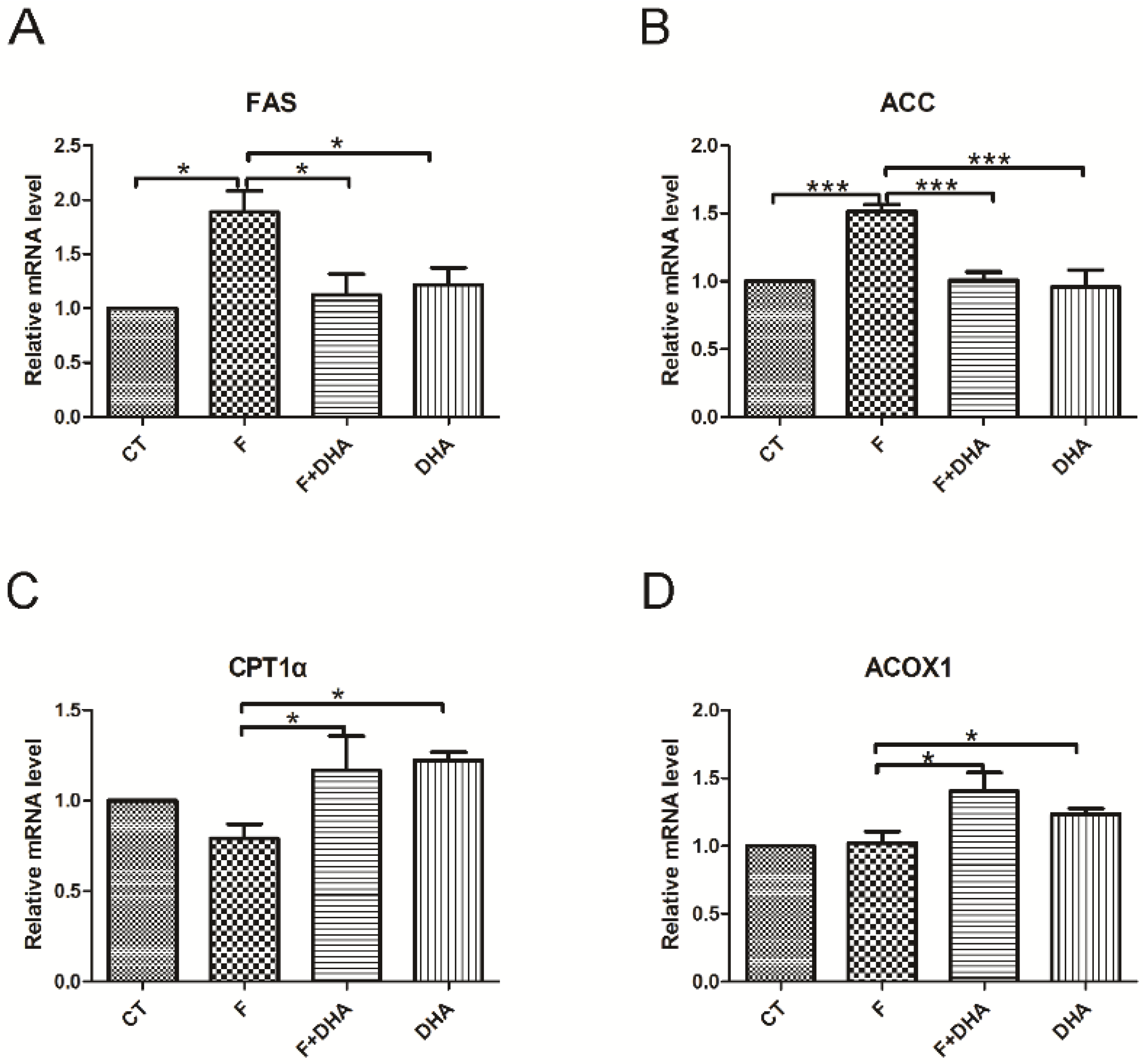
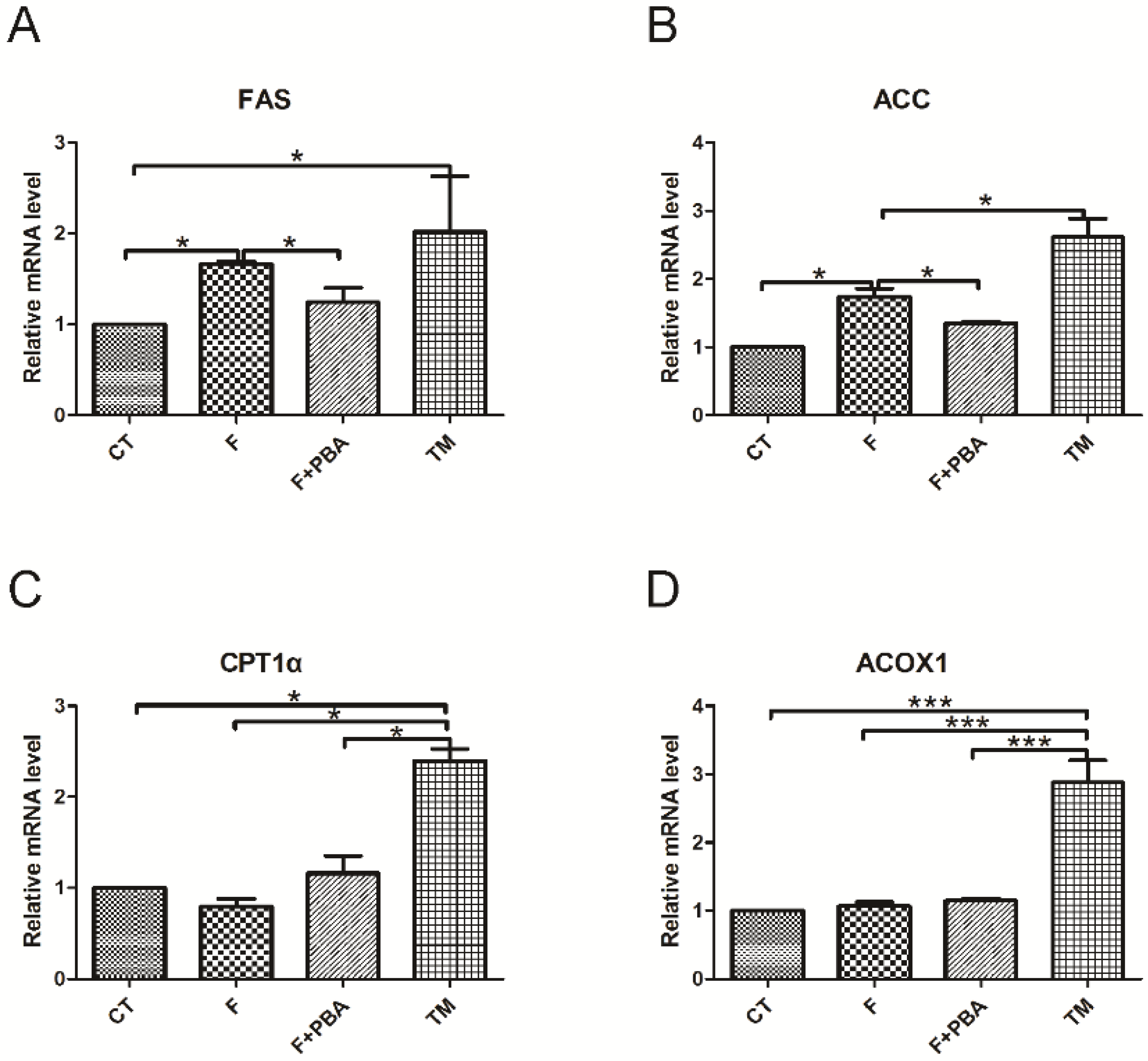
3.2. DHA Attenuates Fructose-Induced Hepatic Steatosis Involving Changes in Expressions of Genes Related to Lipid Metabolism
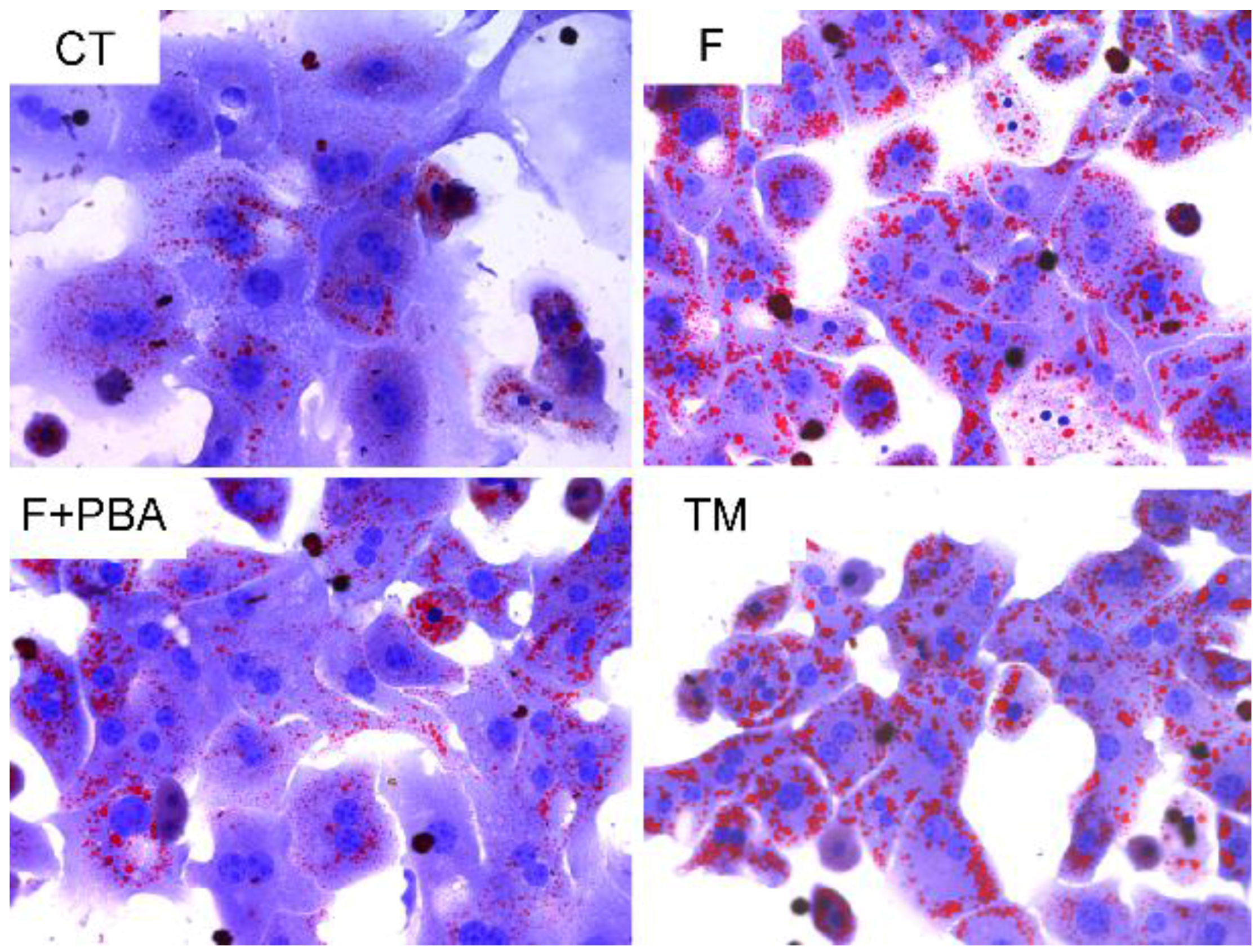
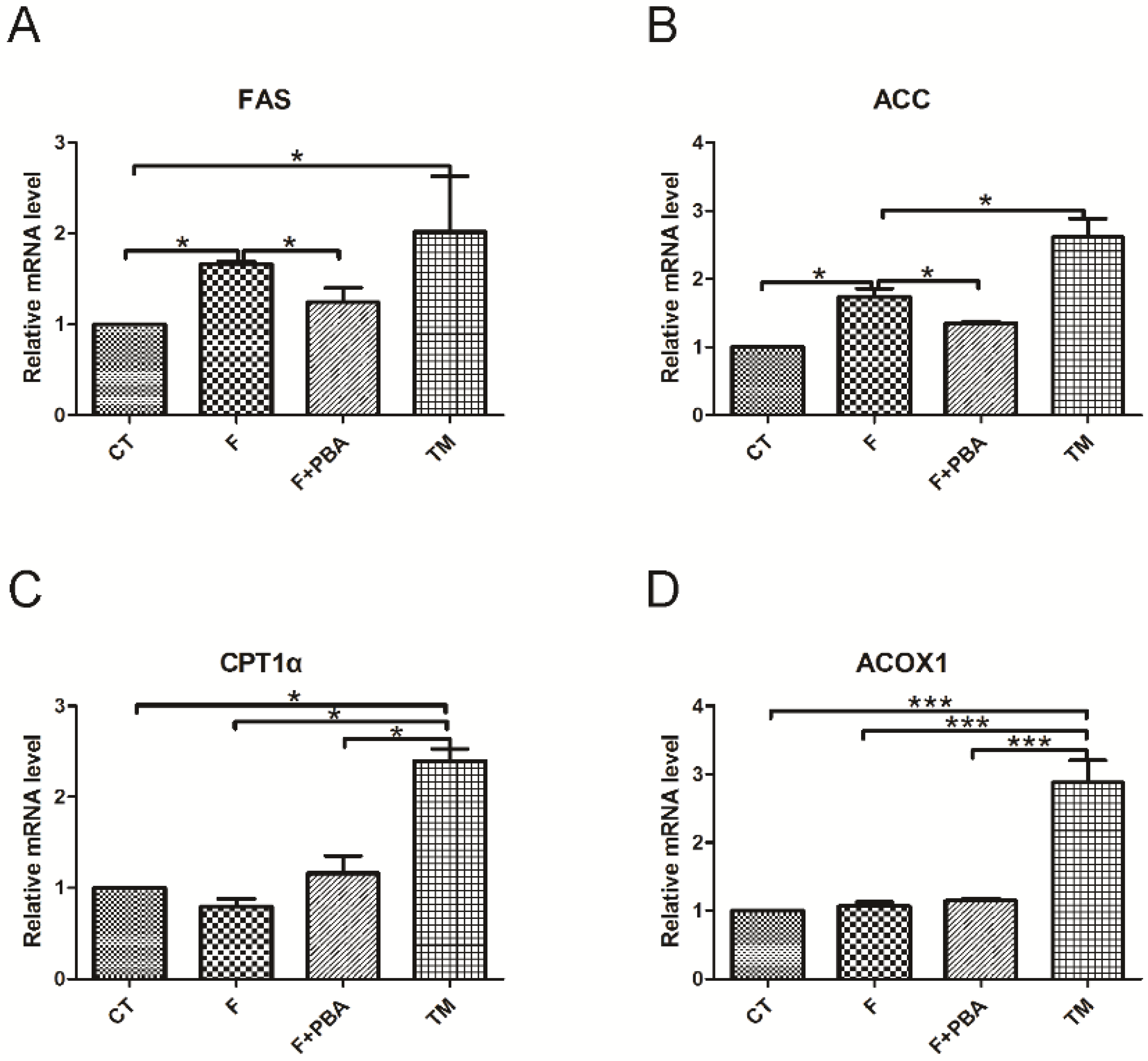
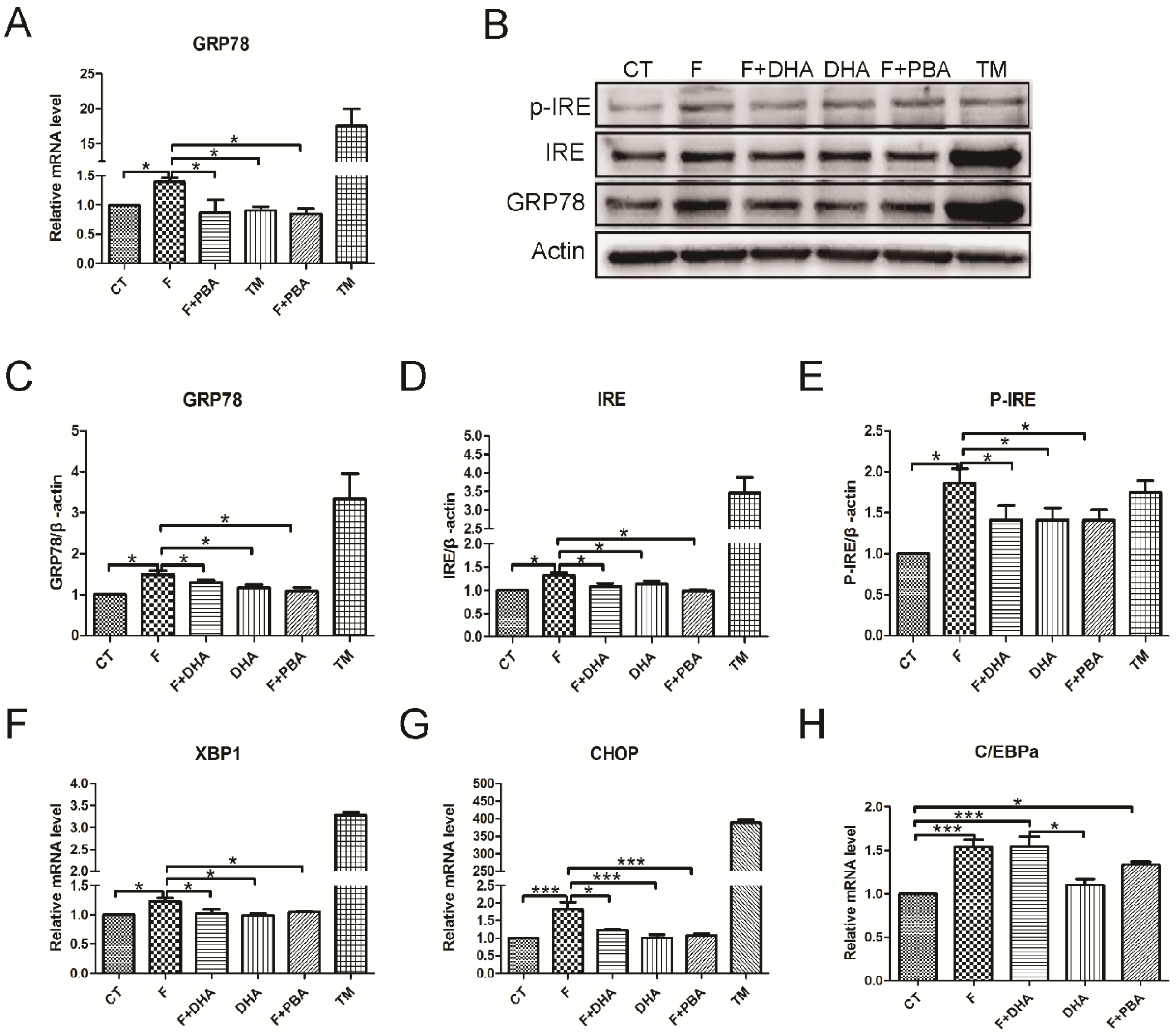
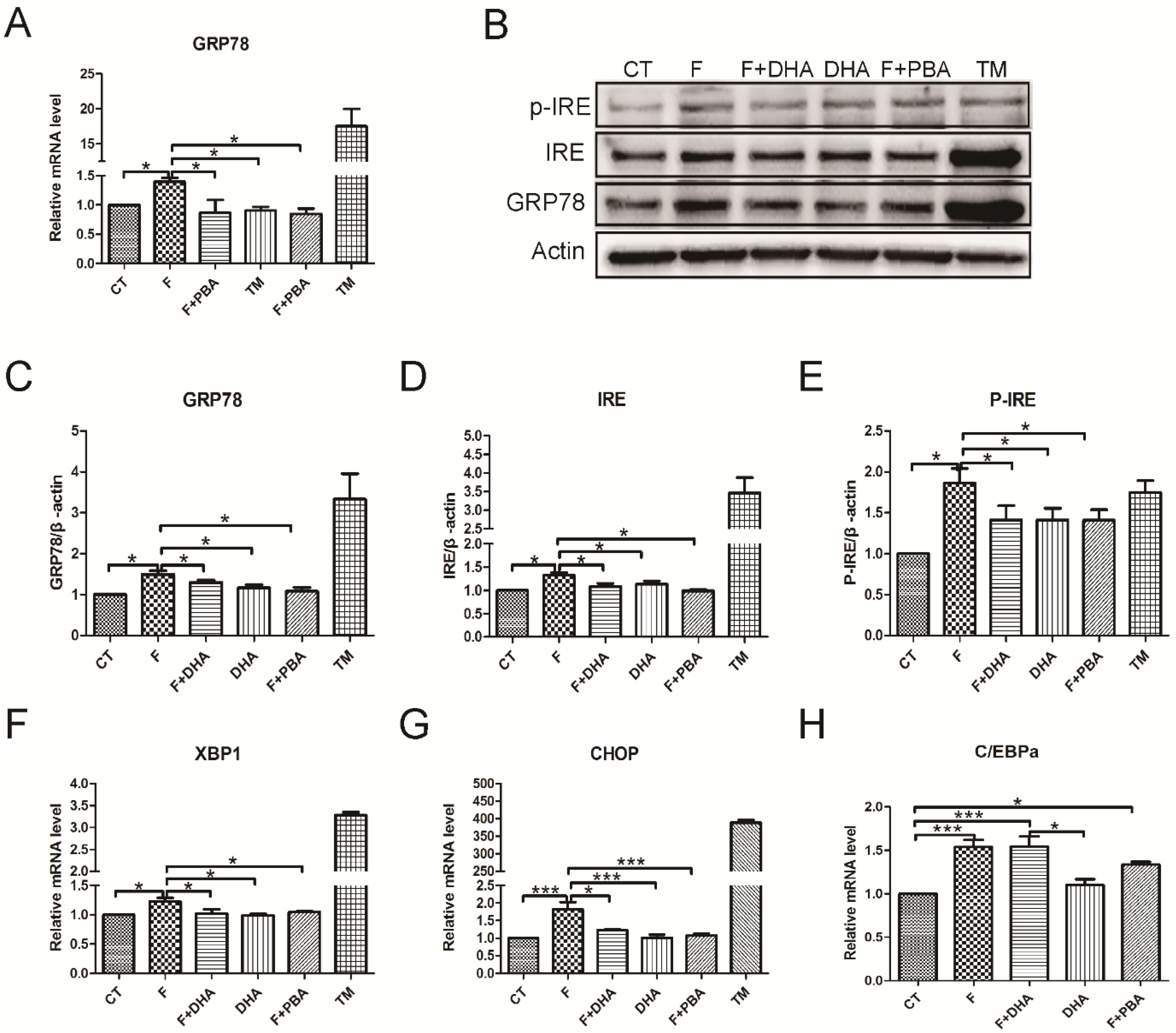
3.3. ER Stress Pathways Mediates Fructose-Induced Lipid Accumulation
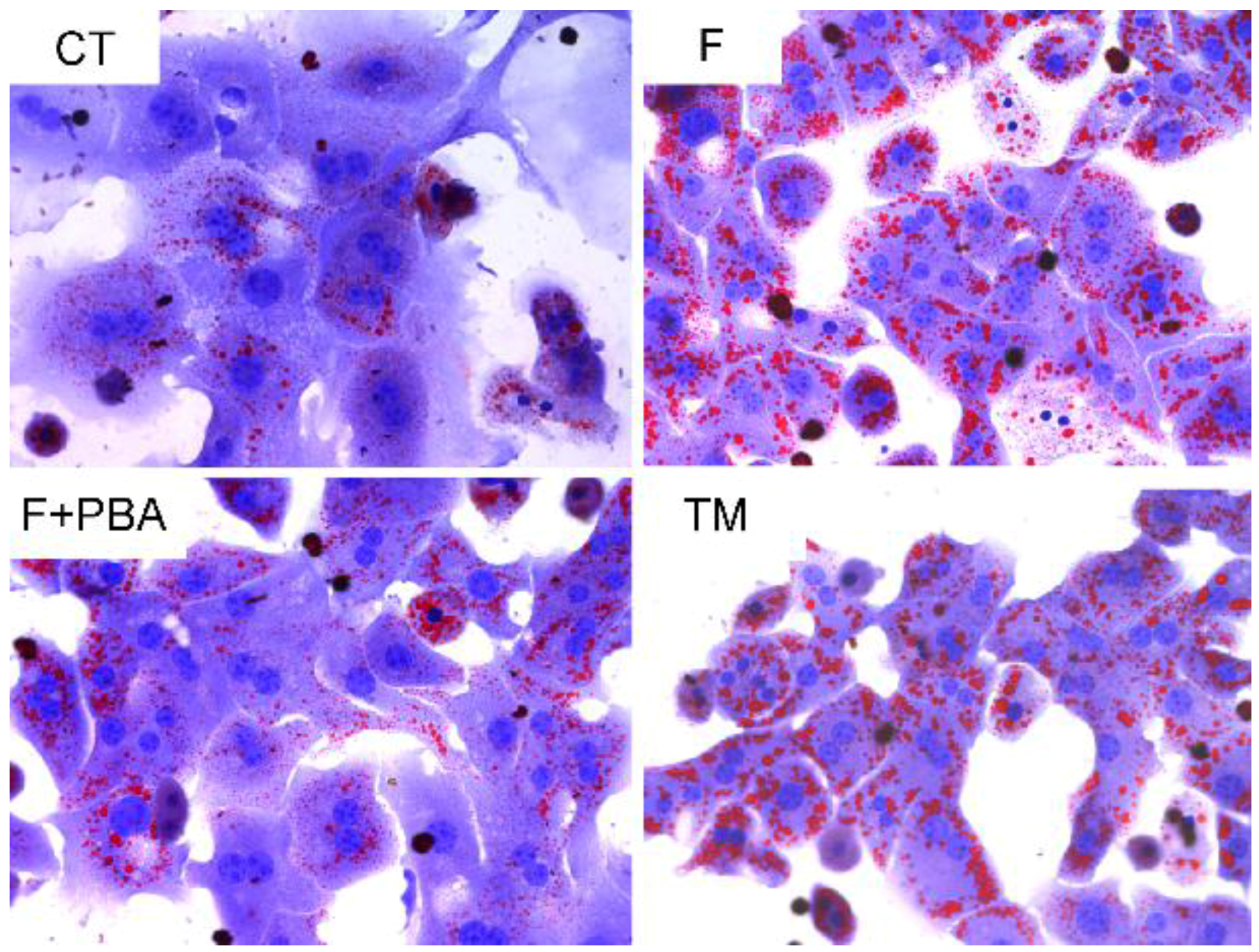
3.4. ER Stress Response Is Involved in the Protective Effects of DHA against Fructose-Induced Hepatic Steatosis
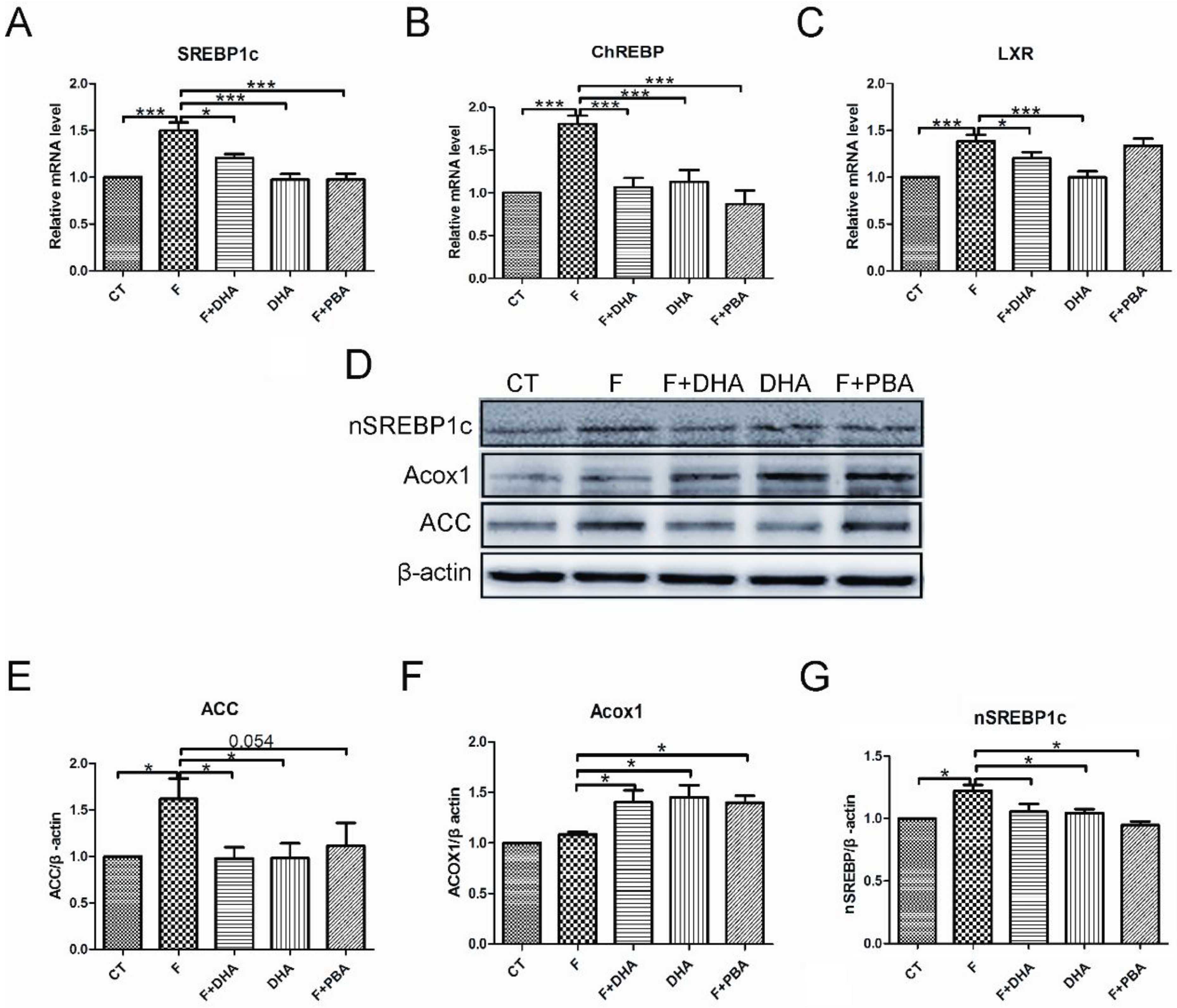
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Amarapurkar, D.N.; Hashimoto, E.; Lesmana, L.A.; Sollano, J.D.; Chen, P.J.; Goh, K.L. How common is non-alcoholic fatty liver disease in the Asia-Pacific region and are there local differences? J. Gastroenterol. Hepatol. 2007, 22, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.; Lee, C.K.; Chan, M.; Seto, W.K.; Lai, C.L.; Yuen, M.F. High prevalence of non-alcoholic fatty liver disease in the Chinese—Results from the Hong Kong liver health census. Liver Int. 2015, 35, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Kaser, S.; Tilg, H. Non-alcoholic steatohepatitis: A microbiota-driven disease. Trends Endocrinol. Metab. 2013, 24, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.G.; Saibara, T.; Chitturi, S.; Kim, B.I.; Sung, J.J.; Chutaputti, A. What are the risk factors and settings for non-alcoholic fatty liver disease in Asia-Pacific? J. Gastroenterol. Hepatol. 2007, 22, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Senturk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Kelishadi, R.; Mansourian, M.; Heidari-Beni, M. Association of fructose consumption and components of metabolic syndrome in human studies: A systematic review and meta-analysis. Nutrition 2014, 30, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): A population based study. J. Hepatol. 2007, 47, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; Skokan, L.E.; Timlin, M.T.; Dingfelder, C.S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 2008, 138, 1039–1046. [Google Scholar] [PubMed]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [PubMed]
- Kawasaki, T.; Igarashi, K.; Koeda, T.; Sugimoto, K.; Nakagawa, K.; Hayashi, S.; Yamaji, R.; Inui, H.; Fukusato, T.; Yamanouchi, T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J. Nutr. 2009, 139, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Yamanouchi, T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2012, 23, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, M.; Jilkova, Z.M.; Kuda, O.; Jelenik, T.; Medrikova, D.; Stankova, B.; Kristinsson, B.; Haraldsson, G.G.; Svensen, H.; Stoknes, I.; et al. Metabolic effects of n-3 PUFA as phospholipids are superior to triglycerides in mice fed a high-fat diet: Possible role of endocannabinoids. PLoS ONE 2012, 7, e38834. [Google Scholar] [CrossRef] [PubMed]
- Itariu, B.K.; Zeyda, M.; Hochbrugger, E.E.; Neuhofer, A.; Prager, G.; Schindler, K.; Bohdjalian, A.; Mascher, D.; Vangala, S.; Schranz, M.; et al. Long-chain n-3 PUFAs reduce adipose tissue and systemic inflammation in severely obese nondiabetic patients: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 96, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- De Castro, G.S.; Deminice, R.; Simoes-Ambrosio, L.M.; Calder, P.C.; Jordao, A.A.; Vannucchi, H. Dietary docosahexaenoic acid and eicosapentaenoic acid influence liver triacylglycerol and insulin resistance in rats fed a high-fructose diet. Mar. Drugs 2015, 13, 1864–1881. [Google Scholar] [CrossRef] [PubMed]
- Devarshi, P.P.; Jangale, N.M.; Ghule, A.E.; Bodhankar, S.L.; Harsulkar, A.M. Beneficial effects of flaxseed oil and fish oil diet are through modulation of different hepatic genes involved in lipid metabolism in streptozotocin-nicotinamide induced diabetic rats. Genes Nutr. 2013, 8, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, M.; Yahagi, N.; Matsuzaka, T.; Najima, Y.; Nakakuki, M.; Nagai, R.; Ishibashi, S.; Osuga, J.; Yamada, N.; Shimano, H. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 2003, 38, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, H.; Tehilla, M.; Attal-Singer, J.; Bruck, R.; Luzzatti, R.; Singer, P. The therapeutic potential of long-chain omega-3 fatty acids in nonalcoholic fatty liver disease. Clin. Nutr. 2011, 30, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Gomez-Pinilla, F. “Metabolic syndrome” in the brain: Deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cognition. J. Physiol. 2012, 590, 2485–2499. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Periz, A.; Horrillo, R.; Ferre, N.; Gronert, K.; Dong, B.; Moran-Salvador, E.; Titos, E.; Martinez-Clemente, M.; Lopez-Parra, M.; Arroyo, V.; et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: A role for resolvins and protectins. FASEB J. 2009, 23, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Harding, H.P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008, 7, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Wu, J.; Back, S.H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.P.; Chan, S.M.; Zeng, X.Y.; Laybutt, D.R.; Iseli, T.J.; Sun, R.Q.; Kraegen, E.W.; Cooney, G.J.; Turner, N.; Ye, J.M. Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance. PLoS ONE 2012, 7, e30816. [Google Scholar] [CrossRef] [PubMed]
- Severgnini, M.; Sherman, J.; Sehgal, A.; Jayaprakash, N.K.; Aubin, J.; Wang, G.; Zhang, L.; Peng, C.G.; Yucius, K.; Butler, J.; et al. A rapid two-step method for isolation of functional primary mouse hepatocytes: Cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology 2012, 64, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.H.; Abu, E.O.; Byrne, C.D. Non-Alcoholic Fatty Liver Disease (NAFLD): New challenge for general practitioners and important burden for health authorities? Prim. Care Diabetes 2010, 4, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, R.; Barroso, E.; Serrano, L.; Coll, T.; Sanchez, R.M.; Merlos, M.; Palomer, X.; Laguna, J.C.; Vazquez-Carrera, M. Atorvastatin prevents carbohydrate response element binding protein activation in the fructose-fed rat by activating protein kinase A. Hepatology 2009, 49, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Loffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Katsumata, A.; Miura, S.; Kamei, Y.; Ezaki, O. Effects of fish oil feeding and fasting on LXRalpha/RXRalpha binding to LXRE in the SREBP-1c promoter in mouse liver. Biochim. Biophys. Acta 2005, 1736, 77–86. [Google Scholar] [PubMed]
- McGarry, J.D. Malonyl-CoA and carnitine palmitoyltransferase I: An expanding partnership. Biochem. Soc. Trans. 1995, 23, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Weis, B.C.; Husti, J.E.; Foster, D.W.; McGarry, J.D. Mitochondrial carnitine palmitoyltransferase I isoform switching in the developing rat heart. J. Biol. Chem. 1995, 270, 8952–8957. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.A.; Mater, M.K.; Thelen, A.P.; Peters, J.M.; Gonzalez, F.J.; Jump, D.B. Evidence against the peroxisome proliferator-activated receptor alpha (PPARalpha) as the mediator for polyunsaturated fatty acid suppression of hepatic L-pyruvate kinase gene transcription. J. Lipid Res. 2000, 41, 742–751. [Google Scholar] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Zheng, Z.; Mendez, R.; Ha, S.W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 2012, 211, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, X.; Zhu, R.M.; Zhang, Y.; Yu, T.; Wang, H.; Zhao, H.; Zhao, M.; Ji, Y.L.; Chen, Y.H.; et al. Endoplasmic reticulum stress is involved in hepatic SREBP-1c activation and lipid accumulation in fructose-fed mice. Toxicol. Lett. 2012, 212, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Peng, C.; Ai, Y.; Wang, H.; Xiao, X.; Li, J. Docosahexaenoic Acid Ameliorates Fructose-Induced Hepatic Steatosis Involving ER Stress Response in Primary Mouse Hepatocytes. Nutrients 2016, 8, 55. https://doi.org/10.3390/nu8010055
Zheng J, Peng C, Ai Y, Wang H, Xiao X, Li J. Docosahexaenoic Acid Ameliorates Fructose-Induced Hepatic Steatosis Involving ER Stress Response in Primary Mouse Hepatocytes. Nutrients. 2016; 8(1):55. https://doi.org/10.3390/nu8010055
Chicago/Turabian StyleZheng, Jinying, Chuan Peng, Yanbiao Ai, Heng Wang, Xiaoqiu Xiao, and Jibin Li. 2016. "Docosahexaenoic Acid Ameliorates Fructose-Induced Hepatic Steatosis Involving ER Stress Response in Primary Mouse Hepatocytes" Nutrients 8, no. 1: 55. https://doi.org/10.3390/nu8010055
APA StyleZheng, J., Peng, C., Ai, Y., Wang, H., Xiao, X., & Li, J. (2016). Docosahexaenoic Acid Ameliorates Fructose-Induced Hepatic Steatosis Involving ER Stress Response in Primary Mouse Hepatocytes. Nutrients, 8(1), 55. https://doi.org/10.3390/nu8010055