Dietary Fructose Reduction Improves Markers of Cardiovascular Disease Risk in Hispanic-American Adolescents with NAFLD

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Study Design
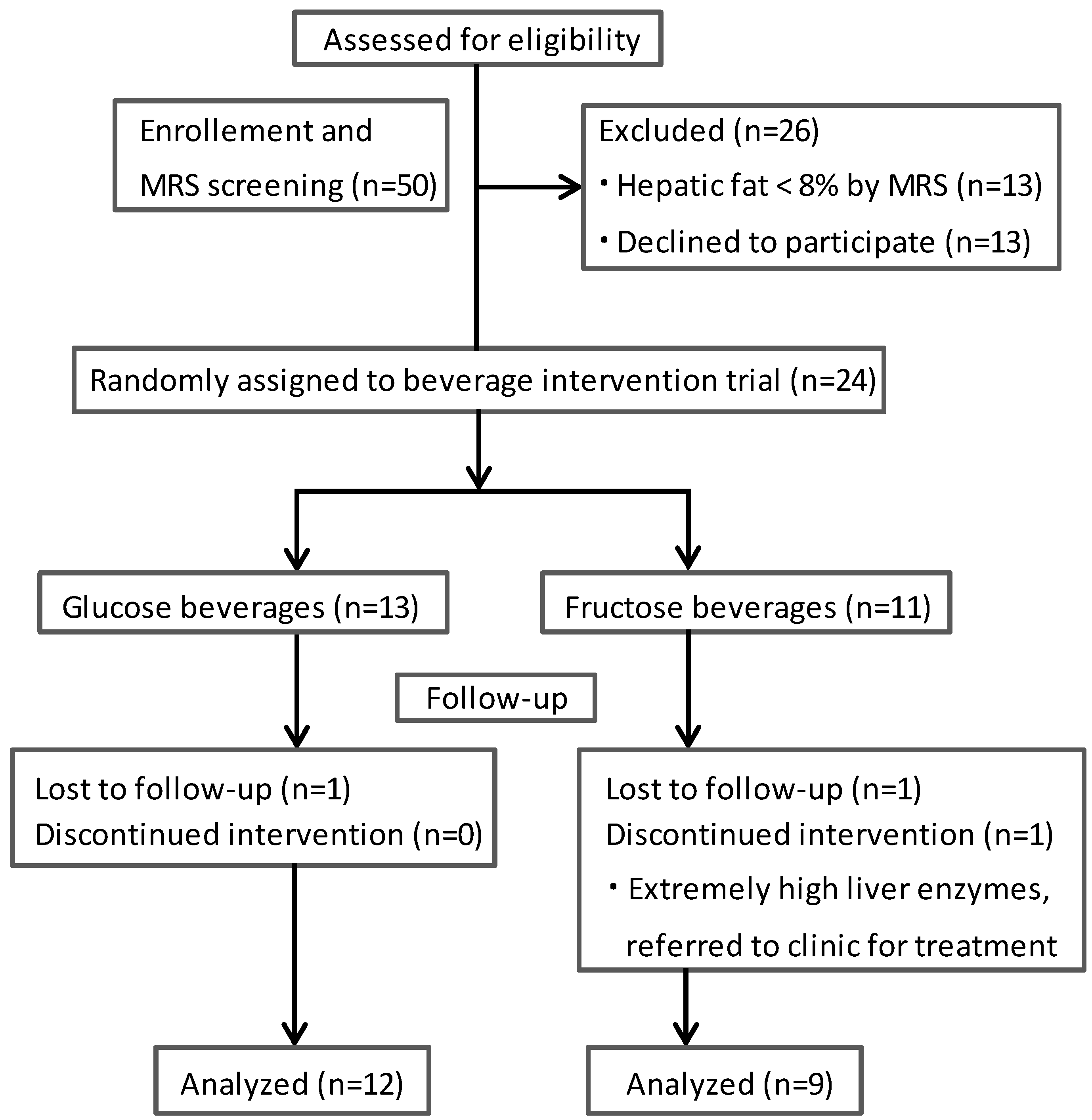
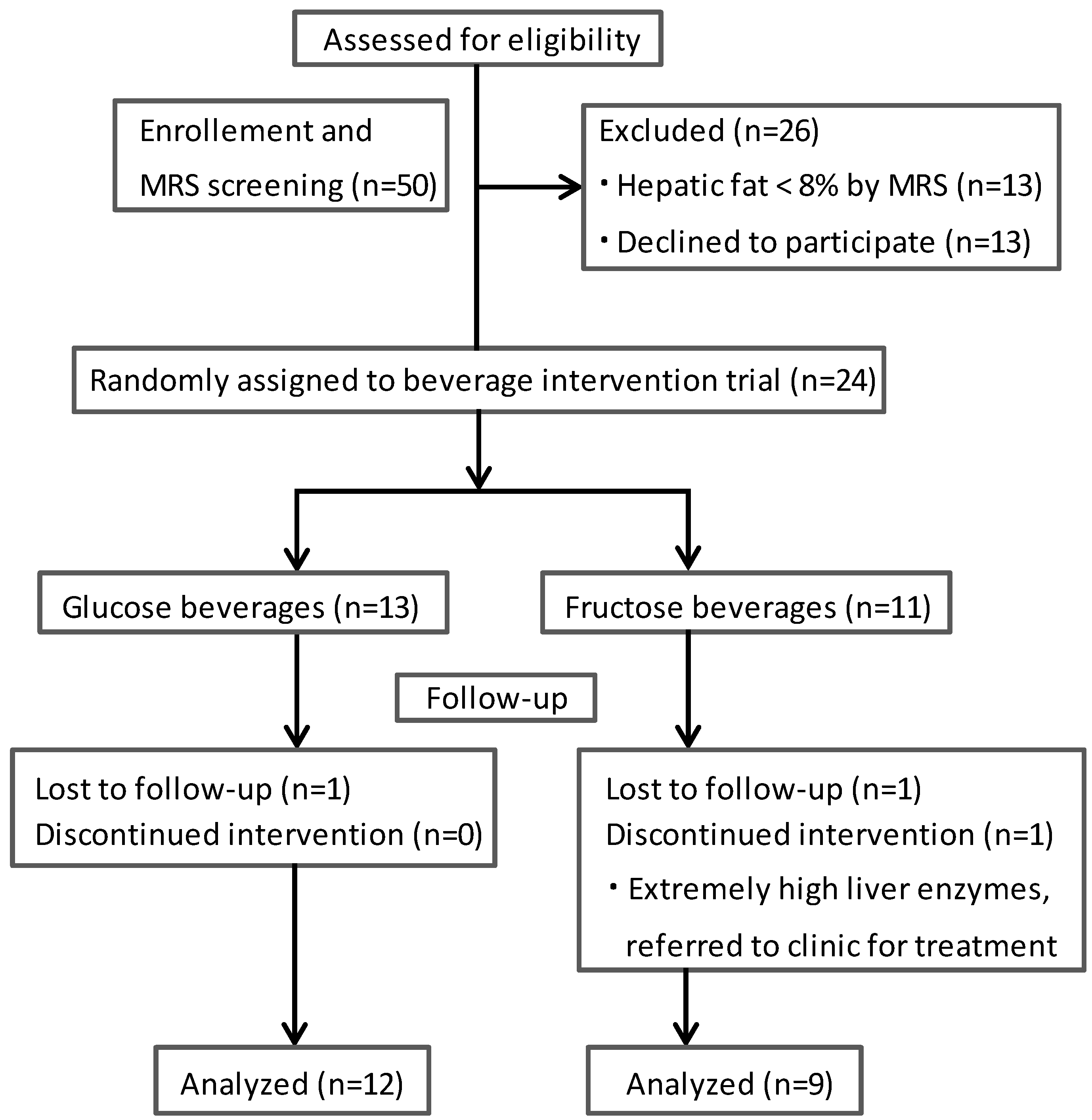
2.2. Determination of Hepatic Fat
2.3. Laboratory Analyses
2.4. Insulin Resistance Index
2.5. Measurement of Oxidative Stress
2.6. Statistical Analyses
3. Results

{kind=link}
{kind=link}
| Parameters | Fructose (n = 9) | Glucose (n = 12) |
|---|---|---|
| Age (years) | 14.2 (0.88) | 13.0 (0.71) |
| Male, n (%) | 3 (33.3) | 8 (66.7) |
| Body weight (kg) | 82.3 (5.62) | 82.0 (4.27) |
| BMI z-score | 2.25 (0.19) | 2.15 (0.09) |
| Hepatic fat (%) | 14.5 (1.79) | 14.0 (1.77) |
| ALT (U/L) | 33.0 (6.74) | 32.7 (5.24) |
| AST (U/L) | 32.4 (3.06) | 33.8 (2.11) |
| Triglycerides (mmol/L) | 1.77 (0.39) | 1.78 (0.20) |
| Cholesterol (mmol/L) | 4.34 (0.24) | 4.40 (0.34) |
| LDL (mmol/L) | 2.79 (0.27) | 2.87 (0.29) |
| HDL (mmol/L) | 1.19 (0.08) | 1.12 (0.06) |
| FFA (mmol/L) | 0.97 (0.08) | 1.11 (0.14) |
| Glucose (mmol/L) | 5.53 (0.28) | 5.04 (0.37) |
| Insulin (mU/L) | 30.4 (4.29) | 36.3 (9.29) |
| hs-CRP (mg/L) | 6.78 (3.16) | 5.21 (1.34) |
| Parameters | Fructose (n = 9) | Glucose (n = 12) | ||||
|---|---|---|---|---|---|---|
| Baseline | Week 4 | p-Value | Baseline | Week 4 | p-Value | |
| Body weight (kg) | 82.3 (5.62) | 83.0 (5.86) | 0.150 | 82.0 (4.27) | 82.5 (4.17) | 0.419 |
| Hepatic fat (%) | 14.5 (1.79) | 13.6 (1.83) | 0.314 | 14.0 (1.77) | 13.8 (1.92) | 0.814 |
| ALT (U/L) | 33.0 (6.74) | 33.4 (4.41) | 0.678 | 32.7 (5.24) | 33.8 (5.69) | 0.562 |
| AST (U/L) | 32.4 (3.06) | 33.3 (3.34) | 0.953 | 33.8 (2.11) | 32.8 (2.10) | 0.531 |
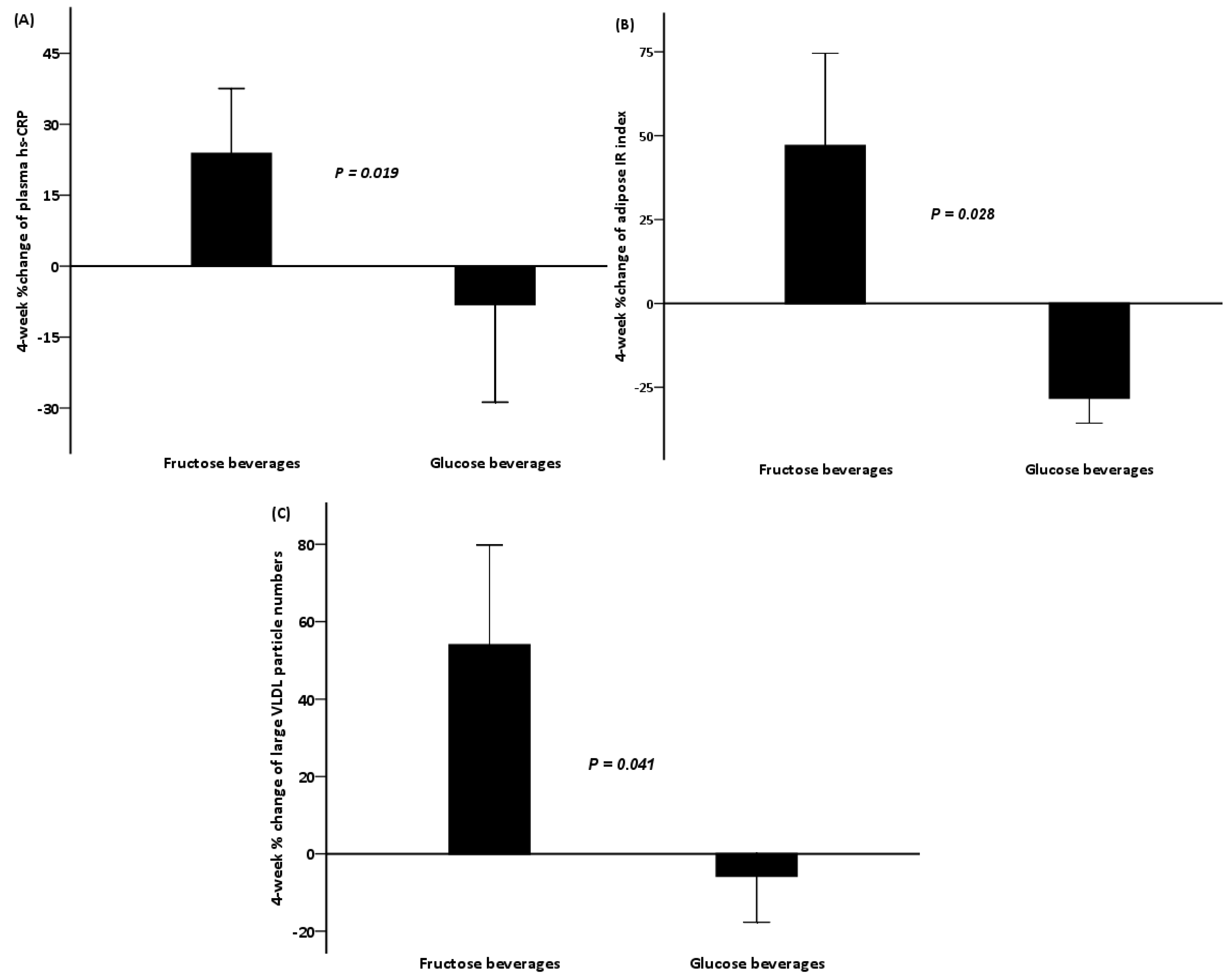
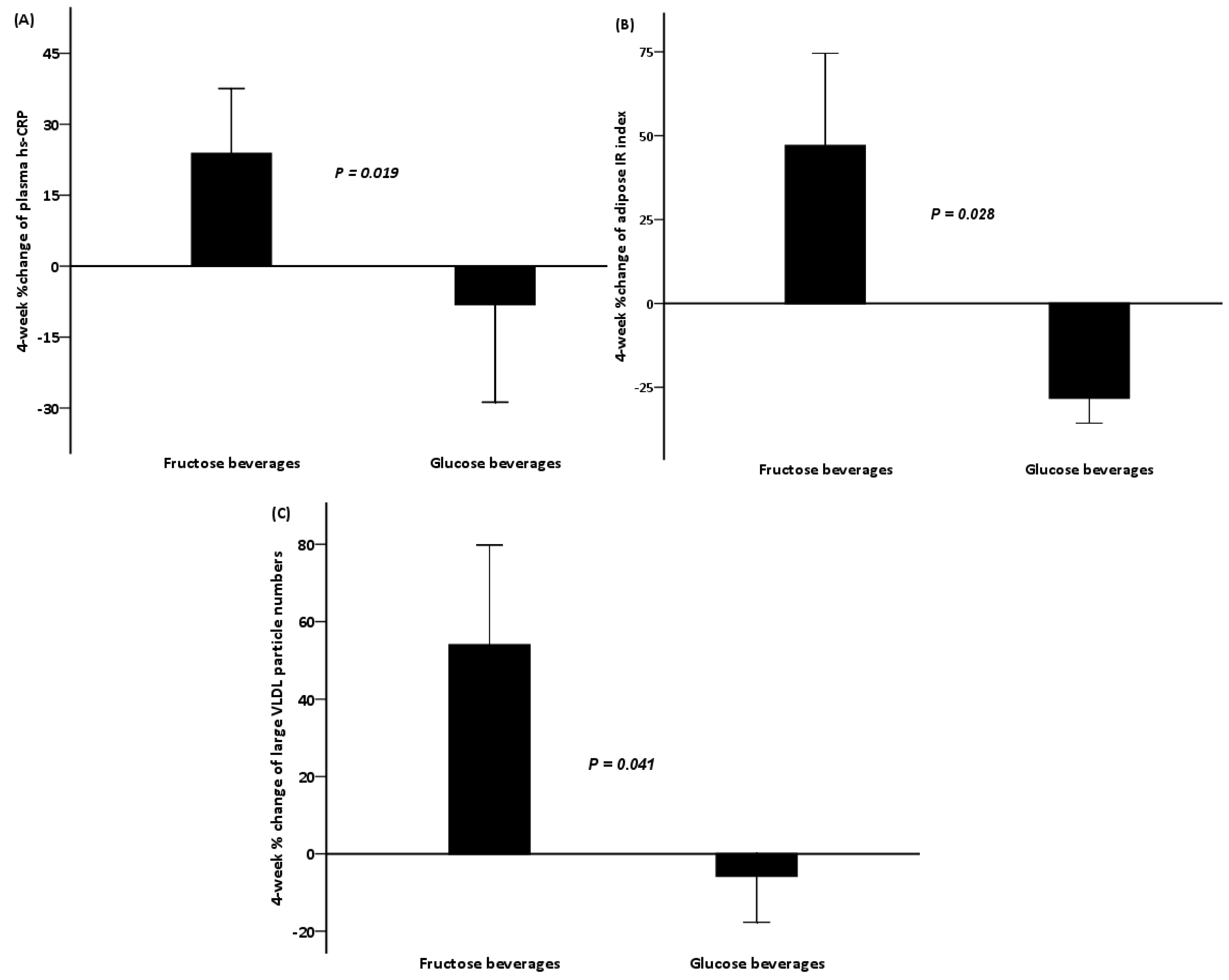
| Triglycerides (mmol/L) | 1.77 (0.39) | 1.15 (0.12) | 0.139 | 1.78 (0.20) | 1.72 (0.23) | 0.754 |
| FFA (mEq/L) | 0.97 (0.08) | 0.90 (0.10) | 0.767 | 1.11 (0.14) | 0.78 (0.07) | 0.027 |
| Glucose (mmol/L) | 5.53 (0.28) | 5.20 (0.24) | 0.374 | 5.01 (0.37) | 5.20 (0.26) | 0.638 |
| Insulin (mU/L) | 30.4 (4.29) | 45.1 (9.78) | 0.260 | 36.3 (9.29) | 29.5 (5.28) | 0.859 |
| Adipose IR | 28.6 (3.78) | 36.1 (5.59) | 0.214 | 34.6 (7.86) | 21.4 (3.82) | 0.004 |
| HOMA-IR | 7.38 (0.97) | 10.7 (2.68) | 0.441 | 8.40 (2.38) | 6.44 (0.99) | 0.754 |
| hs-CRP (mg/L) | 6.78 (3.12) | 7.06 (2.36) | 0.477 | 5.21 (1.34) | 3.99 (1.09) | 0.028 |
| LDL lag time (min) | 18.6 (4.55) | 25.8 (6.13) | 0.084 | 18.5 (3.66) | 37.1 (8.39) | 0.010 |
| Oxidized LDL (mU/L) | 8.82 (0.98) | 7.88 (1.11) | 0.515 | 8.49 (0.97) | 7.06 (0.96) | 0.034 |
| PAI-1 (ng/ml) | 47.3 (2.59) | 49.5 (2.34) | 0.594 | 51.0 (1.90) | 51.2 (2.39) | 0.594 |
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Support
Conflicts of Interest
References
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Argo, C.K.; Caldwell, S.H. Epidemiology and natural history of non-alcoholic steatohepatitis. Clin. Liver Dis. 2009, 13, 511–531. [Google Scholar] [CrossRef] [PubMed]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Berardis, S.; Sokal, E. Pediatric non-alcoholic fatty liver disease: An increasing public health issue. Eur. J. Pediatr. 2014, 173, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Manco, M.; Vania, A.; Nobili, V. Pediatric nonalcoholic fatty liver disease in 2009. J. Pediatr. 2009, 155, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Karpen, S.; Vos, M.B. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J. Pediatr. 2013, 162, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Barshop, N.J.; Sirlin, C.B.; Schwimmer, J.B.; Lavine, J.E. Review article: Epidemiology, pathogenesis and potential treatments of paediatric non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 28, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St. Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, C.; Stal, P.; Askling, J.; Glaumann, H.; Lindberg, G.; Marmur, J.; Hultcrantz, R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 2010, 51, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, N.; Bai, C.; Fang, Y.; Srishord, M.; McCullough, A.; Gramlich, T.; Younossi, Z.M. Long-term follow-up of patients with nonalcoholic fatty liver. Clin. Gastroenterol. Hepatol. 2009, 7, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Anania, C.; Martino, F.; Cantisani, V.; Pascone, R.; Marcantonio, A.; Chiesa, C. Functional and morphological vascular changes in pediatric nonalcoholic fatty liver disease. Hepatology 2010, 52, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Bedogni, G.; Monti, L.; Morino, G.; Natali, G.; Nobili, V. Intima-media thickness and liver histology in obese children and adolescents with non-alcoholic fatty liver disease. Atherosclerosis 2010, 209, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Nobili, V.; Anania, C.; Verdecchia, P.; Chiesa, C. Pediatric nonalcoholic fatty liver disease, metabolic syndrome and cardiovascular risk. World J. Gastroenterol 2011, 17, 3082–3091. [Google Scholar] [PubMed]
- Duffey, K.J.; Popkin, B.M. Shifts in patterns and consumption of beverages between 1965 and 2002. Obesity 2007, 15, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Kit, B.K.; Fakhouri, T.H.; Park, S.; Nielsen, S.J.; Ogden, C.L. Trends in sugar-sweetened beverage consumption among youth and adults in the United States: 1999–2010. Am. J. Clin. Nutr. 2013, 98, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.A.; Auinger, P.; Byrd, R.S. Sugar-Sweetened Beverage Intake Trends in US Adolescents and Their Association with Insulin Resistance-Related Parameters. J. Nutr. Metab. 2010, 2010, 196476. [Google Scholar]
- Wang, Y.C.; Bleich, S.N.; Gortmaker, S.L. Increasing caloric contribution from sugar-sweetened beverages and 100% fruit juices among US children and adolescents, 1988–2004. Pediatrics 2008, 121, e1604–e1614. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among US children and adults: The Third National Health and Nutrition Examination Survey. Medscape. J. Med. 2008, 10, 160. [Google Scholar] [PubMed]
- Diet, nutrition and the prevention of chronic diseases. Available online: http://whqlibdoc.who.int/trs/WHO_TRS_916.pdf (accessed on 28 January 2002).
- Johnson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J.; American Heart Association Nutrition Committee of the Council on Nutrition, Physical Activity, and Metabolism and the Council on Epidemiology and Prevention. Dietary Sugars Intake and Cardiovascular Health: A Scientific Statement from the American Heart Association. Circulation 2009, 120, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Le, N.A.; Liu, S.; Farkas Epperson, M.; Ziegler, T.R.; Welsh, J.A.; Jones, D.P.; McClain, C.J.; Vos, M.B. Children with NAFLD are more sensitive to the adverse metabolic effects of fructose beverages than children without NAFLD. J. Clin. Endocrinol. Metab. 2012, 97, E1088–E1098. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.; Madani, S.; Boukortt, F.; Cherkaoui-Malki, M.; Belleville, J.; Prost, J. Fructose-enriched diet modifies antioxidant status and lipid metabolism in spontaneously hypertensive rats. Nutrition 2006, 22, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo, O.R.; Marra, C.A.; Raschia, A.; Rodriguez, S.; Gagliardino, J.J. Abdominal adipose tissue: Early metabolic dysfunction associated to insulin resistance and oxidative stress induced by an unbalanced diet. Horm. Metab. Res. 2008, 40, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, V.; Anuradha, C.V. Influence of alpha-lipoic acid on lipid peroxidation and antioxidant defence system in blood of insulin-resistant rats. Diabetes Obes. Metab. 2004, 6, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Senturk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Targher, G. Non-alcoholic fatty liver disease, the metabolic syndrome and the risk of cardiovascular disease: The plot thickens. Diabet. Med. 2007, 24, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pineda, N.; Sharma, P.; Xu, Q.; Hu, X.; Vos, M.; Martin, D.R. Measurement of hepatic lipid: High-speed T2-corrected multiecho acquisition at 1H MR spectroscopy—A rapid and accurate technique. Radiology 2009, 252, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.W.; Dumke, K.A.; Goran, M.I. Fructose content in popular beverages made with and without high-fructose corn syrup. Nutrition 2014, 30, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Jeyarajah, E.J.; Cromwell, W.C.; Otvos, J.D. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin. Lab. Med. 2006, 26, 847–870. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Striegl, G.; Puhl, H.; Rotheneder, M. Continuous monitoring of in vitro oxidation of human low density lipoprotein. Free Radic. Res. Commun. 1989, 6, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Innis-Whitehouse, W.; Li, X.; Brown, W.V.; Le, N.A. An efficient chromatographic system for lipoprotein fractionation using whole plasma. J. Lipid Res. 1998, 39, 679–690. [Google Scholar]
- Schwimmer, J.B.; Pardee, P.E.; Lavine, J.E.; Blumkin, A.K.; Cook, S. Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation 2008, 118, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; McClain, C.J. Fructose takes a toll. Hepatology 2009, 50, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; McGillicuddy, F.C.; Anderson, P.D.; Hinkle, C.C.; Shah, R.; Pruscino, L.; Tabita-Martinez, J.; Sellers, K.F.; Rickels, M.R.; Reilly, M.P. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes 2010, 59, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Uffelman, K.D.; Szeto, L.W.; Weller, B.; Steiner, G. Interaction between free fatty acids and insulin in the acute control of very low density lipoprotein production in humans. J. Clin. Investig. 1995, 95, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Boren, J. Fatty liver, insulin resistance, and dyslipidemia. Curr. Diabetes Rep. 2008, 8, 60–64. [Google Scholar] [CrossRef]
- Vos, M.B.; Weber, M.B.; Welsh, J.; Khatoon, F.; Jones, D.P.; Whitington, P.F.; McClain, C.J. Fructose and oxidized low-density lipoprotein in pediatric nonalcoholic fatty liver disease: A pilot study. Arch. Pediatr. Adolesc. Med. 2009, 163, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.E.; Marra, C.A.; Rebolledo, O.R. Glycoxidative stress-induced damage on lipid profile in a fructose-enriched diet model of insulin resistance in rats. Arch. Physiol. Biochem. 2010, 116, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Farina, J.P.; Garcia, M.E.; Alzamendi, A.; Giovambattista, A.; Marra, C.A.; Spinedi, E.; Gagliardino, J.J. Antioxidant treatment prevents the development of fructose-induced abdominal adipose tissue dysfunction. Clin. Sci. 2013, 125, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Maersk, M.; Belza, A.; Stodkilde-Jorgensen, H.; Ringgaard, S.; Chabanova, E.; Thomsen, H.; Pedersen, S.B.; Astrup, A.; Richelsen, B. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: A 6-mo randomized intervention study. Am. J. Clin. Nutr. 2012, 95, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Machann, J.; Kuper, M.A.; Maier, I.B.; Spruss, A.; Konigsrainer, A.; Bischoff, S.C.; Bergheim, I. A moderate weight reduction through dietary intervention decreases hepatic fat content in patients with non-alcoholic fatty liver disease (NAFLD): A pilot study. Eur. J. Nutr. 2013, 52, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [PubMed]
- Lecoultre, V.; Egli, L.; Carrel, G.; Theytaz, F.; Kreis, R.; Schneiter, P.; Boss, A.; Zwygart, K.; Le, K.A.; Bortolotti, M.; et al. Effects of fructose and glucose overfeeding on hepatic insulin sensitivity and intrahepatic lipids in healthy humans. Obesity 2013, 21, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Manco, M.; Devito, R.; Piemonte, F.; Nobili, V. Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Bastelica, D.; Mavri, A.; Morange, P.; Berthet, B.; Grino, M.; Juhan-Vague, I. Plasma PAI-1 levels are more strongly related to liver steatosis than to adipose tissue accumulation. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, A.M.; Lobley, G.E.; Horgan, G.W.; Bremner, D.M.; Fyfe, C.L.; Morrice, P.C.; Duthie, G.G. Effects of a high-protein, low-carbohydrate v. high-protein, moderate-carbohydrate weight-loss diet on antioxidant status, endothelial markers and plasma indices of the cardiometabolic profile. Br. J. Nutr. 2011, 106, 282–291. [Google Scholar]
- Kechagias, S.; Ernersson, A.; Dahlqvist, O.; Lundberg, P.; Lindstrom, T.; Nystrom, F.H.; Fast Food Study Group. Fast-food-based hyper-alimentation can induce rapid and profound elevation of serum alanine aminotransferase in healthy subjects. Gut 2008, 57, 649–654. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jin, R.; Welsh, J.A.; Le, N.-A.; Holzberg, J.; Sharma, P.; Martin, D.R.; Vos, M.B. Dietary Fructose Reduction Improves Markers of Cardiovascular Disease Risk in Hispanic-American Adolescents with NAFLD. Nutrients 2014, 6, 3187-3201. https://doi.org/10.3390/nu6083187
Jin R, Welsh JA, Le N-A, Holzberg J, Sharma P, Martin DR, Vos MB. Dietary Fructose Reduction Improves Markers of Cardiovascular Disease Risk in Hispanic-American Adolescents with NAFLD. Nutrients. 2014; 6(8):3187-3201. https://doi.org/10.3390/nu6083187
Chicago/Turabian StyleJin, Ran, Jean A. Welsh, Ngoc-Anh Le, Jeffrey Holzberg, Puneet Sharma, Diego R. Martin, and Miriam B. Vos. 2014. "Dietary Fructose Reduction Improves Markers of Cardiovascular Disease Risk in Hispanic-American Adolescents with NAFLD" Nutrients 6, no. 8: 3187-3201. https://doi.org/10.3390/nu6083187
APA StyleJin, R., Welsh, J. A., Le, N.-A., Holzberg, J., Sharma, P., Martin, D. R., & Vos, M. B. (2014). Dietary Fructose Reduction Improves Markers of Cardiovascular Disease Risk in Hispanic-American Adolescents with NAFLD. Nutrients, 6(8), 3187-3201. https://doi.org/10.3390/nu6083187