Hepatic Steatosis as a Marker of Metabolic Dysfunction

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Diagnosis and Prevalence of NAFLD
3. Metabolic Dysfunction in NAFLD
4. Glucose Metabolism
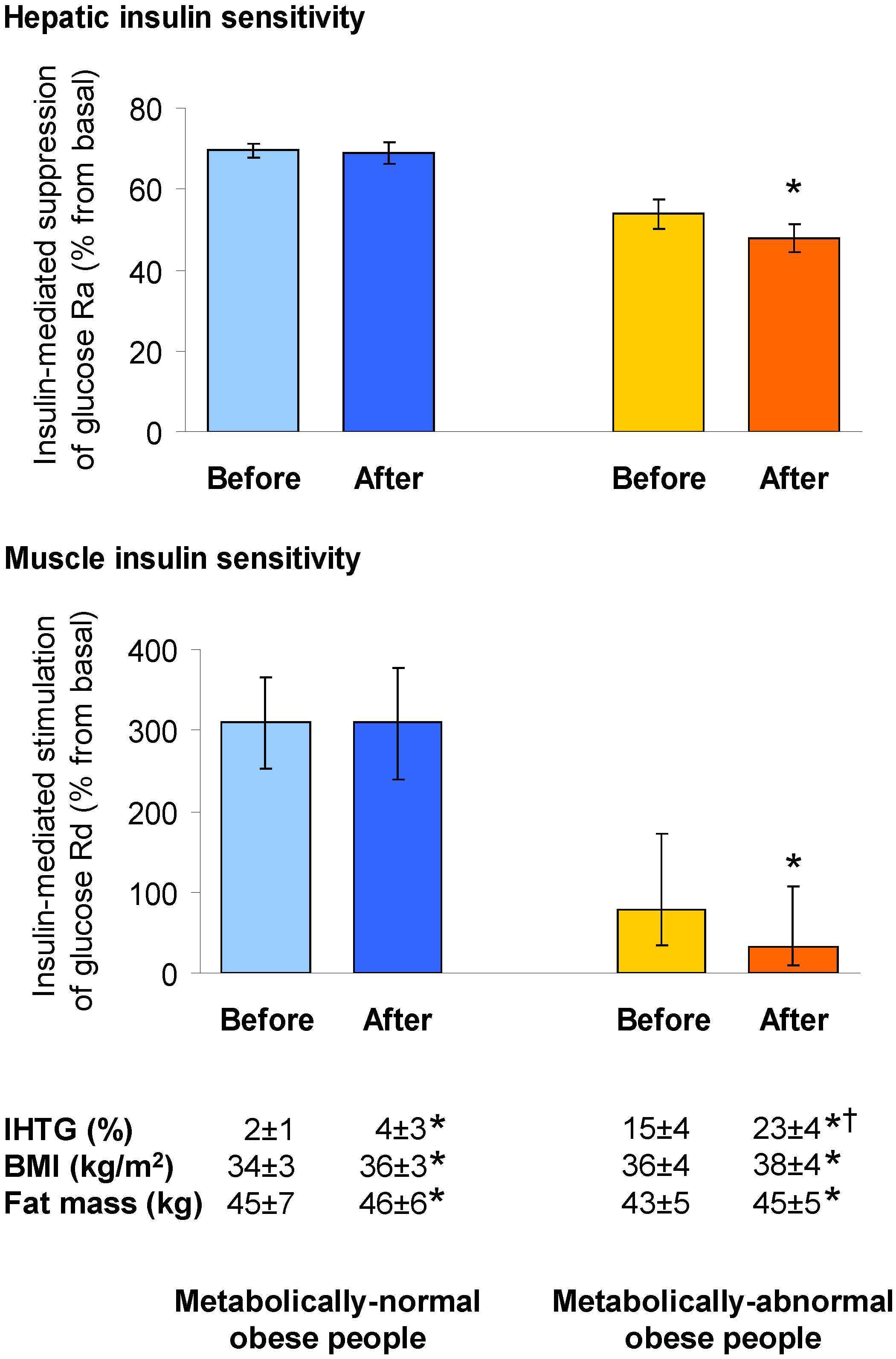
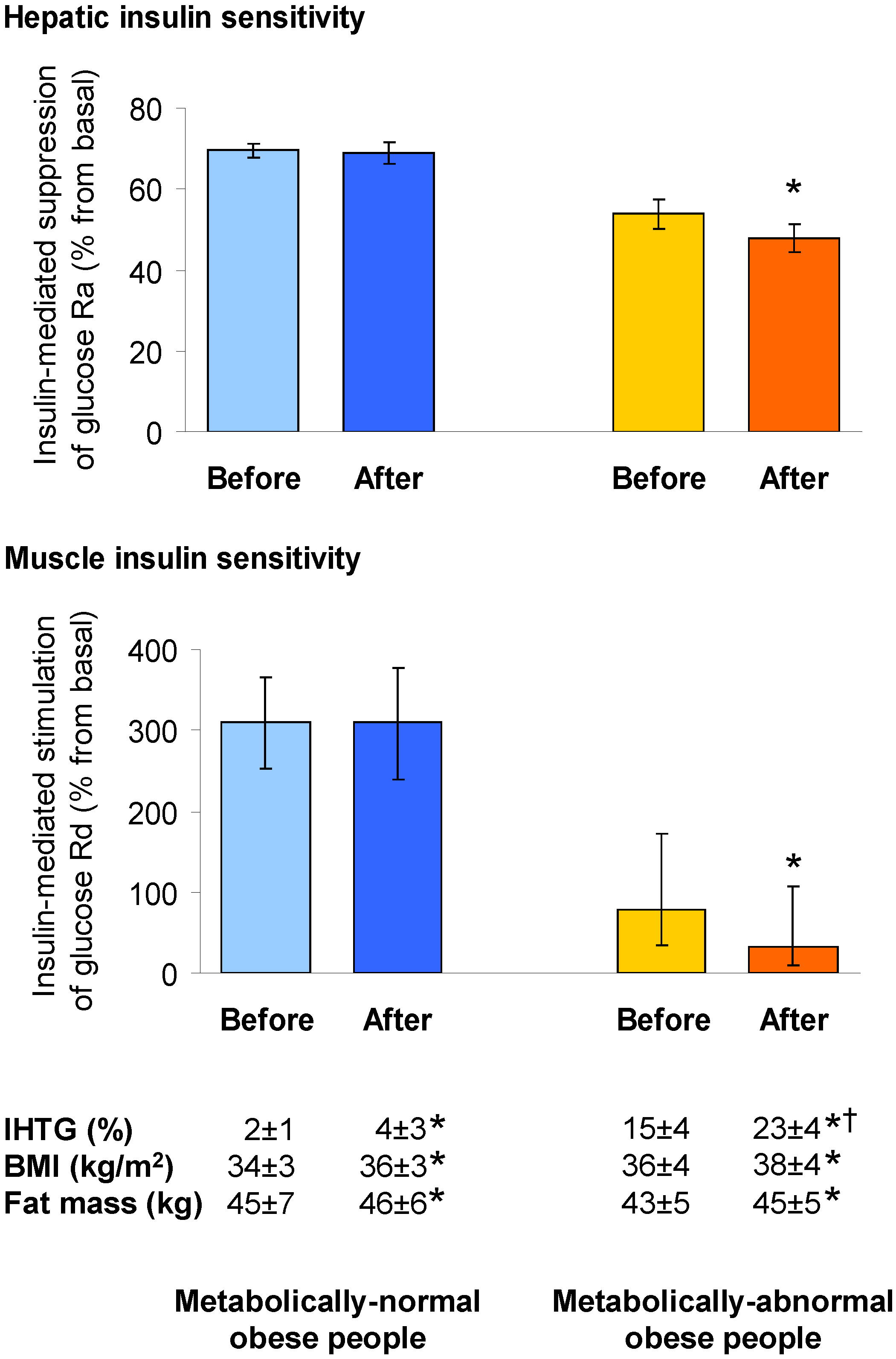
4.1. Hepatic Insulin Action in NAFLD
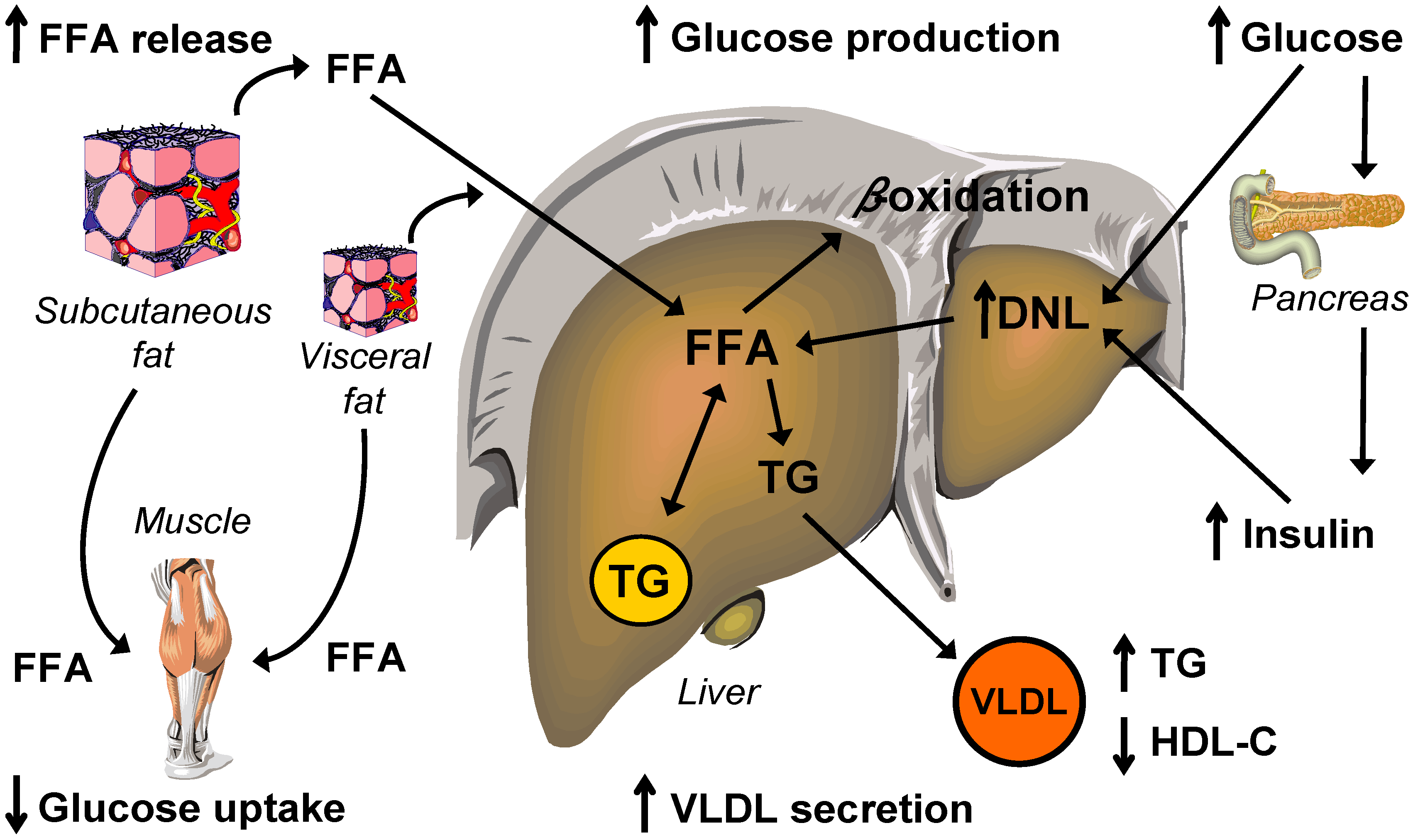
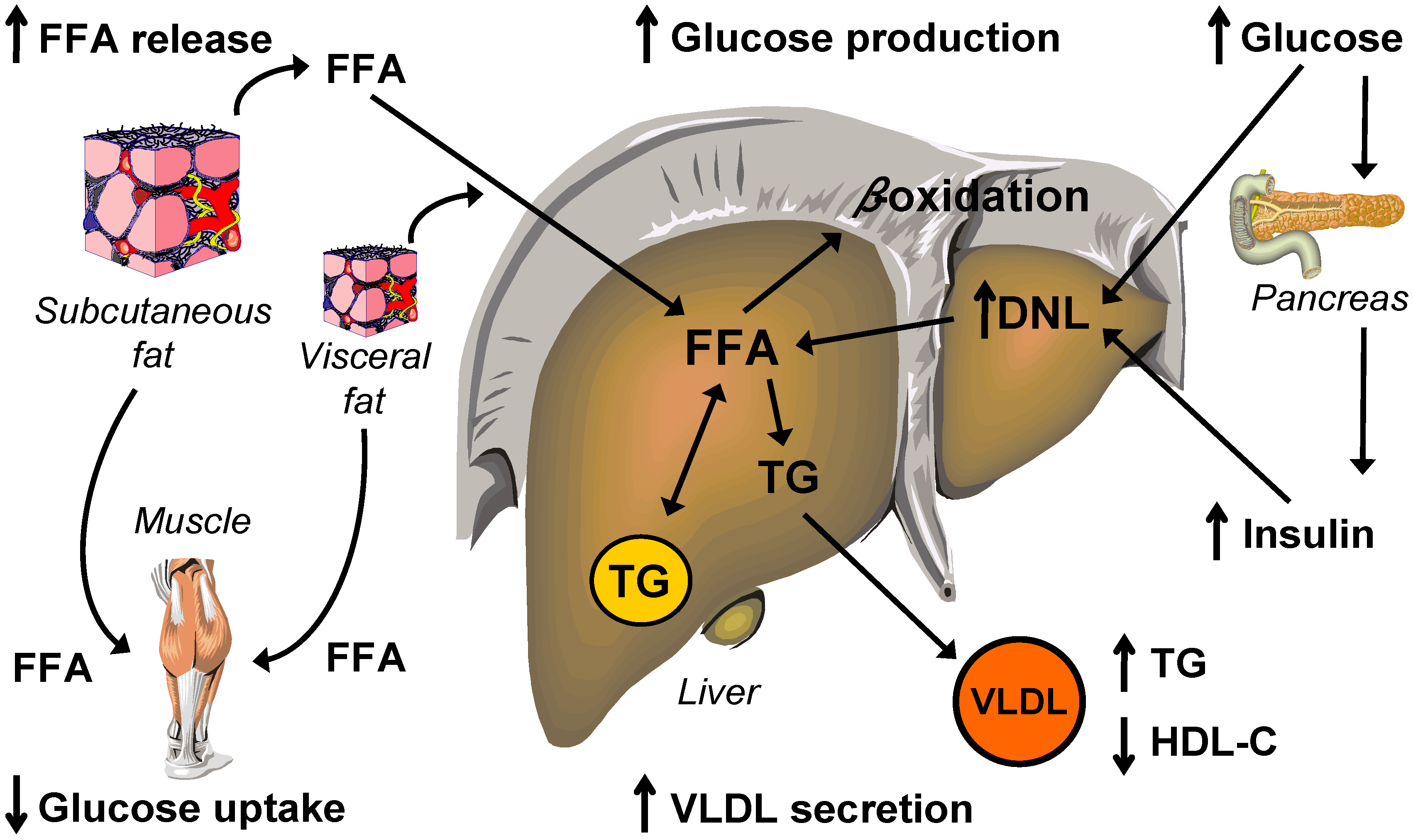
Molecular Mechanisms Linking Hepatic Steatosis to Insulin Resistance
4.2. Muscle Insulin Action in NAFLD
4.3. β-Cell Dysfunction in NAFLD
4.4. Dissociation between Insulin Resistance and NAFLD
5. Lipid and Lipoprotein Metabolism
5.1. Intrahepatic Fatty Acid Availability in NAFLD
5.1.1. Hepatic Fatty Acid Uptake
5.1.2. Hepatic de Novo Lipogenesis
5.2. Intrahepatic Fatty Acid Mobilization in NAFLD
5.2.1. Hepatic Fatty Acid Oxidation and Ketogenesis
5.2.2. Hepatic VLDL-triglyceride Secretion
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Tarantino, G.; Conca, P.; Pasanisi, F.; Ariello, M.; Mastrolia, M.; Arena, A.; Tarantino, M.; Scopacasa, F.; Vecchione, R. Could inflammatory markers help diagnose nonalcoholic steatohepatitis? Eur. J. Gastroenterol. Hepatol. 2009, 21, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (nafld) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.R.; Waters, B.; Patil, S.R.; Reuben, A.; Morelli, J.; Riely, C.A. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am. J. Gastroenterol. 2001, 96, 2957–2961. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Lotan, R.; Shibolet, O.; Webb, M.; Buch, A.; Nitzan-Kaluski, D.; Halpern, Z.; Santo, E.; Oren, R. Non-alcoholic fatty liver disease independently predicts prediabetes during a 7-year prospective follow-up. Liver Int. 2013, 33, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Hoyumpa, A.M., Jr.; Greene, H.L.; Dunn, G.D.; Schenker, S. Fatty liver: Biochemical and clinical considerations. Am. J. Dig. Dis. 1975, 20, 1142–1170. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Brunt, E.M.; Wehmeier, K.R.; Oliver, D.; Bacon, B.R. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the ppar-gamma ligand rosiglitazone. Hepatology 2003, 38, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Feng, J.; Befroy, D.; Dziura, J.; Dalla Man, C.; Cobelli, C.; Shulman, G.I. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in asian-indian men. Proc. Natl. Acad. Sci. USA 2006, 103, 18273–18277. [Google Scholar] [CrossRef] [PubMed]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, C.E.; Everhart, J.E. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterology 2003, 124, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the united states: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D. Common genetic variants and nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of nonalcoholic fatty liver disease in the united states: The third national health and nutrition examination survey, 1988–1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef] [PubMed]
- World Gastroenterology Organisation. Global Guidelines: Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Online Report. 2012. Available online: http://www.worldgastroenterology.org/assets/export/userfiles/2012_NASH%20and%20NAFLD_Final_long.pdf (accessed on 17 June 2015).
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Leite, N.C.; Salles, G.F.; Araujo, A.L.; Villela-Nogueira, C.A.; Cardoso, C.R. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009, 29, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Portillo Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J. Clin. Endocrinol. Metab. 2014. [Google Scholar] [CrossRef] [PubMed]
- Seifter, S.; Englard, S. Energy metabolism. In The Liver: Biology and Pathobiology, 3rd ed.; Arias, I.M., Boyer, J.L., Fausto, N., Jakoby, W.B., Schachter, D.A., Shafritz, D.A., Eds.; Raven Press, Ltd.: New York, NY, USA, 1994; pp. 323–364. [Google Scholar]
- Van den Berghe, G. The role of the liver in metabolic homeostasis: Implications for inborn errors of metabolism. J. Inherit. Metab. Dis. 1991, 14, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.; Kuntz, H.-D. Hepatology: Principles and Practice, 2nd ed.; Springer Medizin Verlag: Heidelberg, Germany, 2006. [Google Scholar]
- Cherrington, A.D. Control of glucose production in vivo by insulin and glucagon. In The Endocrine Pancreas and Regulation of Metabolism (Handbook of Physiology, Section 7: The Endocrine System, Volume ii); Jefferson, L.S., Cherrington, A.D., Eds.; American Physiological Society/Oxford University Press: New York, NY, USA, 2001; Volume II, pp. 759–785. [Google Scholar]
- Boden, G. Carbohydrates and the liver. In Textbook of Hepatology: From Basic Science to Clinical Practice, 3rd ed.; Rodés, J., Benhamou, J.-P., Blei, A.T., Reichen, J., Rizzetto, M., Eds.; Blackwell Publishing: Malden, MA, USA, 2007; pp. 129–133. [Google Scholar]
- Gastaldelli, A.; Miyazaki, Y.; Pettiti, M.; Buzzigoli, E.; Mahankali, S.; Ferrannini, E.; DeFronzo, R.A. Separate contribution of diabetes, total fat mass, and fat topography to glucose production, gluconeogenesis, and glycogenolysis. J. Clin. Endocrinol. Metab. 2004, 89, 3914–3921. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Use of the splanchnic/hepatic balance technique in the study of glucose metabolism. Baillieres Clin. Endocrinol. Metab. 1987, 1, 837–862. [Google Scholar] [CrossRef]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab. 2002, 87, 3023–3028. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Yatsuya, H.; Nihashi, T.; Li, Y.; Hotta, Y.; Matsushita, K.; Muramatsu, T.; Otsuka, R.; Matsunaga, M.; Yamashita, K.; Wang, C.; et al. Independent association of liver fat accumulation with insulin resistance. Obes. Res. Clin. Pract. 2014, 8, e350–e355. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [PubMed]
- Vega, G.L.; Chandalia, M.; Szczepaniak, L.S.; Grundy, S.M. Metabolic correlates of nonalcoholic fatty liver in women and men. Hepatology 2007, 46, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Yoshino, J.; Yoshino, M.; Magkos, F.; Tiemann Luecking, C.; Samovski, D.; Fraterrigo, G.; Okunade, A.L.; Patterson, B.W.; Klein, S. Metabolically normal obese people are protected from adverse effects following weight gain. J. Clin. Investig. 2015, 125, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.S.; Szuszkiewicz-Garcia, M.; Browning, J.D.; Baxter, J.D.; Abate, N.; Malloy, C.R. Influence of liver triglycerides on suppression of glucose production by insulin in men. J. Clin. Endocrinol. Metab. 2015, 100, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.K.; Yoshii, H.; Haber, C.A.; Bogdanovic, E.; Lam, L.; Fantus, I.G.; Giacca, A. Free fatty acid-induced hepatic insulin resistance: A potential role for protein kinase c-delta. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E682–E691. [Google Scholar] [CrossRef] [PubMed]
- Frangioudakis, G.; Burchfield, J.G.; Narasimhan, S.; Cooney, G.J.; Leitges, M.; Biden, T.J.; Schmitz-Peiffer, C. Diverse roles for protein kinase c delta and protein kinase c epsilon in the generation of high-fat-diet-induced glucose intolerance in mice: Regulation of lipogenesis by protein kinase c delta. Diabetologia 2009, 52, 2616–2620. [Google Scholar] [CrossRef] [PubMed]
- Bezy, O.; Tran, T.T.; Pihlajamaki, J.; Suzuki, R.; Emanuelli, B.; Winnay, J.; Mori, M.A.; Haas, J.; Biddinger, S.B.; Leitges, M.; et al. Pkcdelta regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J. Clin. Investig. 2011, 121, 2504–2517. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.W.; Burrington, C.M.; Luo, Y.; Ruhoff, M.S.; Lynch, D.T.; Chaithongdi, N. Pkcdelta is activated in the liver of obese zucker rats and mediates diet-induced whole body insulin resistance and hepatocyte cellular insulin resistance. J. Nutr. Biochem. 2014, 25, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Non alcoholic fatty liver disease, hepatic insulin resistance and type 2 diabetes. Hepatology 2013, 59, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Bansode, R.; Mehta, M.; Mehta, K.D. Loss of protein kinase cbeta function protects mice against diet-induced obesity and development of hepatic steatosis and insulin resistance. Hepatology 2009, 49, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Burrington, C.M.; Davenport, S.K.; Johnson, A.K.; Horsman, M.J.; Chowdhry, S.; Greene, M.W. Pkcdelta regulates hepatic triglyceride accumulation and insulin signaling in lepr(db/db) mice. Biochem. Biophys. Res. Commun. 2014, 450, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446.e2. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Almgren, P.; Tuomi, T.; Groop, L. Contribution of insulin-stimulated glucose uptake and basal hepatic insulin sensitivity to surrogate measures of insulin sensitivity. Diabetes Care 2004, 27, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Ko, E.H.; Kim, J.E.; Kim, E.; Lee, H.; Choi, H.; Yu, J.H.; Kim, H.J.; Seong, J.K.; Kim, K.S.; et al. Nuclear receptor ppargamma-regulated monoacylglycerol o-acyltransferase 1 (mgat1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13656–13661. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Kou, K.; Chen, Z.; Pietka, T.A.; Kumar, M.; Korenblat, K.M.; Lee, K.; Ahn, K.; Fabbrini, E.; Klein, S.; et al. Evidence for regulated monoacylglycerol acyltransferase expression and activity in human liver. J. Lipid Res. 2012, 53, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Soufi, N.; Chambers, K.T.; Chen, Z.; Schweitzer, G.G.; McCommis, K.S.; Erion, D.M.; Graham, M.J.; Su, X.; Finck, B.N. Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes 2014, 63, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Kim, Y.H.; Kim, D.K.; Lee, M.W.; Lee, S.Y.; Park, T.S.; Koo, S.H.; Lee, C.H.; Choi, H.S. Activation of cannabinoid receptor type 1 (cb1r) disrupts hepatic insulin receptor signaling via cyclic amp-response element-binding protein h (crebh)-mediated induction of lipin1 gene. J. Biol. Chem. 2012, 287, 38041–38049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Yang, L.; Fabbrini, E.; Mohammed, B.S.; Eagon, J.C.; Hotamisligil, G.S.; Klein, S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 2009, 58, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of ikk-beta and nf-kappab. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Nishimura, T.; Ishiba, H.; Seko, Y.; Okajima, A.; Fujii, H.; Tochiki, N.; Umemura, A.; Moriguchi, M.; Sumida, Y.; et al. Blockade of interleukin 6 signalling ameliorates systemic insulin resistance through upregulation of glucose uptake in skeletal muscle and improves hepatic steatosis in high-fat diet fed mice. Liver Int. 2015, 35, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Pilli, S.; Gebremariam, A.; Keirns, C.C.; Davis, M.M.; Vijan, S.; Freed, G.L.; Herman, W.H.; Gurney, J.G. Getting heavier, younger: Trajectories of obesity over the life course. Int. J. Obes. (Lond.) 2010, 34, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.A.; Hamer, M.; Sabia, S.; Singh-Manoux, A.; Batty, G.D.; Kivimaki, M. The natural course of healthy obesity over 20 years. J. Am. Coll. Cardiol. 2015, 65, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Gastaldelli, A.; Tiribelli, C.; Agosti, F.; De Col, A.; Fessehatsion, R.; Sartorio, A. Relationship between glucose metabolism and non-alcoholic fatty liver disease severity in morbidly obese women. J. Endocrinol. Investig. 2014, 37, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Cheang, K.L.; Luketic, V.A.; Boyett, S.; Idowu, M.O.; Patidar, K.; Puri, P.; Matherly, S.; Stravitz, R.T.; Sterling, R.K.; et al. Nonalcoholic steatohepatitis (nash) is associated with a decline in pancreatic beta cell (β-cell) function. Dig. Dis. Sci. 2015. [Google Scholar] [CrossRef]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V., Sr.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing dgat in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Minehira, K.; Young, S.G.; Villanueva, C.J.; Yetukuri, L.; Oresic, M.; Hellerstein, M.K.; Farese, R.V., Jr.; Horton, J.D.; Preitner, F.; Thorens, B.; et al. Blocking vldl secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J. Lipid Res. 2008, 49, 2038–2044. [Google Scholar] [CrossRef] [PubMed]
- Grefhorst, A.; Hoekstra, J.; Derks, T.G.; Ouwens, D.M.; Baller, J.F.; Havinga, R.; Havekes, L.M.; Romijn, J.A.; Kuipers, F. Acute hepatic steatosis in mice by blocking beta-oxidation does not reduce insulin sensitivity of very-low-density lipoprotein production. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G592–G598. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Murray, S.F.; Pandey, S.K.; Booten, S.L.; Bao, D.; Song, X.Z.; Kelly, S.; Chen, S.; McKay, R.; Monia, B.P.; et al. Antisense oligonucleotide reduction of dgat2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005, 42, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Fabbrini, E.; Kars, M.; Yue, P.; Schechtman, K.; Schonfeld, G.; Klein, S. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology 2010, 139, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.E.; Lammers, N.M.; Nederveen, A.J.; van der Graaf, M.; Heerschap, A.; Ackermans, M.T.; Sauerwein, H.P.; Stroes, E.S.; Serlie, M.J. Hepatic steatosis does not cause insulin resistance in people with familial hypobetalipoproteinaemia. Diabetologia 2011, 54, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J. Storing up trouble: Does accumulation of intramyocellular triglyceride protect skeletal muscle from insulin resistance? Clin. Exp. Pharmacol. Physiol. 2009, 36, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-coa desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, M.W. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: Possible role in steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G194–G198. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (nafld). Prog. Lipid Res. 2009, 48, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W.; Staffer, S.; Wannhoff, A.; Pathil, A.; Chamulitrat, W. Plasma membrane phospholipase a2 controls hepatocellular fatty acid uptake and is responsive to pharmacological modulation: Implications for nonalcoholic steatohepatitis. FASEB J. 2014, 28, 3159–3170. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Lobello, R.; Scopacasa, F.; Contaldo, F.; Pasanisi, F.; Cirillo, M.; De Caterina, M.; Conca, P.; Terracciano, D.; Gennarelli, N.; et al. The contribution of omental adipose tissue to adipokine concentrations in patients with the metabolic syndrome. Clin. Investig. Med. 2007, 30, E192–E199. [Google Scholar]
- Mittendorfer, B.; Magkos, F.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity 2009, 17, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; deHaseth, D.; Deivanayagam, S.; Mohammed, B.S.; Vitola, B.E.; Klein, S. Alterations in fatty acid kinetics in obese adolescents with increased intrahepatic triglyceride content. Obesity (Silver Spring) 2009, 17, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial tca cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Hazlehurst, J.M.; Hull, D.; Guo, K.; Borrows, S.; Yu, J.; Gough, S.C.; Newsome, P.N.; Tomlinson, J.W. Abdominal subcutaneous adipose tissue insulin resistance and lipolysis in patients with non-alcoholic steatohepatitis. Diabetes Obes. Metab. 2014, 16, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Pardina, E.; Baena-Fustegueras, J.A.; Catalan, R.; Galard, R.; Lecube, A.; Fort, J.M.; Allende, H.; Vargas, V.; Peinado-Onsurbe, J. Increased expression and activity of hepatic lipase in the liver of morbidly obese adult patients in relation to lipid content. Obes. Surg. 2008, 19, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Siren, J.; Hamsten, A.; Fisher, R.M.; Yki-Jarvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human nafld. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef] [PubMed]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sanchez-Campos, S.; Garcia-Mediavilla, M.V.; Fernandez-Bermejo, M.; Lozano-Rodriguez, T.; Vargas-Castrillon, J.; Buque, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase cd36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis c. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Berlanga, A.; Guiu-Jurado, E.; Martinez, S.; Porras, J.A.; Aragones, G.; Sabench, F.; Hernandez, M.; Aguilar, C.; Sirvent, J.J.; et al. Altered fatty acid metabolism-related gene expression in liver from morbidly obese women with non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 22173–22187. [Google Scholar] [CrossRef] [PubMed]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K.; Schwarz, J.M.; Neese, R.A. Regulation of hepatic de novo lipogenesis in humans. Annu. Rev. Nutr. 1996, 16, 523–557. [Google Scholar] [CrossRef] [PubMed]
- Eissing, L.; Scherer, T.; Todter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to chrebp-beta and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef] [PubMed]
- Diraison, F.; Moulin, P.; Beylot, M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Barrows, B.R.; Parks, E.J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Lambert, J.E.; Hovhannisyan, Y.; Ramos-Roman, M.A.; Trombold, J.R.; Wagner, D.A.; Parks, E.J. Palmitoleic acid is elevated in fatty liver disease and reflects hepatic lipogenesis. Am. J. Clin. Nutr. 2015, 101, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Nakamuta, M.; Kohjima, M.; Morizono, S.; Kotoh, K.; Yoshimoto, T.; Miyagi, I.; Enjoji, M. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2005, 16, 631–635. [Google Scholar] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [PubMed]
- Nakamuta, M.; Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Yada, M.; Yada, R.; Takemoto, R.; Fukuizumi, K.; et al. The significance of differences in fatty acid metabolism between obese and non-obese patients with non-alcoholic fatty liver disease. Int. J. Mol. Med. 2008, 22, 663–667. [Google Scholar] [PubMed]
- Timlin, M.T.; Parks, E.J. Temporal pattern of de novo lipogenesis in the postprandial state in healthy men. Am. J. Clin. Nutr. 2005, 81, 35–42. [Google Scholar] [PubMed]
- Tappy, L.; Le, K.A. Does fructose consumption contribute to non-alcoholic fatty liver disease? Clin. Res. Hepatol. Gastroenterol. 2012, 36, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.B.; Gunn, P.J.; Fielding, B.A. The role of dietary sugars and de novo lipogenesis in non-alcoholic fatty liver disease. Nutrients 2014, 6, 5679–5703. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Ma, J.; Patel, K.; Berger, S.; Lau, J.; Lichtenstein, A.H. Fructose, high-fructose corn syrup, sucrose, and nonalcoholic fatty liver disease or indexes of liver health: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2014, 100, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Margherita Mancina, R.; Matikainen, N.; Maglio, C.; Soderlund, S.; Lundbom, N.; Hakkarainen, A.; Rametta, R.; Mozzi, E.; Fargion, S.; Valenti, L.; et al. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to pnpla3 sequence variant. J. Clin. Endocrinol. Metab. 2015. [Google Scholar] [CrossRef]
- Muller, M.J. Hepatic energy and substrate metabolism: A possible metabolic basis for early nutritional support in cirrhotic patients. Nutrition 1998, 14, 30–38. [Google Scholar] [CrossRef]
- Wanders, R.J.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Woeltje, K.F.; Kuwajima, M.; Foster, D.W. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab. Rev. 1989, 5, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D. Mitochondria and energy formation. In Textbook of Hepatology: From Basic Science to Clinical Practice, 3rd ed.; Rodés, J., Benhamou, J.-P., Blei, A.T., Reichen, J., Rizzetto, M., Eds.; Blackwell Publishing: Malden, MA, USA, 2007; pp. 149–165. [Google Scholar]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Song, X.Q.; Mitchell, G.A.; Yamaguchi, S.; Sukegawa, K.; Orii, T.; Kondo, N. Enzymes of ketone body utilization in human tissues: Protein and messenger rna levels of succinyl-coenzyme a (coa):3-ketoacid coa transferase and mitochondrial and cytosolic acetoacetyl-coa thiolases. Pediatr. Res. 1997, 42, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.H.; Bates, M.W.; Page, M.A.; Krebs, H.A. Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem. J. 1971, 121, 41–47. [Google Scholar] [PubMed]
- Eaton, S. Control of mitochondrial beta-oxidation flux. Prog. Lipid Res. 2002, 41, 197–239. [Google Scholar] [CrossRef]
- McGarry, J.D.; Mannaerts, G.P.; Foster, D.W. A possible role for malonyl-coa in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Investig. 1977, 60, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, J.; Guthke, R.; Dahmen, U.; Dirsch, O.; Holzhuetter, H.G.; Schuster, S. A theoretical study of lipid accumulation in the liver—Implications for nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2013, 1841, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain acyl-coa dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Perlegas, P.; Zhao, Y.; Angdisen, J.; Borgerink, H.; Shadoan, M.K.; Wagner, J.D.; Matern, D.; Rinaldo, P.; Cline, J.M. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 2005, 128, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.S.; Kim, J.H.; Jo, N.Y.; Choi, K.M.; Baik, S.H.; Park, J.J.; Kim, J.S.; Byun, K.S.; Bak, Y.T.; Lee, C.H.; et al. Ppar agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J. Gastroenterol. Hepatol. 2008, 23, 102–109. [Google Scholar] [PubMed]
- Stefanovic-Racic, M.; Perdomo, G.; Mantell, B.S.; Sipula, I.J.; Brown, N.F.; O’Doherty, R.M. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E969–E977. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Choi, C.S.; Samuel, V.T.; Liu, Z.X.; Zhang, D.; Wang, A.; Zhang, X.M.; Cline, G.W.; Yu, X.X.; Geisler, J.G.; et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-coa carboxylases 1 and 2. J. Clin. Investig. 2006, 116, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.P.; Kelly, K.B.; Leonard, K.A.; Jacobs, R.L. Creatine reduces hepatic tg accumulation in hepatocytes by stimulating fatty acid oxidation. Biochim. Biophys. Acta 2014, 1841, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Gorski, J.C.; Asghar, M.S.; Asghar, A.; Foresman, B.; Hall, S.D.; Crabb, D.W. Hepatic cytochrome p450 2e1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003, 37, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Seppala-Lindroos, A.; Vehkavaara, S.; Bergholm, R.; Frayn, K.N.; Fielding, B.A.; Yki-Jarvinen, H. Liver fat and lipid oxidation in humans. Liver Int. 2009, 29, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R.; Scholz, R.; Browning, E.T. Control mechanisms of gluconeogenesis and ketogenesis. Ii. Interactions between fatty acid oxidation and the citric acid cycle in perfused rat liver. J. Biol. Chem. 1969, 244, 4617–4627. [Google Scholar] [PubMed]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver atp homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Hartwig, S.; Haas, J.; Lehr, S.; Goeddeke, S.; Susanto, F.; Bohne, L.; Jacob, S.; Koellmer, C.; Nitzgen, U.; et al. Peroxisomes compensate hepatic lipid overflow in mice with fatty liver. Biochim. Biophys. Acta 2015, 1851, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Shelness, G.S.; Sellers, J.A. Very-low-density lipoprotein assembly and secretion. Curr. Opin. Lipidol. 2001, 12, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Elovson, J.; Chatterton, J.E.; Bell, G.T.; Schumaker, V.N.; Reuben, M.A.; Puppione, D.L.; Reeve, J.R., Jr.; Young, N.L. Plasma very low density lipoproteins contain a single molecule of apolipoprotein b. J. Lipid Res. 1988, 29, 1461–1473. [Google Scholar] [PubMed]
- Packard, C.J. Understanding coronary heart disease as a consequence of defective regulation of apolipoprotein b metabolism. Curr. Opin. Lipidol. 1999, 10, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F. Fatty acid regulation of very low density lipoprotein production. Curr. Opin. Lipidol. 1997, 8, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F. Basal very low-density lipoprotein metabolism in response to exercise: Mechanisms of hypotriacylglycerolemia. Prog. Lipid Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Mittendorfer, B. Stable isotope-labeled tracers for the investigation of fatty acid and triglyceride metabolism in humans in vivo. Clin. Lipidol. 2009, 4, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Hernandez-Ono, A.; Ko, C.; Yasunaga, K.; Huang, L.S.; Ginsberg, H.N. Regulation of hepatic apolipoprotein b-lipoprotein assembly and secretion by the availability of fatty acids. I. Differential response to the delivery of fatty acids via albumin or remnant-like emulsion particles. J. Biol. Chem. 2004, 279, 19362–19374. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. “New” Hepatic fat activates pparalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005, 1, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, G.; Patterson, B.W.; Yablonskiy, D.A.; Tanoli, T.S.; Averna, M.; Elias, N.; Yue, P.; Ackerman, J. Fatty liver in familial hypobetalipoproteinemia: Triglyceride assembly into vldl particles is affected by the extent of hepatic steatosis. J. Lipid Res. 2003, 44, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E.; et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Sabesin, S.M.; Frase, S.; Ragland, J.B. Accumulation of nascent lipoproteins in rat hepatic golgi during induction of fatty liver by orotic acid. Lab Investig. 1977, 37, 127–135. [Google Scholar] [PubMed]
- Nassir, F.; Adewole, O.L.; Brunt, E.M.; Abumrad, N.A. Cd36 deletion reduces vldl secretion, modulates liver prostaglandin levels and exacerbates hepatic steatosis in ob/ob mice. J. Lipid Res. 2013, 54, 2988–2997. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Hakkinen, A.; Olofsson, S.O.; Yki-Jarvinen, H.; et al. Overproduction of large vldl particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Gan, S.; Wong, A.T.; Ooi, E.M.; Barrett, P.H. Nonalcoholic fatty liver disease as the transducer of hepatic oversecretion of very-low-density lipoprotein-apolipoprotein b-100 in obesity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Shimano, H.; Hamilton, R.L.; Brown, M.S.; Goldstein, J.L. Disruption of ldl receptor gene in transgenic srebp-1a mice unmasks hyperlipidemia resulting from production of lipid-rich vldl. J. Clin. Investig. 1999, 103, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Lee, H.Y.; Lee, G.H.; Kim, H.K.; Kim, N.Y.; Kim, S.H.; Kim, H.R.; Chae, H.J. Ixeris dentata decreases er stress and hepatic lipid accumulation through regulation of apob secretion. Am. J. Chin. Med. 2014, 42, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Badaloo, A.; Reid, M.; Soares, D.; Forrester, T.; Jahoor, F. Relation between liver fat content and the rate of vldl apolipoprotein b-100 synthesis in children with protein-energy malnutrition. Am. J. Clin. Nutr. 2005, 81, 1126–1132. [Google Scholar] [PubMed]
- Dixon, J.L.; Ginsberg, H.N. Regulation of hepatic secretion of apolipoprotein b-containing lipoproteins: Information obtained from cultured liver cells. J. Lipid Res. 1993, 34, 167–179. [Google Scholar] [PubMed]
- Caviglia, J.M.; Gayet, C.; Ota, T.; Hernandez-Ono, A.; Conlon, D.M.; Jiang, H.; Fisher, E.A.; Ginsberg, H.N. Different fatty acids inhibit apob100 secretion by different pathways: Unique roles for er stress, ceramide, and autophagy. J. Lipid Res. 2011, 52, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Gayet, C.; Ginsberg, H.N. Inhibition of apolipoprotein b100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J. Clin. Investig. 2008, 118, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Fabbrini, E.; Mohammed, B.S.; Patterson, B.W.; Klein, S. Increased whole-body adiposity without a concomitant increase in liver fat is not associated with augmented metabolic dysfunction. Obesity (Silver Spring) 2010, 18, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Shida, T.; Yamagishi, K.; Tanaka, K.; So, R.; Tsujimoto, T.; Shoda, J. Moderate to vigorous physical activity volume is an important factor for managing nonalcoholic fatty liver disease: A retrospective study. Hepatology 2015, 61, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabbrini, E.; Magkos, F. Hepatic Steatosis as a Marker of Metabolic Dysfunction. Nutrients 2015, 7, 4995-5019. https://doi.org/10.3390/nu7064995
Fabbrini E, Magkos F. Hepatic Steatosis as a Marker of Metabolic Dysfunction. Nutrients. 2015; 7(6):4995-5019. https://doi.org/10.3390/nu7064995
Chicago/Turabian StyleFabbrini, Elisa, and Faidon Magkos. 2015. "Hepatic Steatosis as a Marker of Metabolic Dysfunction" Nutrients 7, no. 6: 4995-5019. https://doi.org/10.3390/nu7064995
APA StyleFabbrini, E., & Magkos, F. (2015). Hepatic Steatosis as a Marker of Metabolic Dysfunction. Nutrients, 7(6), 4995-5019. https://doi.org/10.3390/nu7064995