The Ethanol Extract from Lonicera japonica Thunb. Regresses Nonalcoholic Steatohepatitis in a Methionine- and Choline-Deficient Diet-Fed Animal Model

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Extraction
2.2. Animal and Experimental Protocols
2.3. Biochemical Analysis
2.4. Measurement of Hepatic Lipids
2.5. Lipid Peroxidation Assay
2.6. Histological Analysis
2.7. Western Blotting
2.8. Analysis of mRNA Expression of Hepatic Genes

{kind=link}
{kind=link}
| Target Gene | Primers | Sequence |
|---|---|---|
| CYP2E1 | FP | ATGTCATCCCCAAGGGTACA |
| RP | CGGGGAATGACACAGAGTTT | |
| TNFα | FP | CCAGGAGAAAGTCAGCCTCCT |
| RP | TCATACCAGGGCTTGAGCTCA | |
| TGF-β | FP | CCCAGCATCTGCAAAGCTC |
| RP | GTCAATGTACAGCTGCCGCA | |
| α-SMA | FP | TGCTGTCCCTCTATGCCTCT |
| RP | GAAGGAATAGCCACGTCAG | |
| Collagen I | FP | ACAGCCGCTTCACCTACAGC |
| RP | TCAATCACTGTCTTGCCCCA | |
| MMP2 | FP | AACTTTGAGAAGGATGGCAAGT |
| RP | TGCCACCCATGGTAAACAA | |
| MMP9 | FP | CCCCAAAACGGACAAAGAG |
| RP | CTTCAGCACAAAACGGTTGC | |
| DGAT2 | FP | CATGAAGACCCTCATCGCCG |
| RP | GTGACAGAGAAGATGTCTTGG | |
| PPARα | FP | CGTCCTGGCCTTCTAAACGTAG |
| RP | CCTGTAGATCTCCTGCAGTAGCG | |
| β-actin | FP | TGTGATGGTGGGAATGGGTCAG |
| RP | TTTGATGTCACGCACGAT TTCC |
2.9. Statistical Analysis
3. Results
3.1. Effects on Body Weights and Liver Weights in Mice
3.2. Effects of Treatments on Plasma and Hepatic Lipids in Mice
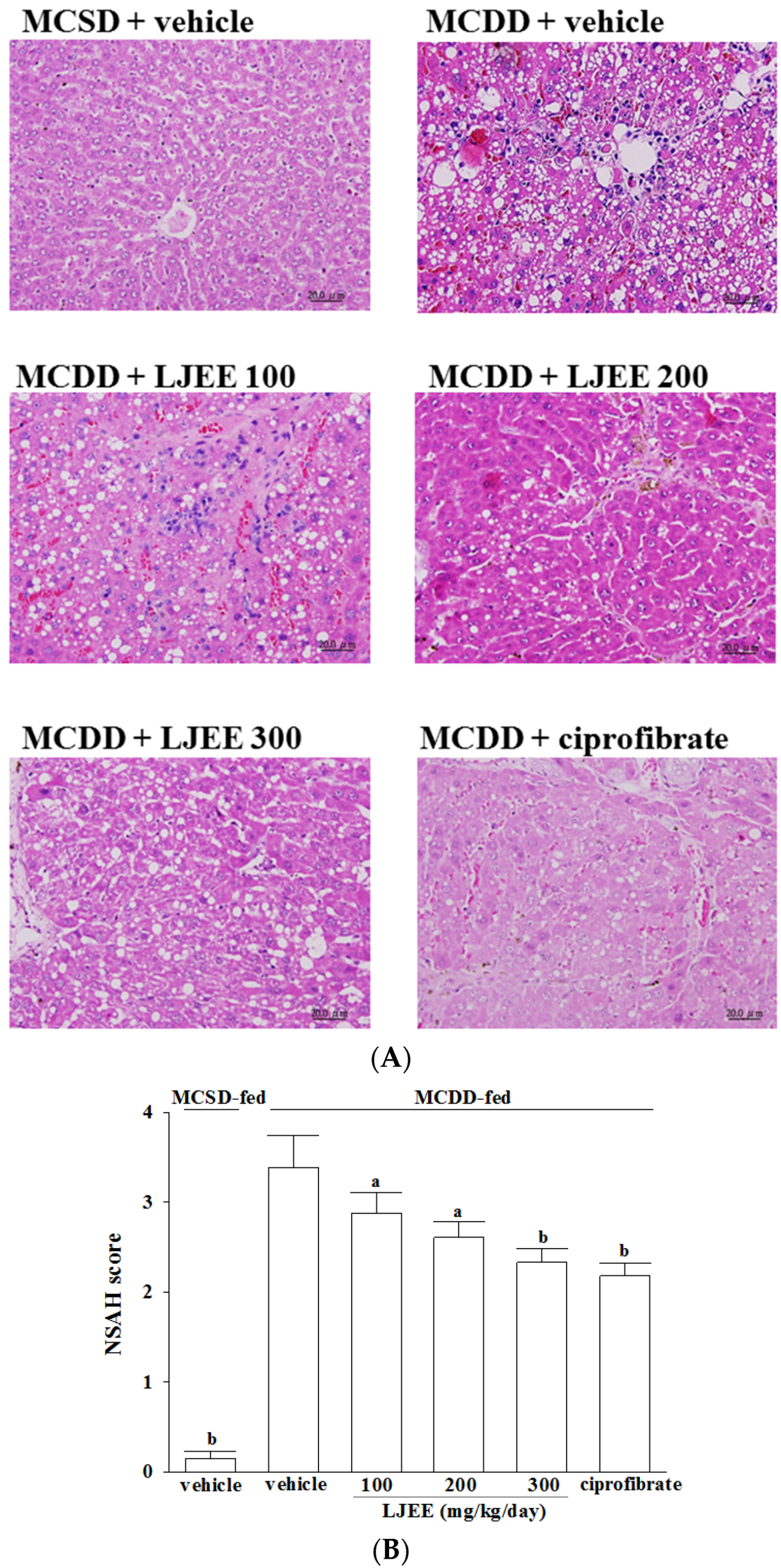
3.3. Effects of Treatments on Liver Injury in Mice
| MCSD | MCDD | |||||
|---|---|---|---|---|---|---|
| Vehicle | Vehicle | LJEE (mg/kg/day) | Ciprofibrate | |||
| 100 | 200 | 300 | (10 mg/kg/day) | |||
| Initial body weight (BW) (g) | 20.71 ± 1.51 | 20.67 ± 1.25 | 20.75± 1.46 | 20.61 ± 1.33 | 20.72 ± 1.57 | 20.78 ± 1.75 |
| Final BW (g) | 28.04 ± 1.72 b | 14.64 ± 1.48 | 14.94 ± 1.59 | 15.23 ± 1.50 | 14.98 ± 1.64 | 15.16 ± 1.76 |
| Liver absolute weight (g) | 1.44 ± 0.11 b | 0.51 ± 0.07 | 0.53 ± 0.09 | 0.57 ± 0.08 | 0.57 ± 0.06 | 0.56 ± 0.05 |
| Liver relative weight (%) | 5.13 ± 0.23 a | 3.48 ± 0.28 | 3.54 ± 0.31 | 3.74 ± 0.28 | 3.80 ± 0.29 | 3.71 ± 0.32 |
| Plasma TC (mg/dL) | 150.82 ± 4.07 b | 24.88 ± 3.91 | 23.87 ± 4.13 | 23.77 ± 3.26 | 25.13 ± 3.83 | 25.96 ± 2.96 |
| Plasma TG (mg/dL) | 95.84 ± 4.26 b | 50.78 ± 3.83 | 55.79 ± 3.46 | 52.29 ± 3.57 | 50.91 ± 4.11 | 54.32 ± 3.62 |
| Plasma ALT (U/L) | 40.26 ± 7.13 b | 303.08 ± 16.23 | 287.85 ± 17.26 a | 253.37 ± 15.78 a | 184.05 ± 14.56 b | 126.94 ± 17.42 b |
| Plasma AST (U/L) | 103.69 ± 9.83 b | 441.62 ± 18.26 | 393.24 ± 17.34 a | 282.46 ± 19.33 a | 224.63 ± 18.14 b | 188.74 ± 15.36 b |
| Hepatic TC (µmol/g liver) | 11.61 ± 0.83 b | 21.61 ± 1.24 | 18.44 ± 1.13 a | 16.67 ± 1.21a | 14.92 ± 1.09 b | 14.27 ± 1.16 b |
| Hepatic TG (µmol/g liver) | 8.72 ± 0.73 b | 17.68 ± 1.14 | 14.74 ± 1.26 | 12.52 ± 1.38 a | 11.18 ± 1.12 a | 10.51 ± 1.07 b |
| Hepatic MDA (µmol/g liver) | 10.23 ± 2.31 b | 393.14 ± 15.26 | 334.16 ± 16.17 a | 314.52 ± 13.27 a | 294.85 ± 17.13 b | 282.96 ± 16.21b |
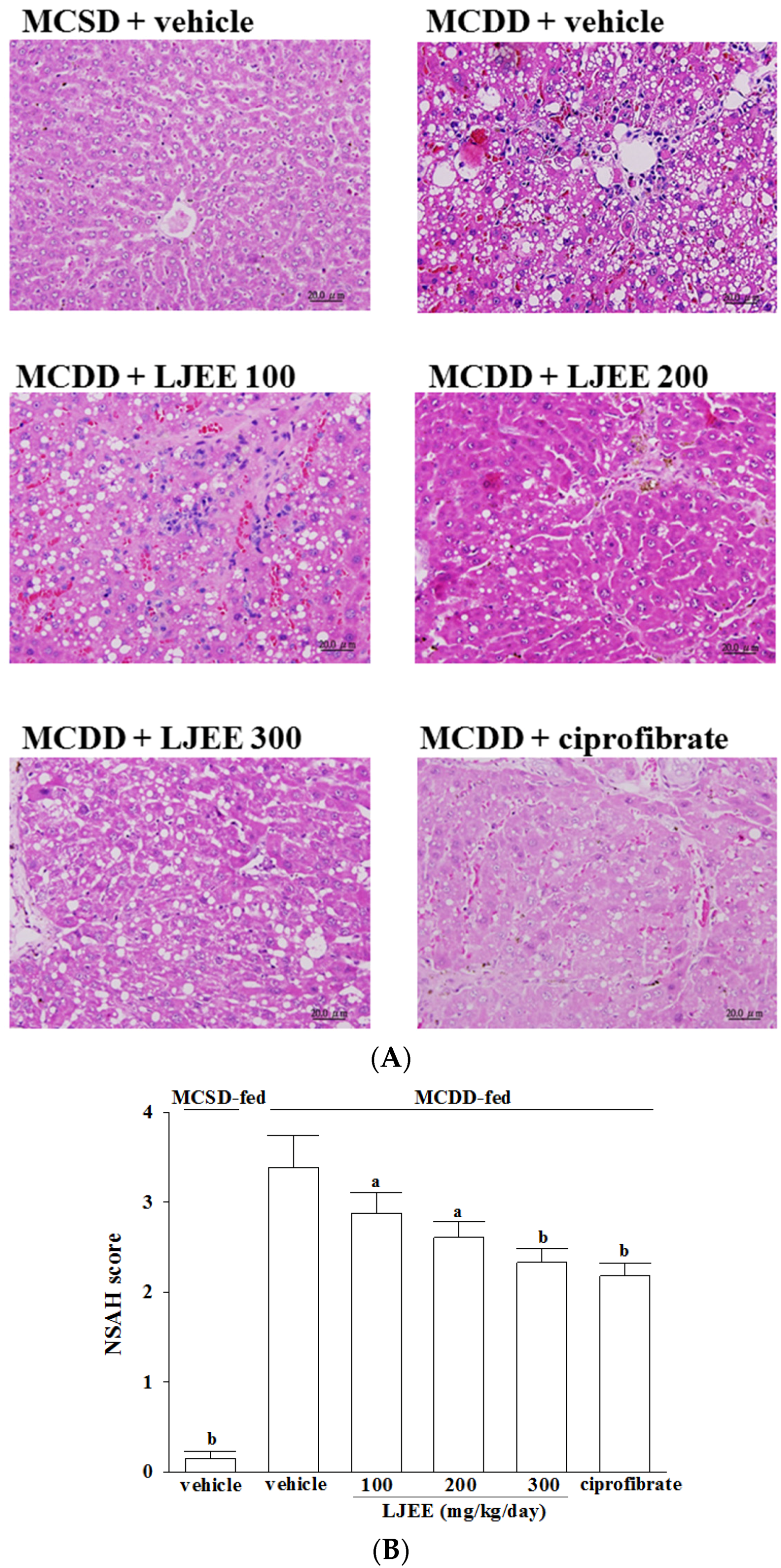
3.4. Effects of Treatments on Oxidative Stress in Livers of Mice
3.5. Effects of Treatments on Hepatic mRNA Expression of NASH-Related Specific Genes in Mice
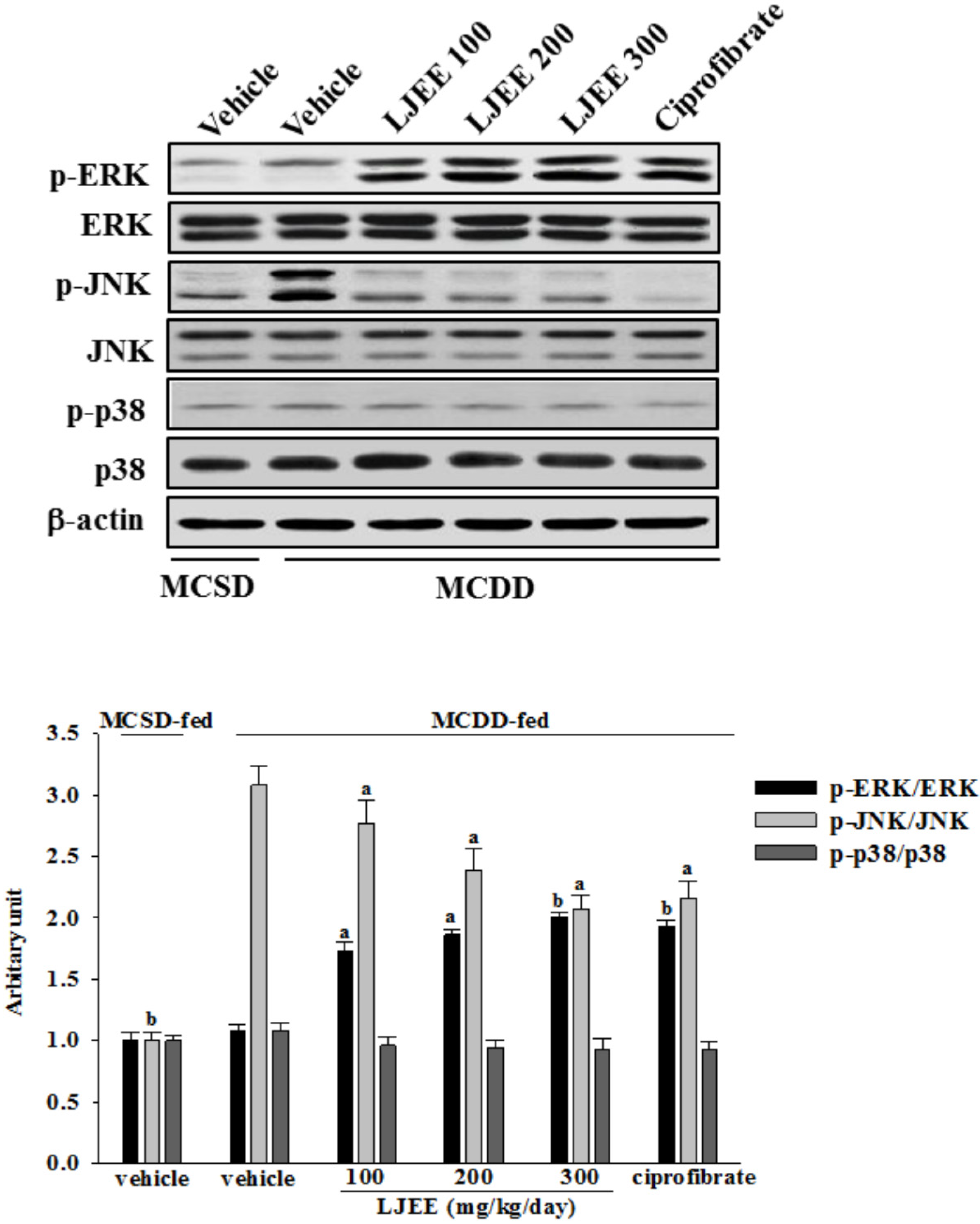
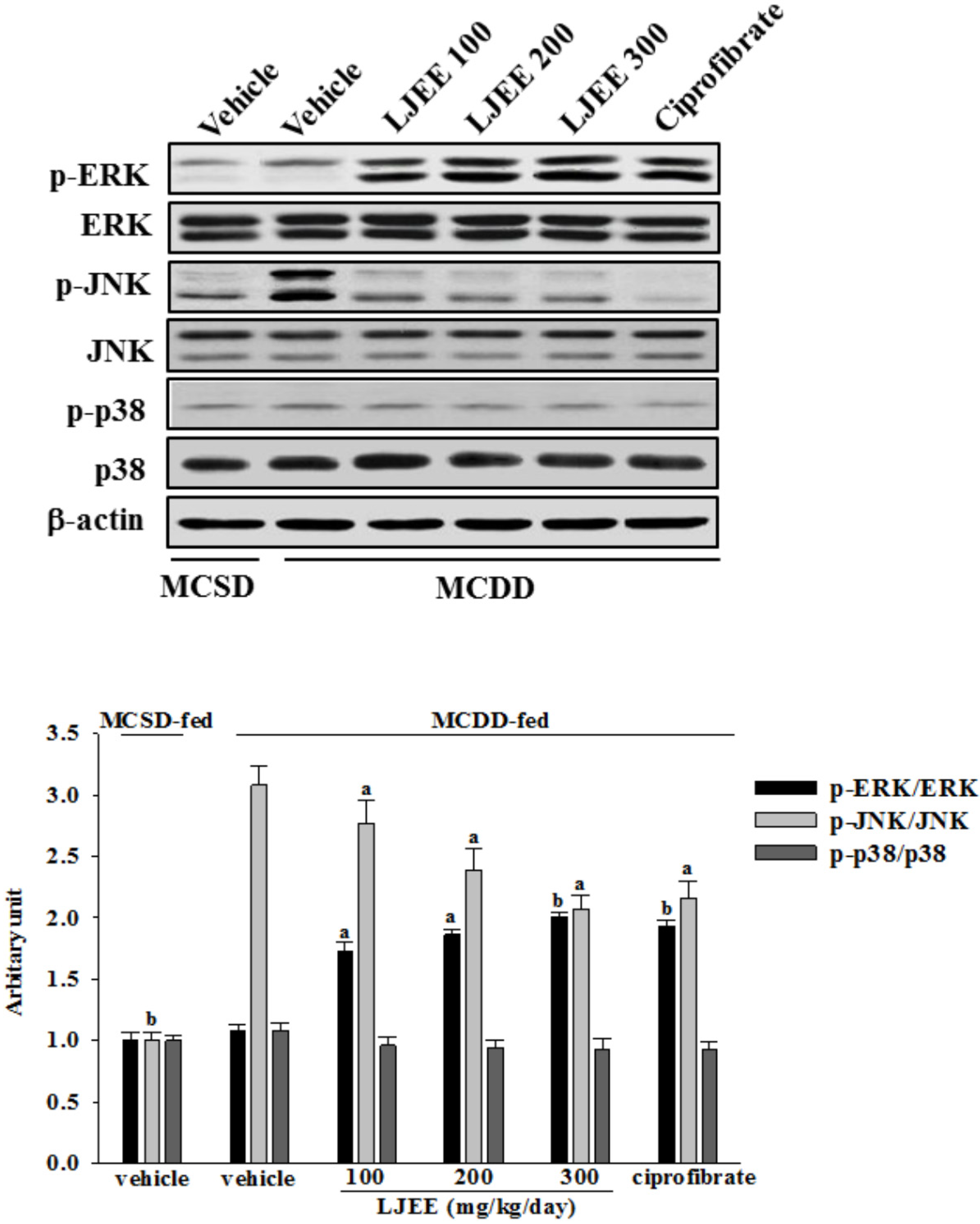
3.6. Effects of Treatments on MAPK Signaling Pathways in Mice
| Relative Expression | MCSD | MCDD | ||||
|---|---|---|---|---|---|---|
| Vehicle | Vehicle | LJEE (mg/kg/day) | Ciprofibrate | |||
| 100 | 200 | 300 | 10 mg/kg/day | |||
| CYP2E1 mRNA | 1.00 ± 0.02 b | 2.71 ± 0.23 | 2.43 ± 0.26 | 2.18 ± 0.13 a | 1.82 ± 0.16 b | 1.94 ± 0.11 b |
| TNF-α mRNA | 1.00 ± 0.05 b | 3.15 ± 0.23 | 2.81 ± 0.28 | 2.63 ± 0.19 a | 2.31 ± 0.17 a | 2.26 ± 0.15 b |
| TGF-β mRNA | 1.00 ± 0.04 b | 2.34 ± 0.13 | 1.93 ± 0.09 | 1.86 ± 0.07 a | 1.73 ± 0.09a | 1.56 ± 0.08 a |
| α-SMA mRNA | 1.00 ± 0.06 b | 3.08 ± 0.19 | 2.77 ± 0.08 | 2.46 ± 0.11 a | 2.13 ± 0.09 b | 2.21 ± 0.13 b |
| Collagen I | 1.00 ± 0.05 b | 3.26 ± 0.17 | 3.04 ± 0.12 | 2.53 ± 0.08 a | 2.02 ± 0.09 b | 1.93 ± 0.12 b |
| MMP2 | 1.00 ± 0.06 b | 2.71 ± 0.16 | 2.43 ± 0.15 | 2.16 ± 0.09 a | 2.01 ± 0.08 b | 1.91 ± 0.14 b |
| MMP9 | 1.00 ± 0.05 b | 3.22 ± 0.26 | 2.89 ± 0.22 | 2.31 ± 0.21 a | 2.12 ± 0.18 b | 1.96 ± 0.11 b |
| DGAT-2 | 1.00 ± 0.05 b | 0.87 ± 0.06 | 0.74 ± 0.05a | 0.63 ± 0.04 a | 0.42 ± 0.04 a | 0.51 ± 0.03 a |
| PPARα | 1.00 ± 0.04 b | 0.47 ± 0.04 | 0.52 ± 0.06 | 0.62 ± 0.05 a | 0.74 ± 0.05 a | 0.78 ± 0.03 a |
4. Discussion
5. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sentürk, H. From fatty liver to fibrosis: A tale of “second hit”. World J. Gastroenterol. 2013, 19, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Amarapurkar, D.N.; Dharod, M.; Gautam, S.; Patel, N. Risk of development of hepatocellular carcinoma in patients with NASH-related cirrhosis. Trop. Gastroenterol. 2013, 34, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Mahady, S.E.; George, J. Management of nonalcoholic steatohepatitis: An evidence-based approach. Clin. Liver Dis. 2012, 16, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Pan, H.; Li, M.; Miao, X.; Ding, H. Lonicera japonica Thunb.: Ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2011, 138, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.Y.; Mei, S.X.; Jiang, B.; Zhou, H.; Sun, H.D. Constituents from Lonicera japonica. Fitoterapia 2000, 71, 713–715. [Google Scholar] [CrossRef]
- Committee for the Pharmacopoeia of PR China. Pharmacopoeia of PR China, Part I; Chinese Medical Science and Technology Press: Beijing, China, 2010. [Google Scholar]
- Committee for the Pharmacopoeia of PR China. Pharmacopoeia of PR China, Part I; Chemical Industrial Press, Guangdong Science and Technology Publishing House: Guangdong, China, 1995. [Google Scholar]
- Jiao, S.G. Research and comprehensive utilization of honeysuckle. Qilu Pharm. Aff. 2009, 28, 487–489. [Google Scholar]
- Sun, C.; Teng, Y.; Li, G.; Yoshioka, S.; Yokota, J.; Miyamura, M.; Fang, H.; Zhang, Y. Metabonomics study of the protective effects of Lonicera japonica extract on acute liver injury in dimethylnitrosamine treated rats. J. Pharm. Biomed. Anal. 2010, 53, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Fang, J.Y.; Hong, T.L.; Wang, T.C.; Zhou, Y.E.; Lin, T.C. Potential antioxidant properties and hepatoprotective effects of an aqueous extract formula derived from three Chinese medicinal herbs against CCl(4)-induced liver injury in rats. Int. Immunopharmacol. 2013, 15, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, D.P.; Zou, X.; Andrew, R.; Morton, N.M.; Livingstone, D.E.; Aucott, R.L.; Nyirenda, M.J.; Iredale, J.P.; Walker, B.R. Metabolic pathways promoting intrahepatic fatty acid accumulation in methionine and choline deficiency: Implications for the pathogenesis of steatohepatitis. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E402–E409. [Google Scholar] [CrossRef] [PubMed]
- Bighetti, E.J.; Patrício, P.R.; Casquero, A.C.; Berti, J.A.; Oliveira, H.C. Ciprofibrate increases cholesteryl ester transfer protein gene expression and the indirect reverse cholesterol transport to the liver. Lipids Health Dis. 2009, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.F.; Liou, S.S.; Chang, C.J.; Liu, I.M. The ethanol extract of Lonicera japonica (Japanese honeysuckle) attenuates diabetic nephropathy by inhibiting p-38 MAPK activity in streptozotocin-induced diabetic rats. Planta Med. 2014, 80, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane-Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–506. [Google Scholar] [PubMed]
- Draper, H.H.; Hadley, M. Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol. 1990, 186, 421–431. [Google Scholar] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, H.; Shimizu, K.; Morikawa, S.; Ogawa, K.; Ezaki, T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar] [PubMed]
- Giboney, P.T. Mildly elevated liver transaminase levels in the asymptomatic patient. Am. Fam. Physician. 2005, 71, 1105–1110. [Google Scholar] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Myers, H.M.; Watkins, S.M.; Brown, B.E.; Feingold, K.R.; Elias, P.M.; Farese, R.V., Jr. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J. Biol. Chem. 2004, 279, 11767–11776. [Google Scholar] [CrossRef] [PubMed]
- Fast, D.G.; Vance, D.E. Nascent VLDL phospholipid composition is altered when phosphatidylcholine biosynthesis is inhibited: Evidence for a novel mechanism that regulates VLDL secretion. Biochim. Biophys. Acta 1995, 1258, 159–168. [Google Scholar] [CrossRef]
- Marcolin, E.; Forgiarini, L.F.; Tieppo, J.; Dias, A.S.; Freitas, L.A.; Marroni, N.P. Methionine- and choline-deficient diet induces hepatic changes characteristic of non-alcoholic steatohepatitis. Arq. Gastroenterol. 2011, 48, 72–79. [Google Scholar] [PubMed]
- Miyashita, T.; Toyoda, Y.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Hepatoprotective effect of tamoxifen on steatosis and non-alcoholic steatohepatitis in mouse models. J. Toxicol. Sci. 2012, 37, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Takahashi, S.; Fang, Z.Z.; Matsubara, T.; Krausz, K.W.; Qu, A.; Gonzalez, F.J. Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta 2014, 1841, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 2003, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Yan, H.; Fei, J.; Jiang, M.; Zhu, H.; Lu, D.; Gao, X. Berberine reduces methylation of the MTTP promoter and alleviates fatty liver induced by a highfat diet in rats. J. Lipid Res. 2010, 51, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xue, C.; Hu, X.; Xu, J.; Li, Z.; Wang, J.; Yanagita, T.; Xue, Y.; Wang, Y. Dietary sea cucumber cerebroside alleviates orotic acid-induced excess hepatic adipopexis in rats. Lipids Health Dis. 2012, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the potent PPARα agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.J. PPARα expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Caro, A.A.; Cederbaum, A.I. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Leclercq, I.; Farrell, G.C. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1135–G1139. [Google Scholar]
- Ucar, F.; Sezer, S.; Erdogan, S.; Akyol, S.; Armutcu, F.; Akyol, O. The relationship between oxidative stress and nonalcoholic fatty liver disease: Its effects on the development of nonalcoholic steatohepatitis. Redox. Rep. 2013, 18, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Knittel, T.; Müller, L.; Saile, B.; Ramadori, G. Effect of tumour necrosis factor-alpha on proliferation, activation and protein synthesis of rat hepatic stellate cells. J. Hepatol. 1997, 27, 1067–1080. [Google Scholar] [CrossRef]
- Bissell, D.M.; Roulot, D.; George, J. Transforming growth factor beta and the liver. Hepatology 2001, 34, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Gastaldelli, A.; Svegliati Baroni, G.; Tell, G.; Tiribelli, C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 2008, 14, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.P.; Tuan, T.L.; Hughes, M.; Wu, H.; Garner, W.L. Transforming growth factor-beta- and tumor necrosis factor-alpha-mediated induction and proteolytic activation of MMP-9 in human skin. J. Biol. Chem. 2001, 276, 22341–22350. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [PubMed]
- Wang, X.; Martindale, J.L.; Liu, Y.; Holbrook, N.J. The cellular response to oxidative stress: Influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J. 1998, 333, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, S.; Yazdanparast, R. Inhibition of JNK along with activation of ERK1/2 MAPK pathways improve steatohepatitis among the rats. Clin. Nutr. 2010, 29, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Liu, H.; Wang, Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology 2003, 37, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzeng, T.-F.; Tzeng, Y.-C.; Cheng, Y.-J.; Liou, S.-S.; Liu, I.-M. The Ethanol Extract from Lonicera japonica Thunb. Regresses Nonalcoholic Steatohepatitis in a Methionine- and Choline-Deficient Diet-Fed Animal Model. Nutrients 2015, 7, 8670-8684. https://doi.org/10.3390/nu7105423
Tzeng T-F, Tzeng Y-C, Cheng Y-J, Liou S-S, Liu I-M. The Ethanol Extract from Lonicera japonica Thunb. Regresses Nonalcoholic Steatohepatitis in a Methionine- and Choline-Deficient Diet-Fed Animal Model. Nutrients. 2015; 7(10):8670-8684. https://doi.org/10.3390/nu7105423
Chicago/Turabian StyleTzeng, Thing-Fong, Yu-Cheng Tzeng, Yu-Jou Cheng, Shorong-Shii Liou, and I-Min Liu. 2015. "The Ethanol Extract from Lonicera japonica Thunb. Regresses Nonalcoholic Steatohepatitis in a Methionine- and Choline-Deficient Diet-Fed Animal Model" Nutrients 7, no. 10: 8670-8684. https://doi.org/10.3390/nu7105423
APA StyleTzeng, T.-F., Tzeng, Y.-C., Cheng, Y.-J., Liou, S.-S., & Liu, I.-M. (2015). The Ethanol Extract from Lonicera japonica Thunb. Regresses Nonalcoholic Steatohepatitis in a Methionine- and Choline-Deficient Diet-Fed Animal Model. Nutrients, 7(10), 8670-8684. https://doi.org/10.3390/nu7105423