Chronic Iron Deficiency as an Emerging Risk Factor for Osteoporosis: A Hypothesis

Abstract
:1. Iron in Human Health
1.1. Prevalence and Risk Factors of Iron Deficiency Anemia
1.2. Functional Consequences of Iron Deficiency
1.3. Evaluation of Iron Status
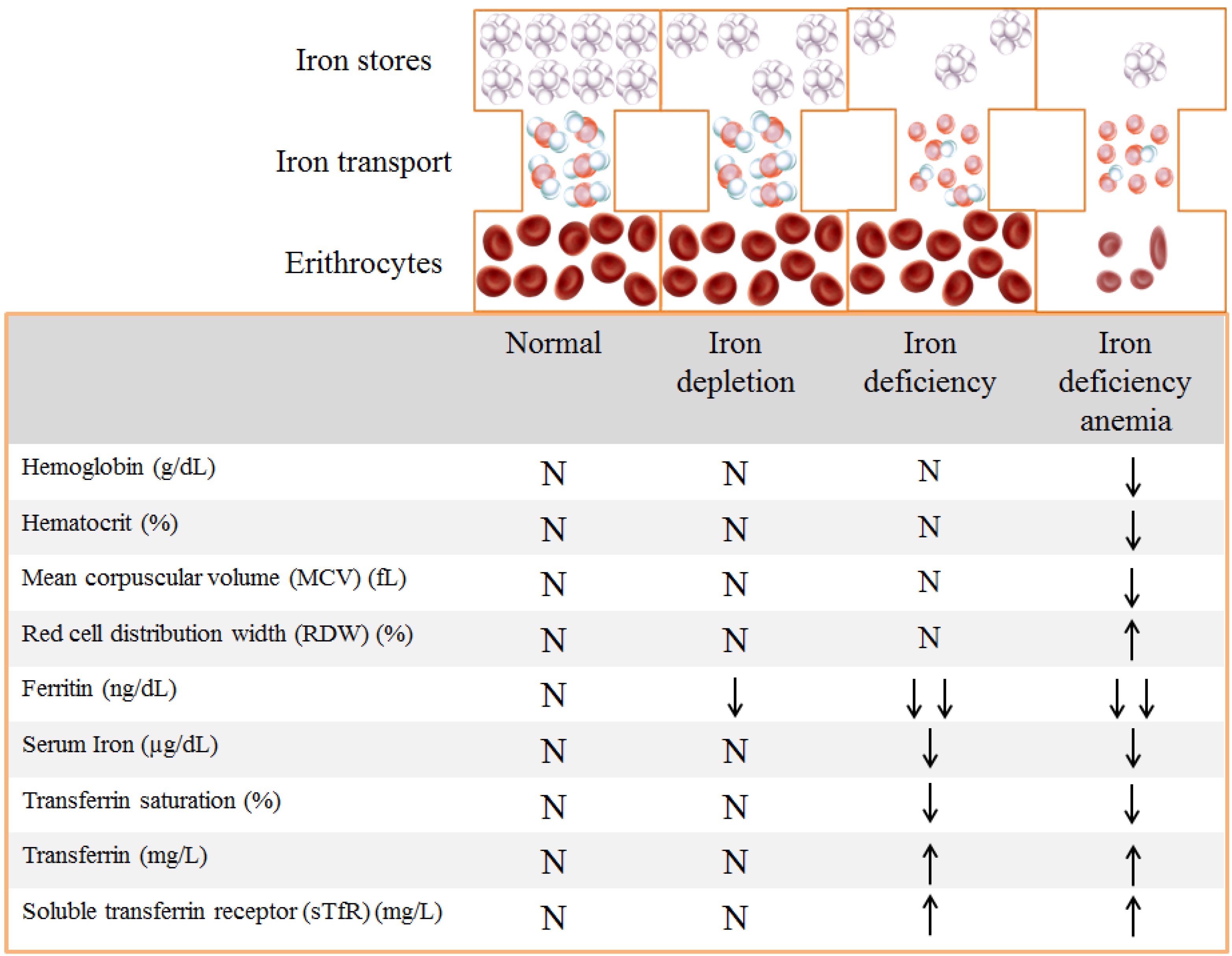

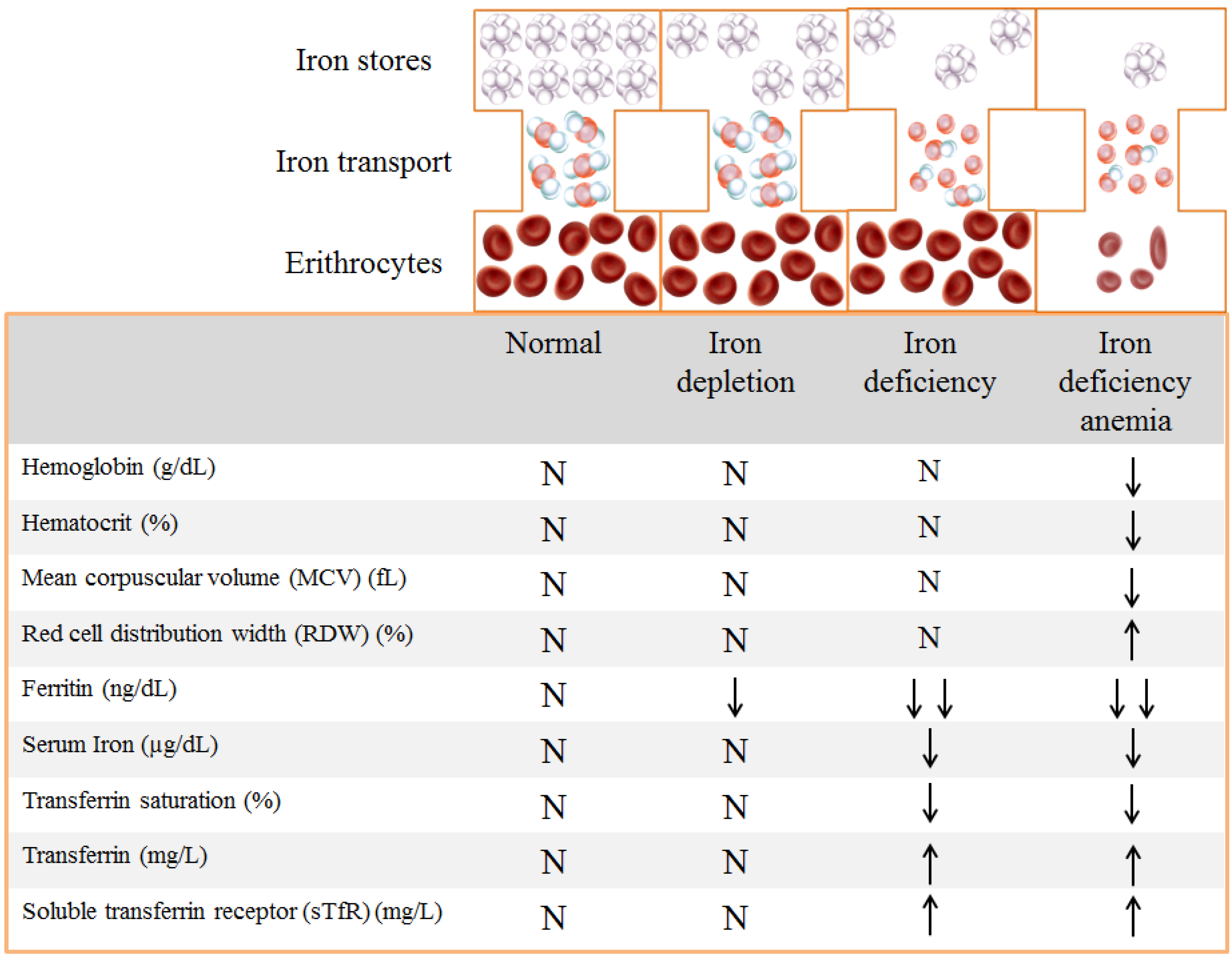{kind=link}
{kind=link}
{kind=link}
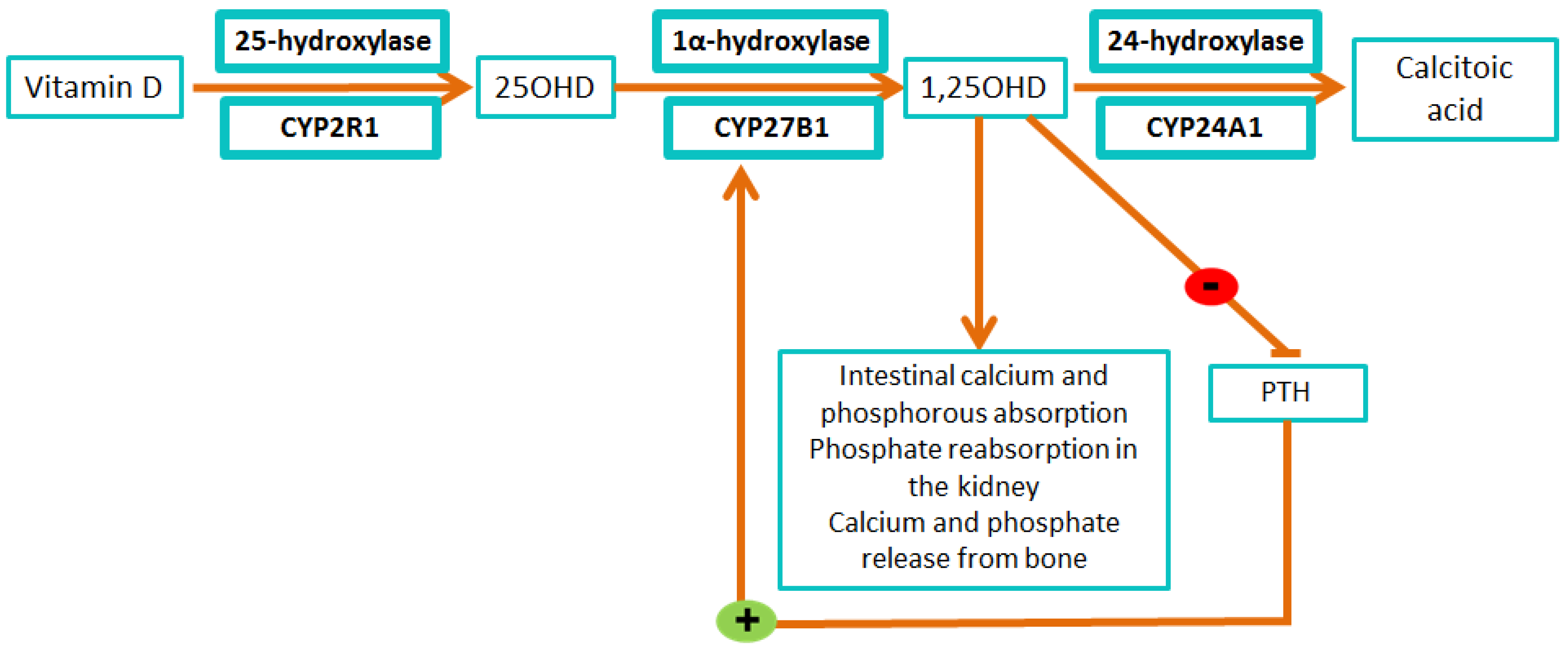
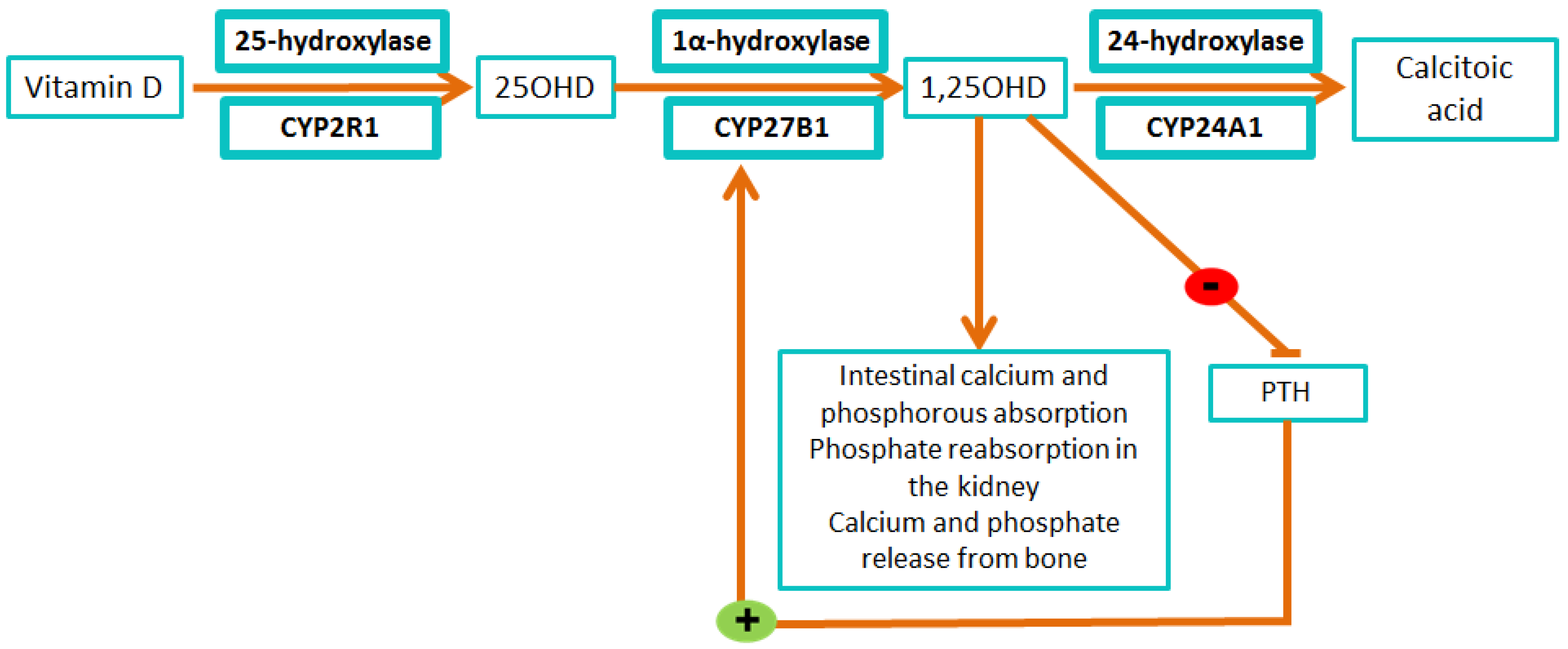{kind=link}
| Population | Non-Anemia | Anemia | ||
|---|---|---|---|---|
| Mild | Moderate | Severe | ||
| Children 6–59 months | >110 | 100–109 | 70–99 | <70 |
| Children 5–11 years | >115 | 110–114 | 80–109 | <80 |
| Children 12–14 years | >120 | 110–119 | 80–109 | <80 |
| Non-pregnant women (>15 years of age) | >120 | 110–119 | 80–109 | <80 |
| Pregnant women | >110 | 100–109 | 70–99 | <70 |
| Men (>15 years of age) | >130 | 110–129 | 80–109 | <80 |
2. Bone Metabolism
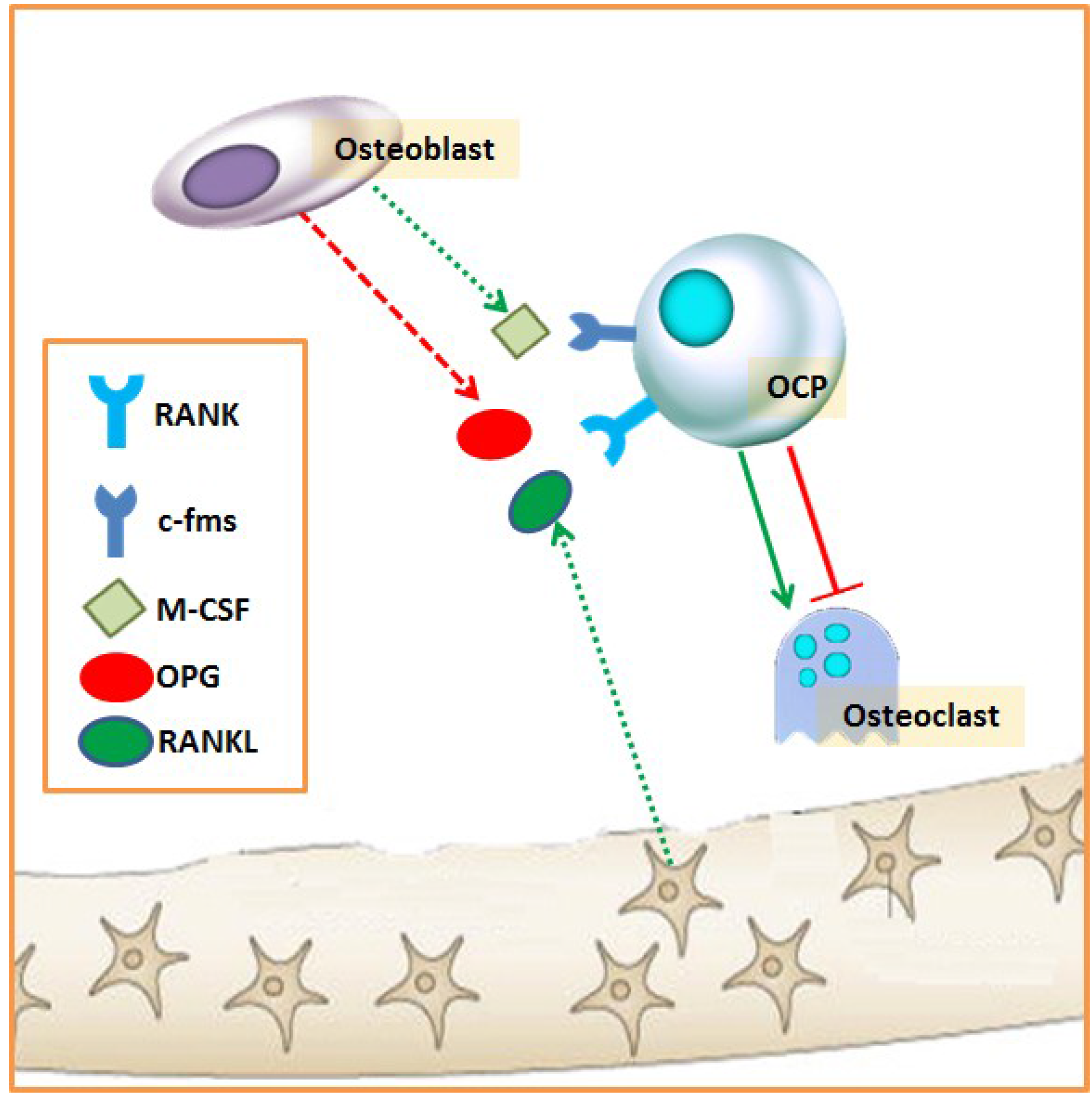
2.1. Bone Remodeling
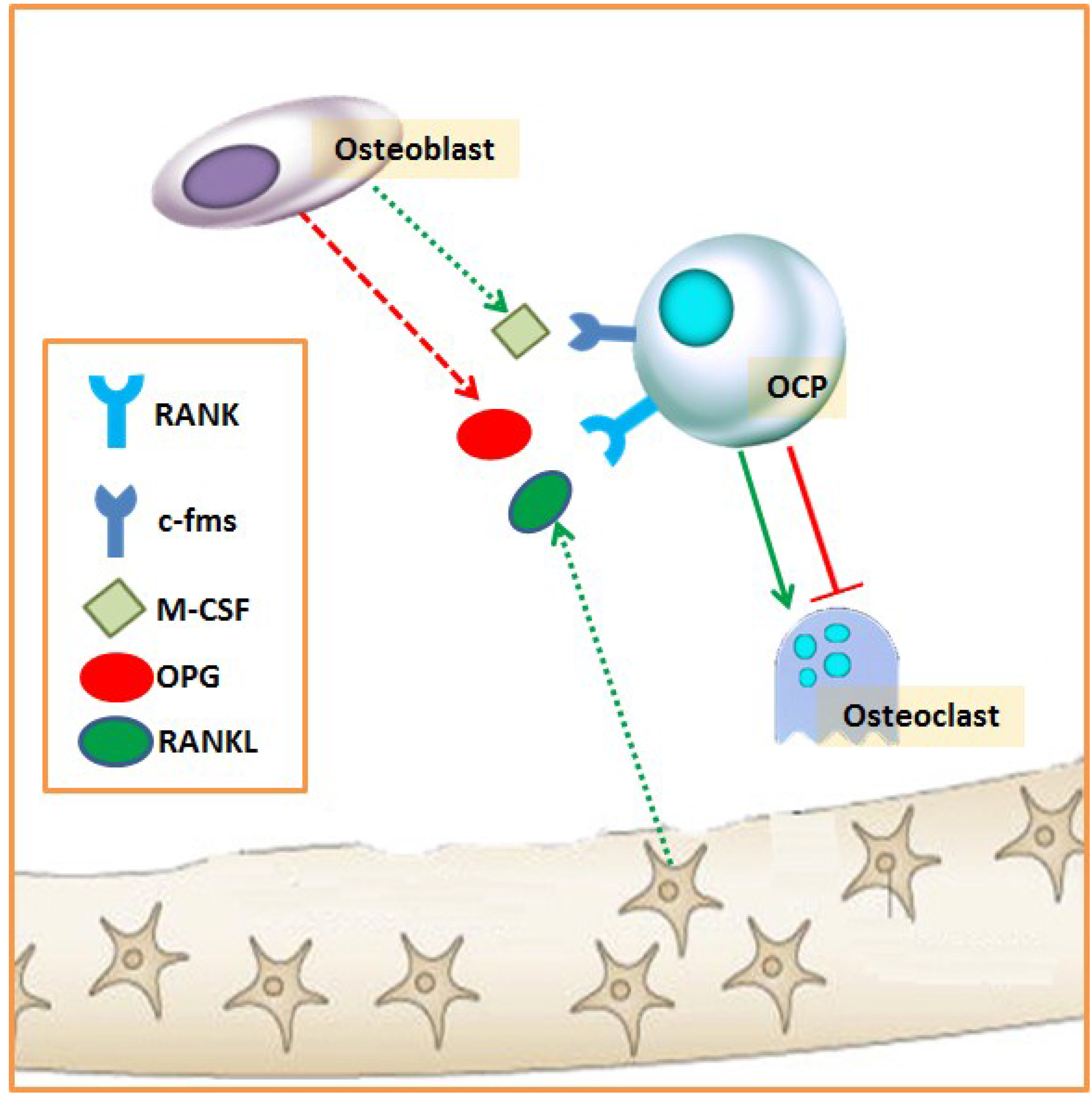
2.2. Biochemical Markers of Bone Remodeling
| Bone Resorption Marker | Bone Formation Marker |
|---|---|
| C-terminal cross-linked telopeptide of type I collagen (CTx) | Bone alkaline phosphatase (ALP) |
| N-telopeptide cross-linked of type 1 collagen (NTx) | Osteocalcin (OC) |
| Pyridinoline (PYD) | N-terminal propeptide of type I procollagen (P1NP) |
| Deoxypyridinoline (DPD) | C-terminal propeptide of type I procollagen (P1CP) |
| Hydroxyproline (HYP) | |
| Tartrat-resistant acid phosphatase type 5b (TRAP 5b) |
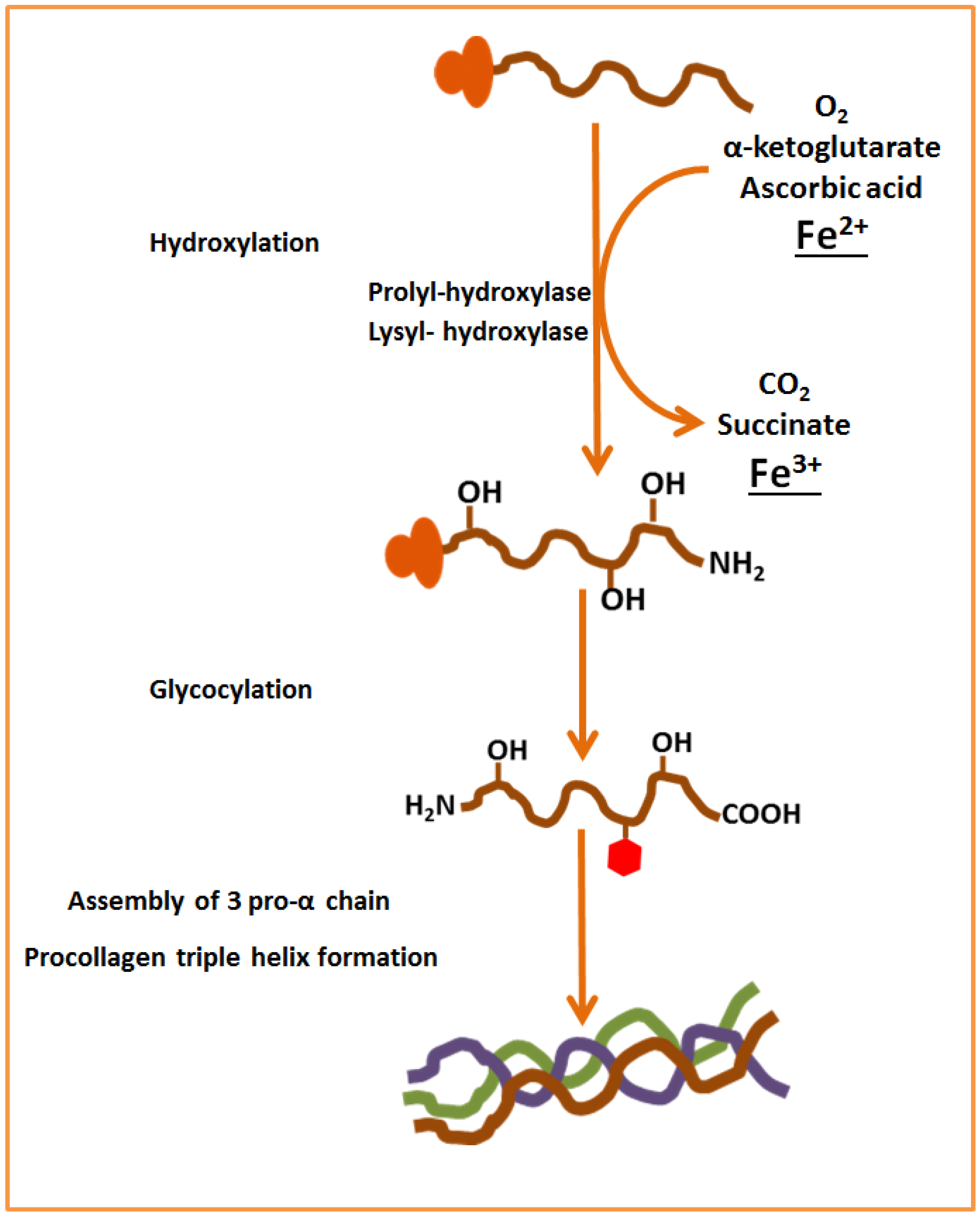
3. Role of Iron in Bone Metabolism
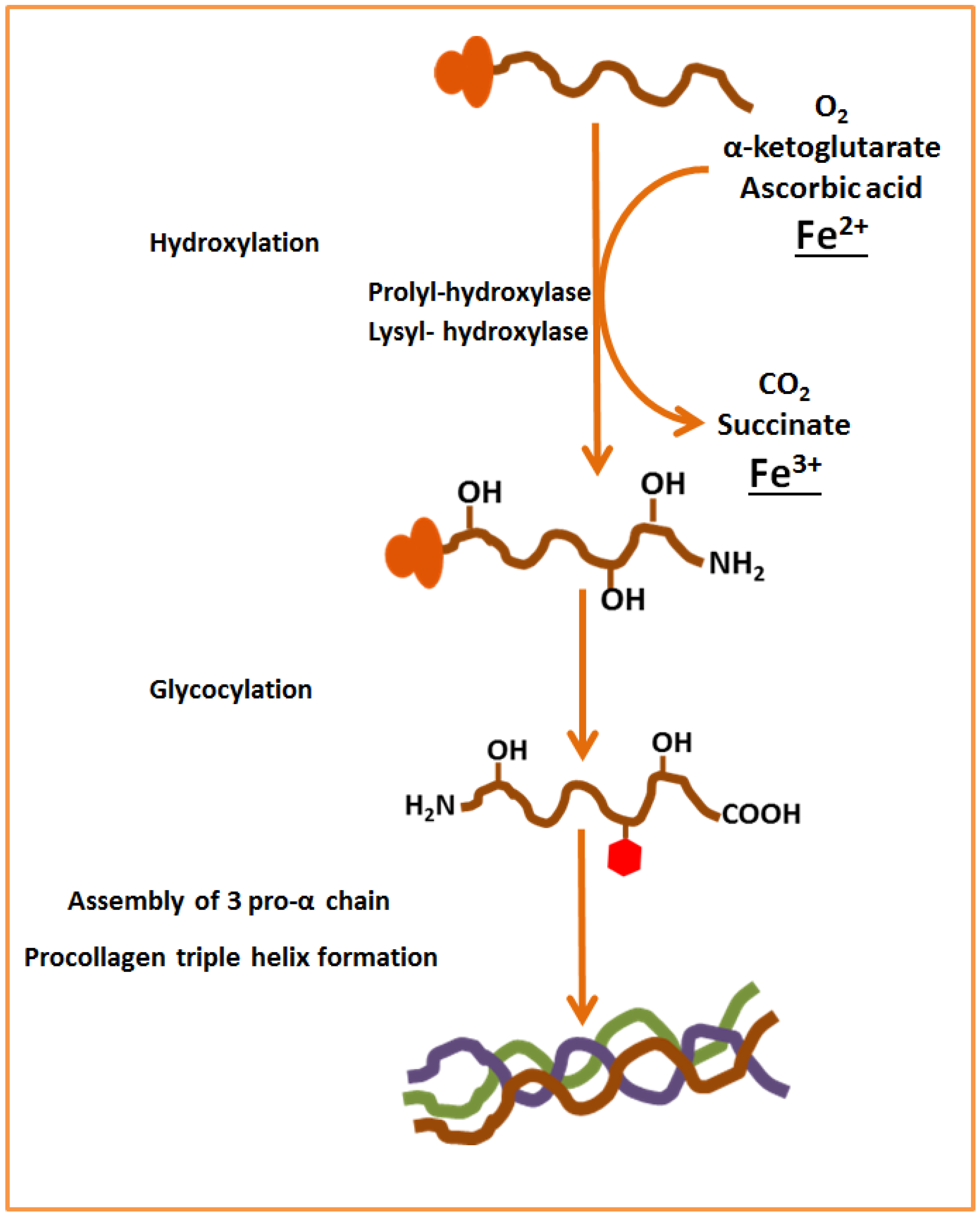
4. Relationship between Body Iron Levels and Bone Metabolism
4.1. Iron Overload and Bone Loss
| Subjects | Main Results | Ref. | |
|---|---|---|---|
| Hemochromatosis | Men, n = 38 (13% hypogonadal) | 79% osteopenic; 34% osteoporotic | [61] |
| Hemochromatosis | Adults, n = 87 | 41% osteopenic; 25% osteoporotic, Lower BMD and decrease in bone formation in patients with higher tissue iron | [75] |
| Thalassemia | Adults, n = 80. BMD | Negative association between ferritin levels and BMD | [76] |
| Thalassemia | All, n = 702; n = 312, BMD | Thalassemia major 17% fracture; thalassemia intermedia 12% | [77] |
| Thalassemia | Adults, n = 41 | 41% osteoporosis | [78] |
| Thalassemia | Children, n = 18 | 61% low bone mass | [79] |
| Sickle cell anemia | Adults, n = 17 | 47% osteopenia; hepatic iron and serum ferritin higher in osteopenic than non-osteopenic patients | [80] |
| Methods | Bone Determinations | Effects of Iron Deficiency on Bone | Ref. |
|---|---|---|---|
| Animal studies | |||
| Weanling female rats: Control diet Calcium restricted diet Iron deficient diet Pair-fed to the iron-deficient group | OC, DPD, serum 1,25OHD, BMD, and BMC (total and femur), femur strength | Decreased: BMD, BMC, and femur bone strength No change: DPD, OC, and 1,25OHD | [83] |
| Weanling male rats: Control diet Iron-deficient diet | OC, CTx, DPD, BMC, and BMD (femur and lumbar vertebra) | Decreased: OC, DPD, BMC, and BMD Increased: CTx | [84] |
| Weanling male rats: Control diet Iron-free diet Pair-fed to the iron-deficient group | PTH, serum 1,25OHD, IGF-I, OC, BMC, and BMD (femur) | Decreased: OC, DPD, Serum IGF-I, BMC, and BMD Increased: CTx, No change: PTH and IGF-I | [85] |
| Weanling male rats Control diet Low-Fe diet | P1NP, TRACP 5b, CTx, PTH, 25OHD, and BMC (sternum and femur) | Decreased: P1NP Increased: PTH, TRACP 5b, and CTX No change: 25OHD, Ca, and P content in sternum | [86] |
| Human studies | |||
| Healthy postmenopausal women (n = 242; 40–66 years) | Dietary Fe and Ca, BMD | Dietary iron positively associated with BMD | [87] |
| Healthy postmenopausal women (n = 228; 40–65 years) | BMD at different sites | Dietary iron positively associated with BMD only in women using hormone replacement therapy | [88] |
| Osteoporotic postmenopausal women (n = 455; 66 ± 10 years) | BMD, ALP, OC, 25OHD | Negative correlation between transferrin and BMD | [89] |
| Mild iron deficient women (n = 41; 18–35 years) | 25OHD, PTH, ALP, NTx | Positive association between 25OHD and transferrin saturation | [90] |
| Non-anemic women (n = 165; 18–35 years) | 25OHD, PTH, P1NP, NTx | Negative correlations: ferritin and NTx; transferrin and P1NP | [91] |
4.2. Iron Deficiency and Bone Loss
| n | Transferrin | Ferritin Log | |
|---|---|---|---|
| NTx log | 220 | 0.047 | −0.237 * |
| P1NP | 193 | −0.254 * | 0.055 |
| Parameter | Iron Deficiency Anemia (n = 42) | Iron Deficiency (n = 94) | Iron Sufficiency (n = 84) | ANOVA p |
|---|---|---|---|---|
| 25OHD (nmol/L) | 56.0 ± 25.0 | 50.7 ± 25.7 | 55.1 ± 21.0 | NS |
| P1NP (ng/mL) | 40.4 ± 15.9 | 54.6 ± 21.7 | 50.8 ± 19.9 | NS |
| PTH pg/mL | 39.8 ± 14.1 | 40.5 ± 16.5 | 38.1 ± 15.4 | NS |
| NTx (nmol BCF/mmol creatinine) | 44.5 ± 26.1 | 64.5 ± 35.2 * | 45.3 ± 38.8 | 0.001 |
5. Mechanisms of the Relationship between Iron Deficiency and Bone Loss
6. Hypothesis and Directions for Further Research
Acknowledgments
Author Contributions
Abbreviations
| ALP | Alkaline phosphatase |
| BMD | Bone mineral density |
| BMC | bone mineral content |
| BMI | body mass index |
| CTx | C-terminal cross-linked telopeptide of type I collagen |
| DPD | deoxypyridinoline |
| NTx | N-telopeptide cross-linked of type 1 collagen |
| 25OHD | 25-hydroxyvitamin D |
| OC | osteocalcin |
| P1NP | N-terminal propeptide of type I procollagen |
| PYR | pyridinoline |
Conflicts of Interest
References
- Denic, S.; Agarwal, M.M. Nutritional iron deficiency: An evolutionary perspective. Nutrition 2007, 23, 603–614. [Google Scholar]
- Lieu, P.T.; Heiskala, M.; Peterson, P.A.; Yang, Y. The roles of iron in health and disease. Mol. Asp. Med. 2001, 22, 1–87. [Google Scholar]
- Lynch, S. Iron metabolism. In The Guidebook Nutritional Anemia; Badham, J., Zimmerman, M.B., Kraemer, K., Eds.; SIGHT AND LIFE Press: Basel, Switzerland, 2007; pp. 17–18. [Google Scholar]
- Worldwide Prevalence of Anaemia 1993–2005. WHO Global Database on Anaemia. WHO Library Cataloguing-in-Publication Data: 2008. Available online: http://whqlibdoc.who.int/publications/2008/9789241596657_eng.pdf (accessed on 19 January 2015).
- Zimmermann, M.B.; Hurrell, R.F. Nutritional iron deficiency. Lancet 2007, 370, 511–520. [Google Scholar]
- McArthur, J.O.; Petocz, P.; Caterson, I.D.; Samman, S. A randomized controlled trial in young women of the effects of consuming pork meat or iron supplements on nutritional status and feeling of well-being. J. Am. Coll. Nutr. 2012, 31, 175–184. [Google Scholar]
- Navas-Carretero, S.; Perez-Granados, A.M.; Sarria, B.; Carbajal, A.; Pedrosa, M.M.; Roe, M.A.; Fairweather-Tait, S.J.; Vaquero, M.P. Oily fish increases iron bioavailability of a phytate rich meal in young iron deficient women. J. Am. Coll. Nutr. 2008, 27, 96–101. [Google Scholar]
- Navas-Carretero, S.; Perez-Granados, A.M.; Sarria, B.; Vaquero, M.P. Iron absorption from meat pate fortified with ferric pyrophosphate in iron-deficient women. Nutrition 2009, 25, 20–24. [Google Scholar]
- Hurrell, R.; Egli, I. Iron bioavailability and dietary reference values. Am. J. Clin. Nutr. 2010, 91, 1461S–1467S. [Google Scholar]
- Vaquero, M.P.; Blanco-Rojo, R.; Toxqui, L. Nutrición y anemia. In Manual Práctico de Nutrición y Salud; Carbajal-Azcona, A., Martínez-Roldán, C., Eds.; Exlibris, S.L.: Madrid, Spain, 2012; pp. 367–376. [Google Scholar]
- Toxqui, L.; Perez-Granados, A.M.; Blanco-Rojo, R.; Wright, I.; Gonzalez-Vizcayno, C.; Vaquero, M.P. Effects of an iron or iron and vitamin d-fortified flavored skim milk on iron metabolism: A randomized controlled double-blind trial in iron-deficient women. J. Am. Coll. Nutr. 2013, 32, 312–320. [Google Scholar]
- Monsen, E.R.; Hallberg, L.; Layrisse, M.; Hegsted, D.M.; Cook, J.D.; Mertz, W.; Finch, C.A. Estimation of available dietary iron. Am. J. Clin. Nutr. 1978, 31, 134–141. [Google Scholar]
- Blanco-Rojo, R.; Toxqui, L.; Lopez-Parra, A.M.; Baeza-Richer, C.; Perez-Granados, A.M.; Arroyo-Pardo, E.; Vaquero, M.P. Influence of diet, menstruation and genetic factors on iron status: A cross-sectional study in Spanish women of childbearing age. Int. J. Mol. Sci. 2014, 15, 4077–4087. [Google Scholar]
- Toxqui, L.; Perez-Granados, A.M.; Blanco-Rojo, R.; Wright, I.; Vaquero, M.P. A simple and feasible questionnaire to estimate menstrual blood loss: Relationship with hematological and gynecological parameters in young women. BMC Women’s Health 2014, 14, 71. [Google Scholar]
- Harvey, L.J.; Armah, C.N.; Dainty, J.R.; Foxall, R.J.; John Lewis, D.; Langford, N.J.; Fairweather-Tait, S.J. Impact of menstrual blood loss and diet on iron deficiency among women in the uk. Br. J. Nutr. 2005, 94, 557–564. [Google Scholar]
- Goddard, A.F.; McIntyre, A.S.; Scott, B.B. Guidelines for the management of iron deficiency anaemia. British society of gastroenterology. Gut 2000, 46 (Suppl. 3–4), IV1–IV5. [Google Scholar]
- Sarria, B.; Navas-Carretero, S.; Lopez-Parra, A.M.; Perez-Granados, A.M.; Arroyo-Pardo, E.; Roe, M.A.; Teucher, B.; Vaquero, M.P.; Fairweather-Tait, S.J. The G277S transferrin mutation does not affect iron absorption in iron deficient women. Eur. J. Nutr. 2007, 46, 57–60. [Google Scholar]
- Benyamin, B.; McRae, A.F.; Zhu, G.; Gordon, S.; Henders, A.K.; Palotie, A.; Peltonen, L.; Martin, N.G.; Montgomery, G.W.; Whitfield, J.B.; et al. Variants in TF and HFE explain approximately 40% of genetic variation in serum-transferrin levels. Am. J. Hum. Genet. 2009, 84, 60–65. [Google Scholar]
- Blanco-Rojo, R.; Baeza-Richer, C.; Lopez-Parra, A.M.; Perez-Granados, A.M.; Brichs, A.; Bertoncini, S.; Buil, A.; Arroyo-Pardo, E.; Soria, J.M.; Vaquero, M.P. Four variants in transferrin and hfe genes as potential markers of iron deficiency anaemia risk: An association study in menstruating women. Nutr. Metab. 2011, 8, 69. [Google Scholar]
- Blanco-Rojo, R.; Bayele, H.K.; Srai, S.K.; Vaquero, M.P. Intronic snp rs3811647 of the human transferrin gene modulates its expression in hepatoma cells. Nutr Hosp. 2012, 27, 2142–2145. [Google Scholar]
- Baeza-Richer, C.; Blanco-Rojo, R.; Lopez-Parra, A.M.; Brichs, A.; Bertoncini, S.; Perez-Granados, A.M.; Buil, A.; Soria, J.M.; Arroyo-Pardo, E.; Vaquero, M.P. Identification of a novel quantitative trait nucleotype related to iron status in a calcium channel gene. Dis. Mark. 2013, 34, 121–129. [Google Scholar]
- Iron Deficiency Anaemia Assessment, Prevention, and Control. A Guide for Programme Managers; United Nations Children’s Fund; United Nations University; World Health Organization. Available online: http://www.who.int/nutrition/publications/micronutrients/anaemia_iron_deficiency/WHO_NHD_01.3/en/ (accessed on 19 January 2015).
- Jauregui-Lobera, I. Iron deficiency and cognitive functions. Neuropsychiatr. Dis. Treat. 2014, 10, 2087–2095. [Google Scholar]
- Jonker, F.A.; Boele van Hensbroek, M. Anaemia, iron deficiency and susceptibility to infections. J. Infect. 2014, 69 (Suppl. 1), S23–S27. [Google Scholar]
- Buratti, P.; Gammella, E.; Rybinska, I.; Cairo, G.; Recalcati, S. Recent advances in iron metabolism: Relevance for health, exercise, and performance. Med. Sci. Sports Exerc. 2014, in press. [Google Scholar]
- Gibson, R.S. Assesment of iron status. In Principles of Nutritional Assessment; Gibson, R.S., Ed.; Oxford University Press: New York, NY, USA, 2005; pp. 446–469. [Google Scholar]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Toxqui, L.; de Piero, A.; Courtois, V.; Bastida, S.; Sanchez-Muniz, F.J.; Vaquero, M.P. Iron deficiency and overload. Implications in oxidative stress and cardiovascular health. Nutr. Hosp. 2010, 25, 350–365. [Google Scholar]
- Ganz, T. Iron homeostasis: Fitting the puzzle pieces together. Cell Metab. 2008, 7, 288–290. [Google Scholar]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar]
- Proff, P.; Romer, P. The molecular mechanism behind bone remodelling: A review. Clin. Oral Investig. 2009, 13, 355–362. [Google Scholar]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar]
- Boyce, B.F.; Rosenberg, E.; de Papp, A.E.; Duong le, T. The osteoclast, bone remodelling and treatment of metabolic bone disease. Eur. J. Clin. Investig. 2012, 42, 1332–1341. [Google Scholar]
- Kular, J.; Tickner, J.; Chim, S.M.; Xu, J. An overview of the regulation of bone remodelling at the cellular level. Clin. Biochem. 2012, 45, 863–873. [Google Scholar]
- Reynaga-Montecinos, B.; Zeni, N. Biochemical markers of bone remodelling. Clinical utility. Acta Bioquím. Clín. Latinoam. 2009, 43, 177–193. (in Spanish). [Google Scholar]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar]
- Schaffler, M.B.; Cheung, W.Y.; Majeska, R.; Kennedy, O. Osteocytes: Master orchestrators of bone. Calcif. Tissue Int. 2014, 94, 5–24. [Google Scholar]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar]
- Martin, T.J.; Sims, N.A. RANKL/OPG; critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, in press. [Google Scholar]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar]
- Hadjidakis, D.J.; Androulakis, II. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar]
- Matsuo, K.; Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209. [Google Scholar]
- Seeman, E. Bone modeling and remodeling. Crit. Rev. Eukaryot. Gene Expr. 2009, 19, 219–233. [Google Scholar]
- Lorget, F.; Kamel, S.; Mentaverri, R.; Wattel, A.; Naassila, M.; Maamer, M.; Brazier, M. High extracellular calcium concentrations directly stimulate osteoclast apoptosis. Biochem. Biophys. Res. Commun. 2000, 268, 899–903. [Google Scholar]
- Baim, S.; Miller, P.D. Assessing the clinical utility of serum ctx in postmenopausal osteoporosis and its use in predicting risk of osteonecrosis of the jaw. J. Bone Min. Res. 2009, 24, 561–574. [Google Scholar]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Ann. Rev. Biochem. 2009, 78, 929–958. [Google Scholar]
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [Google Scholar]
- Tuderman, L.; Myllyla, R.; Kivirikko, K.I. Mechanism of the prolyl hydroxylase reaction. 1. Role of co-substrates. Eur. J. Biochem. 1977, 80, 341–348. [Google Scholar]
- De Jong, L.; Kemp, A. Stoicheiometry and kinetics of the prolyl 4-hydroxylase partial reaction. Biochim. Biophys. Acta 1984, 787, 105–111. [Google Scholar]
- Pikuleva, I.A.; Waterman, M.R. Cytochromes P450: Roles in diseases. J. Biol. Chem. 2013, 288, 17091–17098. [Google Scholar]
- Sakaki, T.; Kagawa, N.; Yamamoto, K.; Inouye, K. Metabolism of vitamin D3 by cytochromes P450. Front. Biosci. 2005, 10, 119–134. [Google Scholar]
- Dong, Q.; Miller, W.L. Vitamin D 25-hydroxylase deficiency. Mol. Genet. Metab. 2004, 83, 197–198. [Google Scholar]
- Holick, M.F. Vitamin D status: Measurement, interpretation, and clinical application. Ann. Epidemiol. 2009, 19, 73–78. [Google Scholar]
- Holick, M.F.; Chen, T.C. Vitamin D deficiency: A worldwide problem with health consequences. Am. J. Clin. Nutr. 2008, 87, 1080S–1086S. [Google Scholar]
- Fu, G.K.; Lin, D.; Zhang, M.Y.; Bikle, D.D.; Shackleton, C.H.; Miller, W.L.; Portale, A.A. Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin d-dependent rickets type 1. Mol. Endocrinol. 1997, 11, 1961–1970. [Google Scholar]
- Schlingmann, K.P.; Kaufmann, M.; Weber, S.; Irwin, A.; Goos, C.; John, U.; Misselwitz, J.; Klaus, G.; Kuwertz-Broking, E.; Fehrenbach, H.; et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N. Engl. J. Med. 2011, 365, 410–421. [Google Scholar]
- Jones, G.; Prosser, D.E.; Kaufmann, M. Cytochrome P450-mediated metabolism of vitamin D. J. Lipid Res. 2014, 55, 13–31. [Google Scholar]
- Vogiatzi, M.G.; Macklin, E.A.; Fung, E.B.; Cheung, A.M.; Vichinsky, E.; Olivieri, N.; Kirby, M.; Kwiatkowski, J.L.; Cunningham, M.; Holm, I.A.; et al. Bone disease in thalassemia: A frequent and still unresolved problem. J. Bone Miner. Res. 2009, 24, 543–557. [Google Scholar]
- Weinberg, E.D. Role of iron in osteoporosis. Pediatr. Endocrinol. Rev. 2008, 6 (Suppl. 1), 81–85. [Google Scholar]
- Guggenbuhl, P.; Deugnier, Y.; Boisdet, J.F.; Rolland, Y.; Perdriger, A.; Pawlotsky, Y.; Chales, G. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos. Int. 2005, 16, 1809–1814. [Google Scholar]
- Lee, K.S.; Jang, J.S.; Lee, D.R.; Kim, Y.H.; Nam, G.E.; Han, B.D.; Do Han, K.; Cho, K.H.; Kim, S.M.; Choi, Y.S.; et al. Serum ferritin levels are positively associated with bone mineral density in elderly Korean men: The 2008–2010 Korea National Health and Nutrition Examination Surveys. J. Bone Miner. Metab. 2014, 32, 683–690. [Google Scholar]
- Kim, B.J.; Ahn, S.H.; Bae, S.J.; Kim, E.H.; Lee, S.H.; Kim, H.K.; Choe, J.W.; Koh, J.M.; Kim, G.S. Iron overload accelerates bone loss in healthy postmenopausal women and middle-aged men: A 3-year retrospective longitudinal study. J. Bone Miner. Res. 2012, 27, 2279–2290. [Google Scholar]
- Guggenbuhl, P.; Brissot, P.; Loreal, O. Miscellaneous non-inflammatory musculoskeletal conditions. Haemochromatosis: The bone and the joint. Best Pract. Res. Clin. Rheumatol. 2011, 25, 649–664. [Google Scholar]
- Buyukbese, M.A.; Cetinus, E.; Cetinkaya, A.; Aras, S. Ferritin levels in postmenopausal women do not seem to play a significant role in osteoporosis. South. Med. J. 2005, 98, 845. [Google Scholar]
- Terpos, E.; Voskaridou, E. Treatment options for thalassemia patients with osteoporosis. Ann. N. Y. Acad. Sci. 2010, 1202, 237–243. [Google Scholar]
- De Sanctis, V.; Soliman, A.T.; Elsedfy, H.; Yassin, M.; Canatan, D.; Kilinc, Y.; Sobti, P.; Skordis, N.; Karimi, M.; Raiola, G.; et al. Osteoporosis in thalassemia major: An update and the I-CET 2013 recommendations for surveillance and treatment. Pediatr. Endocrinol. Rev. 2013, 11, 167–180. [Google Scholar]
- Voskaridou, E.; Terpos, E. New insights into the pathophysiology and management of ossteoporosis in patients with beta thalasaemia. Br. J. Haematol. 2004, 127, 127–139. [Google Scholar]
- Nakchbandi, I.A. Osteoporosis and fractures in liver disease: Relevance, pathogenesis and therapeutic implications. World J. Gastroenterol. 2014, 20, 9427–9438. [Google Scholar]
- Miller, R.G.; Segal, J.B.; Ashar, B.H.; Leung, S.; Ahmed, S.; Siddique, S.; Rice, T.; Lanzkron, S. High prevalence and correlates of low bone mineral density in young adults with sickle cell disease. Am. J. Hematol. 2006, 81, 236–241. [Google Scholar]
- Gupta, R.; Marouf, R.; Adekile, A. Pattern of bone mineral density in sickle cell disease patients with the high-Hb F phenotype. Acta Haematol. 2010, 123, 64–70. [Google Scholar]
- Sarrai, M.; Duroseau, H.; D’Augustine, J.; Moktan, S.; Bellevue, R. Bone mass density in adults with sickle cell disease. Br. J. Haematol. 2007, 136, 666–672. [Google Scholar]
- Reynolds, J. A re-evaluation of the “fish vertebra” sign in sickle cell hemoglobinopathy. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1966, 97, 693–707. [Google Scholar]
- Voskaridou, E.; Stoupa, E.; Antoniadou, L.; Premetis, E.; Konstantopoulos, K.; Papassotiriou, I.; Terpos, E. Osteoporosis and osteosclerosis in sickle cell/beta-thalassemia: The role of the rankl/osteoprotegerin axis. Haematologica 2006, 91, 813–816. [Google Scholar]
- Valenti, L.; Varenna, M.; Fracanzani, A.L.; Rossi, V.; Fargion, S.; Sinigaglia, L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos. Int. 2009, 20, 549–555. [Google Scholar]
- Ebrahimpour, L.; Akhlaghpoor, S.; Azarkayvan, A.; Salehi, M.; Morteza, A.; Alinaghi, R. Correlation between bone mineral densitometry and liver/heart iron overload evaluated by quantitative T2* MRI. Hematology 2012, 17, 297–301. [Google Scholar]
- Vogiatzi, M.G.; Macklin, E.A.; Fung, E.B.; Vichinsky, E.; Olivieri, N.; Kwiatkowski, J.; Cohen, A.; Neufeld, E.; Giardina, P.J. Prevalence of fractures among the thalassemia syndromes in North America. Bone 2006, 38, 571–575. [Google Scholar]
- Chan, Y.L.; Pang, L.M.; Chik, K.W.; Cheng, J.C.; Li, C.K. Patterns of bone diseases in transfusion-dependent homozygous thalassaemia major: Predominance of osteoporosis and desferrioxamine-induced bone dysplasia. Pediatr. Radiol. 2002, 32, 492–497. [Google Scholar]
- Vogiatzi, M.G.; Autio, K.A.; Schneider, R.; Giardina, P.J. Low bone mass in prepubertal children with thalassemia major: Insights into the pathogenesis of low bone mass in thalassemia. J. Pediatr. Endocrinol. Metab. 2004, 17, 1415–1421. [Google Scholar]
- Shah, F.T.; Chatterjee, R.; Owusu-Asante, M.; Porter, J.B. Adults with severe sickle cell anaemia and iron overload have a high incidence of osteopenia and osteoporosis. Blood. 2004, 104, 468A–468A. [Google Scholar]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar]
- Mahachoklertwattana, P.; Sirikulchayanonta, V.; Chuansumrit, A.; Karnsombat, P.; Choubtum, L.; Sriphrapradang, A.; Domrongkitchaiporn, S.; Sirisriro, R.; Rajatanavin, R. Bone histomorphometry in children and adolescents with beta-thalassemia disease: Iron-associated focal osteomalacia. J. Clin. Endocrinol. Metab. 2003, 88, 3966–3972. [Google Scholar]
- Medeiros, D.M.; Stoecker, B.; Plattner, A.; Jennings, D.; Haub, M. Iron deficiency negatively affects vertebrae and femurs of rats independently of energy intake and body weight. J. Nutr. 2004, 134, 3061–3067. [Google Scholar]
- Katsumata, S.; Tsuboi, R.; Uehara, M.; Suzuki, K. Dietary iron deficiency decreases serum osteocalcin concentration and bone mineral density in rats. Biosci. Biotechnol. Biochem. 2006, 70, 2547–2550. [Google Scholar]
- Katsumata, S.; Katsumata-Tsuboi, R.; Uehara, M.; Suzuki, K. Severe iron deficiency decreases both bone formation and bone resorption in rats. J. Nutr. 2009, 139, 238–243. [Google Scholar]
- Diaz-Castro, J.; Lopez-Frias, M.R.; Campos, M.S.; Lopez-Frias, M.; Alferez, M.J.; Nestares, T.; Ojeda, M.L.; Lopez-Aliaga, I. Severe nutritional iron-deficiency anaemia has a negative effect on some bone turnover biomarkers in rats. Eur. J. Nutr. 2012, 51, 241–247. [Google Scholar]
- Harris, M.M.; Houtkooper, L.B.; Stanford, V.A.; Parkhill, C.; Weber, J.L.; Flint-Wagner, H.; Weiss, L.; Going, S.B.; Lohman, T.G. Dietary iron is associated with bone mineral density in healthy postmenopausal women. J. Nutr. 2003, 133, 3598–3602. [Google Scholar]
- Maurer, J.; Harris, M.M.; Stanford, V.A.; Lohman, T.G.; Cussler, E.; Going, S.B.; Houtkooper, L.B. Dietary iron positively influences bone mineral density in postmenopausal women on hormone replacement therapy. J. Nutr. 2005, 135, 863–869. [Google Scholar]
- D’Amelio, P.; Cristofaro, M.A.; Tamone, C.; Morra, E.; Di Bella, S.; Isaia, G.; Grimaldi, A.; Gennero, L.; Gariboldi, A.; Ponzetto, A.; et al. Role of iron metabolism and oxidative damage in postmenopausal bone loss. Bone 2008, 43, 1010–1015. [Google Scholar]
- Blanco-Rojo, R.; Perez-Granados, A.M.; Toxqui, L.; Zazo, P.; de la Piedra, C.; Vaquero, M.P. Relationship between vitamin d deficiency, bone remodelling and iron status in iron-deficient young women consuming an iron-fortified food. Eur. J. Nutr. 2013, 52, 695–703. [Google Scholar]
- Toxqui, L.; Perez-Granados, A.M.; Blanco-Rojo, R.; Wright, I.; de la Piedra, C.; Vaquero, M.P. Low iron status as a factor of increased bone resorption and effects of an iron and vitamin D-fortified skimmed milk on bone remodelling in young spanish women. Eur. J. Nutr. 2014, 53, 441–448. [Google Scholar]
- Zhao, G.Y.; Zhao, L.P.; He, Y.F.; Li, G.F.; Gao, C.; Li, K.; Xu, Y.J. A comparison of the biological activities of human osteoblast hFOB1.19 between iron excess and iron deficiency. Biol. Trace Elem. Res. 2012, 150, 487–495. [Google Scholar]
- Díaz-Castro, J.; Ramirez Lopez-Frias, M.; Campos, M.S.; Lopez-Frias, M.; Alferez, M.J.; Nestares, T.; Ortega, E.; Lopez-Aliaga, I. Goat milk during iron repletion improves bone turnover impaired by severe iron deficiency. J. Dairy Sci. 2011, 94, 2752–2761. [Google Scholar]
- Kim, B.J.; Lee, S.H.; Koh, J.M.; Kim, G.S. The association between higher serum ferritin level and lower bone mineral density is prominent in women ≥45 years of age (KNHANES 2008–2010). Osteoporos. Int. 2013, 24, 2627–2637. [Google Scholar]
- Wright, I.; Blanco-Rojo, R.; Fernandez, M.C.; Toxqui, L.; Moreno, G.; Perez-Granados, A.M.; de la Piedra, C.; Remacha, A.F.; Vaquero, M.P. Bone remodelling is reduced by recovery from iron-deficiency anaemia in premenopausal women. J. Physiol. Biochem. 2013, 69, 889–896. [Google Scholar]
- Blanco-Rojo, R.; Perez-Granados, A.M.; Toxqui, L.; Gonzalez-Vizcayno, C.; Delgado, M.A.; Vaquero, M.P. Efficacy of a microencapsulated iron pyrophosphate-fortified fruit juice: A randomised, double-blind, placebo-controlled study in Spanish iron-deficient women. Br. J. Nutr. 2011, 105, 1652–1659. [Google Scholar]
- Arnett, T.R.; Gibbons, D.C.; Utting, J.C.; Orriss, I.R.; Hoebertz, A.; Rosendaal, M.; Meghji, S. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J. Cell. Physiol. 2003, 196, 2–8. [Google Scholar]
- Shiozawa, Y.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, Z.; Song, J.; Wang, J.; Lee, C.H.; Sud, S.; et al. Erythropoietin couples hematopoiesis with bone formation. PLoS ONE 2010, 5, e10853. [Google Scholar]
- Ilich-Ernst, J.Z.; McKenna, A.A.; Badenhop, N.E.; Clairmont, A.C.; Andon, M.B.; Nahhas, R.W.; Goel, P.; Matkovic, V. Iron status, menarche, and calcium supplementation in adolescent girls. Am. J. Clin. Nutr. 1998, 68, 880–887. [Google Scholar]
- Hiram-Bab, S.; Liron, T.; Deshet-Unger, N.; Mittelman, M.; Gassmann, M.; Rauner, M.; Franke, K.; Wielockx, B.; Neumann, D.; Gabet, Y. Erythropoietin directly stimulates osteoclast precursors and induces bone loss. FASEB J. 2015, in press.. [Google Scholar]
- Lee, M.Y.; Fukunaga, R.; Lee, T.J.; Lottsfeldt, J.L.; Nagata, S. Bone modulation in sustained hematopoietic stimulation in mice. Blood 1991, 77, 2135–2141. [Google Scholar]
- Okito, A.; Nakahama, K.I.; Akiyama, M.; Ono, T.; Morita, I. Involvement of the G-protein-coupled receptor 4 in RANKL expression by osteoblasts in an acidic environment. Biochem. Biophys. Res. Commun. 2015. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toxqui, L.; Vaquero, M.P. Chronic Iron Deficiency as an Emerging Risk Factor for Osteoporosis: A Hypothesis. Nutrients 2015, 7, 2324-2344. https://doi.org/10.3390/nu7042324
Toxqui L, Vaquero MP. Chronic Iron Deficiency as an Emerging Risk Factor for Osteoporosis: A Hypothesis. Nutrients. 2015; 7(4):2324-2344. https://doi.org/10.3390/nu7042324
Chicago/Turabian StyleToxqui, Laura, and M. Pilar Vaquero. 2015. "Chronic Iron Deficiency as an Emerging Risk Factor for Osteoporosis: A Hypothesis" Nutrients 7, no. 4: 2324-2344. https://doi.org/10.3390/nu7042324
APA StyleToxqui, L., & Vaquero, M. P. (2015). Chronic Iron Deficiency as an Emerging Risk Factor for Osteoporosis: A Hypothesis. Nutrients, 7(4), 2324-2344. https://doi.org/10.3390/nu7042324