Does Vitamin C Deficiency Promote Fatty Liver Disease Development?



Abstract
:1. Introduction
2. Obesity, Systemic Inflammation and NAFLD
2.1. Obesity and Systemic Inflammation
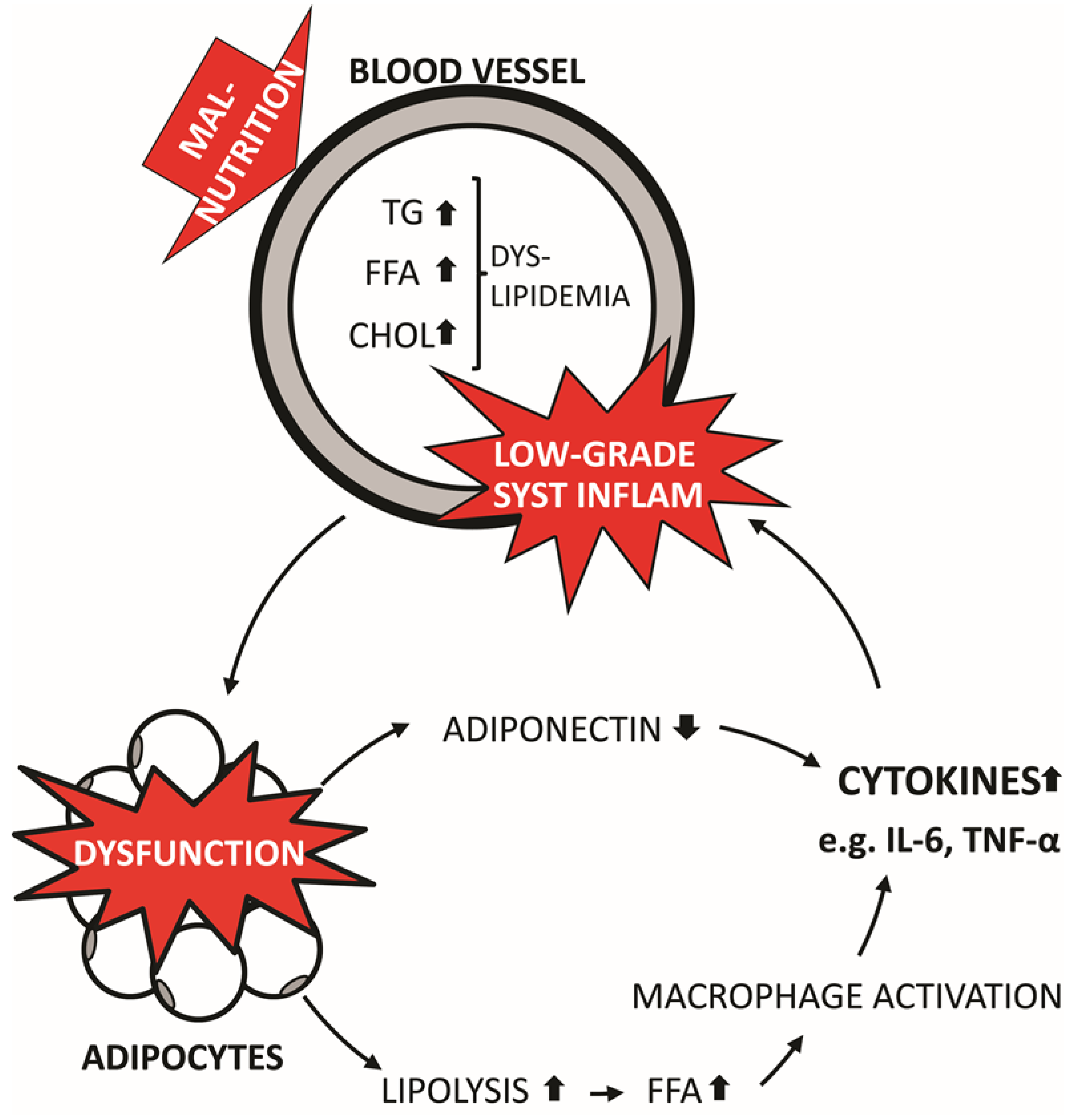
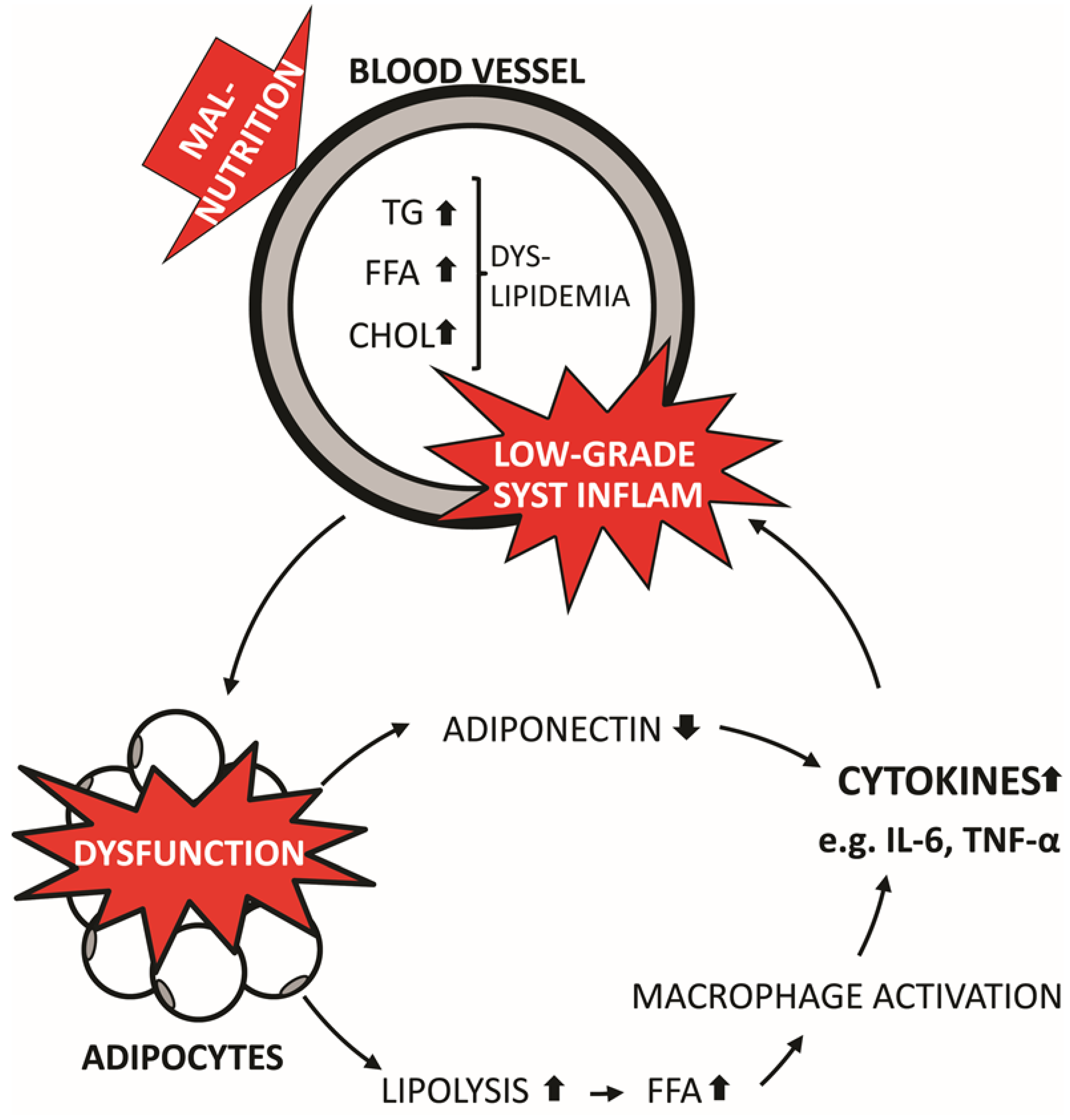
2.2. NAFLD
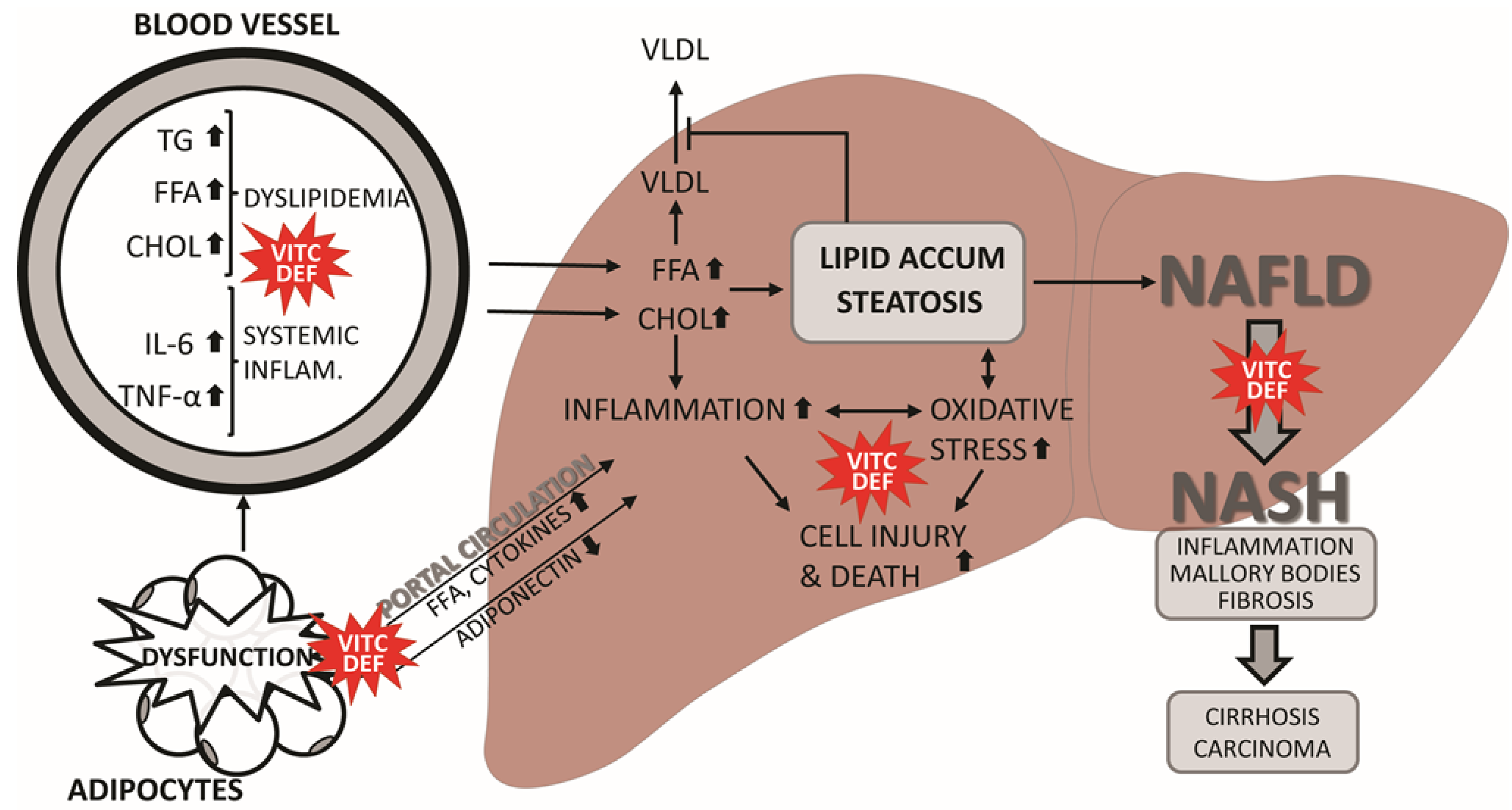
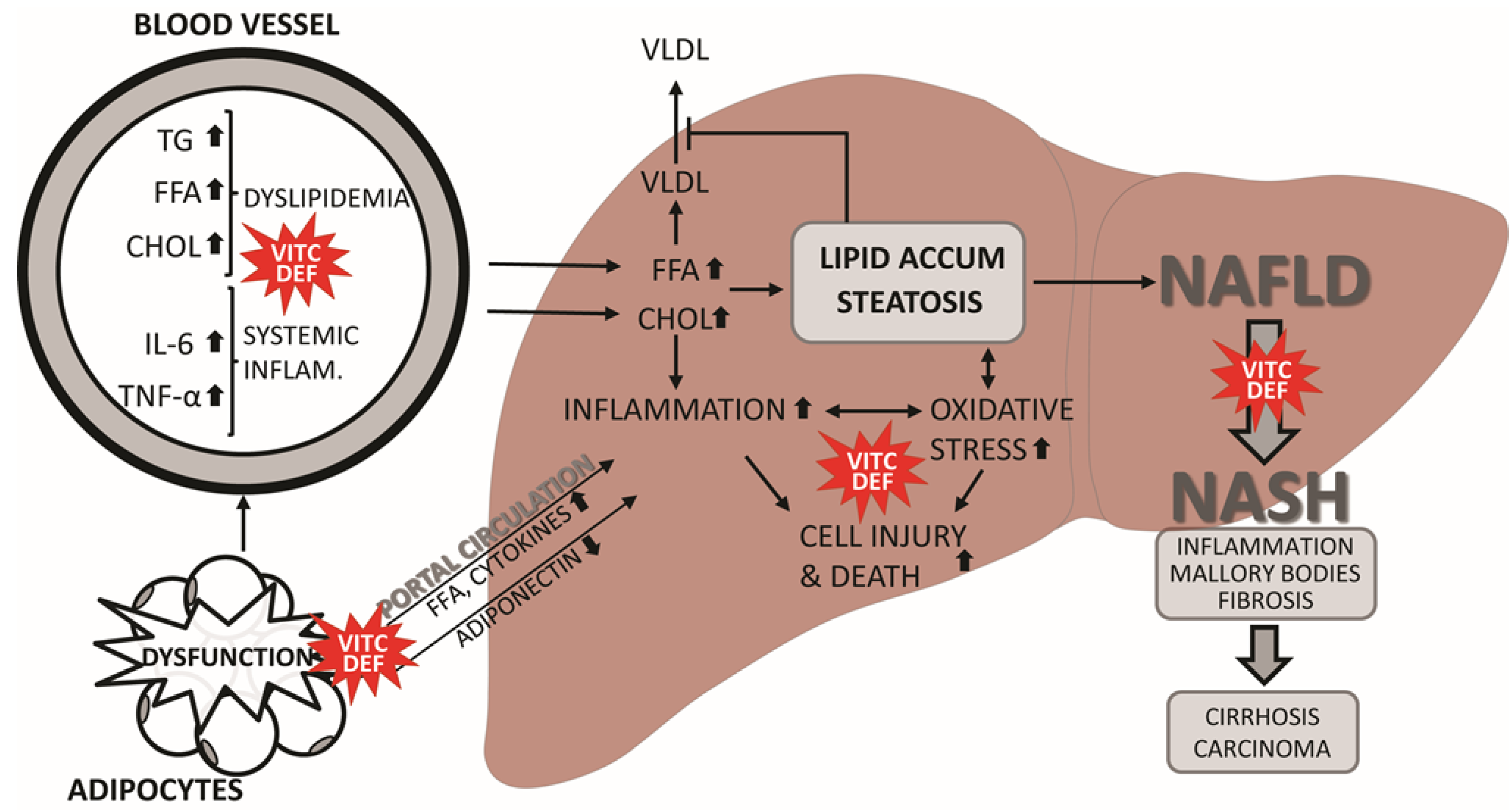
3. Does Vitamin C Deficiency Promote NAFLD?
3.1. Animal Studies of Vitamin C Deficiency

{kind=link}
{kind=link}
| Species | Age/Weight | Design | Outcome | Ref. |
|---|---|---|---|---|
| Guinea Pigs | 200–300 g | Control (VitC 5 mg VitC/day) Deficient (0.5 mg VitC/day) High VitC (100 mg VitC/day) ± 100 mg chol/day Duration: 16 weeks | VitC deficiency, liver: TG ↑ Chol ↑ PL ↔ VitC deficiency + chol, liver: TG ↑ Chol ↑ PL ↔ Histology reveal fat infiltration, local necrosis and proliferation of connective tissue | [86] |
| Guinea Pigs | 400–450 g | Control (1000 mg VitC/kg diet) VitC deficient (0 mg VitC/kg diet) VitC high (25,000 mg VitC/kg) Duration: 4 weeks | VitC deficiency, liver: Chol ↑ TG ↑ VitC deficiency, serum: Chol ↑ TG ↑ HDL ↓ LDL ↑ VLDL ↓ | [82] |
| Guinea Pigs | 300–450 g | Control (10 mg VitC/day) Deficient (0.5 mg VitC/day) Duration: 22, 28 or 31 weeks | VitC deficiency, liver/serum/thoracic aorta: Chol ↑ | [84] |
| Guinea Pigs | ? | Control (660 mg VitC/kg diet) Deficiency (33 mg VitC/kg diet) High (13,200 mg VitC/kg diet) Duration: 5 weeks | VitC deficiency, liver: TBARS ↑ MDA ↑ protein carbonyls ↑ | [92] |
| Guinea Pigs | 250–300 g | Control (25 mg VitC/kg/day) Deficiency (0 mg VitC/kg/day) Duration: 21 days | VitC deficiency, liver: TBARS ↑ | [93] |
| Guinea Pigs | 600–700 g | Control (500 mg VitC/kg diet) Deficiency (50 mg VitC/kg diet) Duration: 6 weeks | VitC deficiency, liver: TAG ↑ CE ↑ FC ↔ | [83] |
| Guinea Pigs | 12 weeks | High or low fat diet with different VitC: Low (100 mg VitC/kg diet) High (691 mg VitC/kg diet) Duration: 6 months | High fat diet, liver: VitC ↓ | [87] |
| Mice, SMP30−/− and WT | 30 days | SMP30−/− or WT ±1.5 mg/L VitC in water Duration: 57 days | VitC deficiency, liver: protein carbonyl ↑ SOD-activity ↑ Cu Zn-SOD protein expression ↑ TBARS ↔, CAT protein expression ↓ | [94] |
| Mice, Gulo−/− or WT | 20–21 weeks | 16 weeks on western diet, then: Control (0.33 g VitC/L in drinking water) Deficiency (0 g VitC/L in drinking water) Duration: 3 weeks | VitC deficiency: GSH ↔ MDA ↑ F2- an F4-isoprostanes ↔ | [95] |
| Mice, Gulo−/− | Newborn | Control Gulo+/+ (0 mg VitC) Deficiency Gulo−/− (0 mg VitC) Duration: 18 days | Gulo−/−, liver: MDA ↑ protein carbonyl ↑ sulfhydryls ↔ GSH ↑ | [96] |
| Rats, ODS or WT | 6 weeks | ODS fed 0, 50, 300, 3000 mg VitC/kg diet ± 0.5% chol and 0.25% cholic acid WT fed 0 mg VitC/kg diet ± 0.5% chol and 0.25% cholic acid Duration: 19 days | VitC deficiency, liver: Chol ↑/↔ total lipids ↑/↔ VitC deficiency, serum: Chol ↔ HDL-C ↔ VitC deficiency + chol/cholic acid, liver: Chol ↑, total lipids ↔ VitC deficiency + chol/cholic acid, serum: Chol ↑, HDL-C ↑ | [88] |
| Rats, ODS | 6 weeks | Control 300 mg VitC/kg diet Deficient 0 mg VitC/kg diet Duration: 14 days | VitC deficiency, liver: CINC-1 ↑ Apo-A1 mRNA ↓ ApoE mRNA ↔ | [97] |
| Rats, ODS or WT | 9 weeks | Control (30 mg VitC/L in drinking water ± 0.5% chol Deficiency 0 mg/L in drinking water Duration: 3 weeks | VitC deficiency, liver: Chol ↔ TG ↔ PL ↔ VitC deficiency, serum: Chol ↑ Total lipoprotein ↓ HDL ↓IDL ↓ LDL ↑ VLDL ↓ VitC deficiency + chol did not affect serum levels further. Chol feeding, regardless of VitC status, increase hepatic lipids | [89] |
3.2. Animal Studies of VitC Intervention and NAFLD
3.3. Epidemiological Studies of VitC Intake and NAFLD
| Species | Age/Weight | Design | Outcome | Ref. |
|---|---|---|---|---|
| Guinea Pigs | 300 g | Normal or atherogenic diet Control (10 mg/kg/day) Deficiency (1 mg/kg/day) High (25 mg/kg/day) VitC administration: Oral Duration: 4 months | Compared to control (normal diet): VitC deficiency, liver: TC ↑ TG ↑ PL ↓ serum: TC ↔ TG ↔ PL ↔ High VitC, liver: TC ↔ TG ↓ PL ↑ serum: TC ↔ TG ↔ PL ↔ Compared to control (atherogenic diet): VitC deficiency, liver: TC ↑ TG ↑ PL ↓ serum: TC ↑ TG ↑ PL ↔ High VitC, liver: TC ↓TG ↔ PL ↓ serum: TC ↓ TG ↓ PL ↓ | [103] |
| Rats | 250–300 g | Control (MCD diet for 10 weeks, then 8 additional weeks of MCD diet + vehicle) Treatment (MCD diet for 10 weeks, then 8 additional weeks of MCD diet + VitC 30 mg/kg/day) VitC administration: Oral Duration: 18 weeks | VitC, liver: ballooning ↓ inflammation ↓ steatosis ↔ SOD ↑ CAT ↑ protein carbonyls ↓ VitC, serum: AST ↓ ALT ↓ ALP ↓ γGT ↓ TC ↓ HDL ↑ LDL ↓ | [100] |
| Rats | 250–300 g | Control (MCD diet + vehicle) Treatment (MCD diet + VitC 30 mg/kg/day) VitC administration: Oral Duration: 10 weeks | VitC, liver: ballooning ↓ SOD ↑ CAT ↑ GR ↑ GPx ↑ TBARS ↓ protein carbonyls ↓ VitC, serum: AST ↓ ALT ↓ ALP ↓ γGT ↓ TC ↓ HDL ↑ LDL ↓ | [101] |
| Rats | 300–350 g | Control (CD diet + vehicle) Treatment (CD diet +30 mg VitC/kg/day) VitC administration: Oral Duration: 4 weeks | VitC, liver: prevents steatosis and reduces oxidative stress VitC, plasma: AST ↔ TG ↔ | [102] |
| Design | Groups | Outcome | Ref. |
|---|---|---|---|
| Cross-Sectional | Adults Healthy controls (n = 25) NASH (n = 25) | NASH patients consumes less dietary VitC (p = 0.0001) | [77] |
| Cross-Sectional | Adults NAFLD patients (n = 96) | Dietary VitC intake was below recommended levels | [78] |
| Cross-Sectional | Adults Male healthy controls (n = 63) Male NAFLD (n = 103) Women healthy controls (n = 116) Women NAFLD (n = 66) | Dietary intake of VitC was not different in men and women with NAFLD compared with control (p = 0.666) Intake of VitC correlated negatively with the odds-ratio of NAFLD for male patients (OR: 4.23, p-trend = 0.014) | [108] |
| Cross-Sectional | Children Steatosis (n = 39) Borderline Z3 (n = 27) Borderline Z1 (n = 36) NASH (n = 47) | Dietary VitC intake was similar in all groups (p = 0.15) and above recommended levels High grade of steatosis was not associated with lower dietary VitC intake (p = 0.97) Amount of hepatocyte ballooning increases with lower dietary VitC levels (p = 0.05) | [109] |
| Cross-Sectional | Children NAFLD (n = 38) | Dietary VitC intake was in agreement with recommended levels | [110] |
| Cross-Sectional | Adults Healthy control (n = 27) Steatosis (n = 33) NASH (n = 41) | Plasma concentrations of VitC did not differ between groups (p > 0.05) Dietary intake of VitC did not differ between groups (p > 0.05) | [111] |
| Prospective | Adults Healthy controls (n = 23) NAFLD (n = 29) | Plasma VitC concentrations were not different between groups (p = 0.65) Plasma VitC concentrations did not correlate with inflammatory grade (p = 0.56) or fibrosis stage (p = 0.53) | [112] |
| Cross-Sectional | Adults Fatty liver disease (n = 38) NASH (n = 67) | Plasma VitC concentrations were lower in NASH patients (p = 0.001) | [113] |
3.4. Clinical Intervention Studies with VitC and NAFLD
| Design | Groups and Intervention | Outcome | Ref. | |
|---|---|---|---|---|
| 12 months, double-blinded, randomized clinical trial | Children Lifestyle intervention + placebo (n = 43) Lifestyle intervention + VitC (500 mg/day) and VitE (600 IU/day) (n = 45) | No differences between groups (p > 0.05) | [117] | |
| 24 months 12 month double-blinded followed by 12 month open-label, randomized clinical trial | Children Lifestyle intervention + placebo (n = 28) Lifestyle intervention + VitC (500 mg/day) and VitE (600 IU/day) (n = 25) | No differences between placebo and VitE/VitC groups (p > 0.05) Compared to baseline, treatment improved steatosis grade (p < 0.001), lobular inflammation (p < 0.001), hepatocyte ballooning (p < 0.001) and NAFLD activity score (p > 0.001), but not portal inflammation (p = 0.1) and fibrosis stage (p = 0.6). | [118] | |
| 6 month, double-blinded, randomized clinical trial | Adults Lifestyle intervention + placebo (n = 22) Lifestyle intervention + VitC (1000 mg/day) and VitE (1000 IU/day) (n = 23) | No differences between placebo and VitC/VitE groups (p > 0.05) Compared to baseline, VitC/VitE treatment improves fibrosis (p = 0.002) | [119] | |
| 12 months, pilot study No control group | Adults VitC (300 mg/day) and VitE (300 mg/day (n = 23) | Treatment decreased serum ALT (p < 0.0001) and hs-CRP (p < 0.005) and improved steatosis (6/10), necroinflammation (8/10), fibrosis (4/10). | [123] | |
| 6 month, open-label, randomized study | Adults Ursodeoxycholic acid (10 mg/kg/day) (n = 29) VitC (500 mg/day) and VitE (600 IU/day) (n = 27) | No differences between ursodeoxycholic acid and VitC/VitE treatment (p > 0.05) | [124] | |
| 4 years, randomized clinical trial | Adults NAFLD Placebo (n = 36) Treatment (1000 mg VitC/day, 1000 IU VitE/day, 20 mg atorvastatin/day) (n = 44) | Adults Normal liver Placebo (n = 190) Treatment (1000 mg VitC/day, 1000 IU VitE/day, 20 mg atorvastatin/day (n = 185) | Treatment reduced risk of having moderate to severe hepatic steatosis (OR = 0.36, p < 0.017) | [125] |
4. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar] [PubMed]
- Jankovic, A.; Korac, A.; Srdic-Galic, B.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Vucetic, M.; Markelic, M.; Velickovic, K.; Golic, I.; et al. Differences in the redox status of human visceral and subcutaneous adipose tissues—Relationships to obesity and metabolic risk. Metabolism 2014, 63, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Uzun, H.; Zengin, K.; Taskin, M.; Aydin, S.; Simsek, G.; Dariyerli, N. Changes in leptin, plasminogen activator factor and oxidative stress in morbidly obese patients following open and laparoscopic swedish adjustable gastric banding. Obes. Surg. 2004, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Lemieux, I.; Despres, J.P. Obesity, inflammation, and cardiovascular risk. Clin. Pharmacol. Ther. 2010, 87, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, K.; Abrams, G.A. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 3540–3553. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Horoz, M.; Bolukbas, C.; Bolukbas, F.F.; Sabuncu, T.; Aslan, M.; Sarifakiogullari, S.; Gunaydin, N.; Erel, O. Measurement of the total antioxidant response using a novel automated method in subjects with nonalcoholic steatohepatitis. BMC Gastroenterol. 2005, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, V.R.; Moreira, E.A.; Wilhelm-Filho, D.; de Miranda, J.X.; Beninca, J.P.; Vigil, S.V.; Moratelli, A.M.; Garlet, T.R.; de Souza Meirelles, M.S.; Vannucchi, H.; et al. Proinflammatory and oxidative stress markers in patients submitted to roux-en-y gastric bypass after 1 year of follow-up. Eur. J. Clin. Nutr. 2012, 66, 891–899. [Google Scholar]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Kivirikko, K.I.; Myllyla, R. Post-translational processing of procollagens. Ann. N. Y. Acad. Sci. 1985, 460, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Hodges, R.E. Serum levels of vitamin C in relation to dietary and supplemental intake of vitamin C in smokers and nonsmokers. Ann. N. Y. Acad. Sci. 1987, 498, 144–152. [Google Scholar] [CrossRef]
- Simon, J.A.; Hudes, E.S.; Tice, J.A. Relation of serum ascorbic acid to mortality among us adults. J. Am. Coll. Nutr. 2001, 20, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Hampl, J.S.; Taylor, C.A.; Johnston, C.S. Vitamin C deficiency and depletion in the United States: The third national health and nutrition examination survey, 1988 to 1994. Am. J. Public Health 2004, 94, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Wrieden, W.L.; Hannah, M.K.; Bolton-Smith, C.; Tavendale, R.; Morrison, C.; Tunstall-Pedoe, H. Plasma vitamin C and food choice in the third glasgow monica population survey. J. Epidemiol. Commun. Health 2000, 54, 355–360. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Poulsen, H.E. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br. J. Nutr. 2010, 103, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency increase lifestyle-associated vascular disease progression? Evidence based on experimental and clinical studies. Antioxid. Redox Signal. 2013, 19, 2084–2104. [Google Scholar] [CrossRef] [PubMed]
- Aasheim, E.T.; Hofso, D.; Hjelmesaeth, J.; Birkeland, K.I.; Bohmer, T. Vitamin status in morbidly obese patients: A cross-sectional study. Am. J. Clin. Nutr. 2008, 87, 362–369. [Google Scholar] [PubMed]
- Canoy, D.; Wareham, N.; Welch, A.; Bingham, S.; Luben, R.; Day, N.; Khaw, K.T. Plasma ascorbic acid concentrations and fat distribution in 19,068 british men and women in the european prospective investigation into cancer and nutrition norfolk cohort study. Am. J. Clin. Nutr. 2005, 82, 1203–1209. [Google Scholar] [PubMed]
- Johnston, C.S.; Beezhold, B.L.; Mostow, B.; Swan, P.D. Plasma vitamin C is inversely related to body mass index and waist circumference but not to plasma adiponectin in nonsmoking adults. J. Nutr. 2007, 137, 1757–1762. [Google Scholar] [PubMed]
- Mah, E.; Matos, M.D.; Kawiecki, D.; Ballard, K.; Guo, Y.; Volek, J.S.; Bruno, R.S. Vitamin C status is related to proinflammatory responses and impaired vascular endothelial function in healthy, college-aged lean and obese men. J. Am. Diet. Assoc. 2011, 111, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Bano, G. Glucose homeostasis, obesity and diabetes. Best Pract. Res. Clin. Obstet. Gynaecol. 2013, 27, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 2013, 339, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Wisse, B.E. The inflammatory syndrome: The role of adipose tissue cytokines in metabolic disorders linked to obesity. J. Am. Soc. Nephrol. 2004, 15, 2792–2800. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Farnier, C.; Krief, S.; Blache, M.; Diot-Dupuy, F.; Mory, G.; Ferre, P.; Bazin, R. Adipocyte functions are modulated by cell size change: Potential involvement of an integrin/erk signalling pathway. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, S.D.; van Citters, G.W.; Kirkman, E.L.; Bergman, R.N. Extreme insulin resistance of the central adipose depot in vivo. Diabetes 2002, 51, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [PubMed]
- Elnakish, M.T.; Hassanain, H.H.; Janssen, P.M.; Angelos, M.G.; Khan, M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: Important role of rac/nadph oxidase. J. Pathol. 2013, 231, 290–300. [Google Scholar] [PubMed]
- Ford, E.S.; Mokdad, A.H.; Giles, W.H.; Brown, D.W. The metabolic syndrome and antioxidant concentrations: Findings from the third national health and nutrition examination survey. Diabetes 2003, 52, 2346–2352. [Google Scholar] [CrossRef] [PubMed]
- Lissner, L.; Lindroos, A.K.; Sjostrom, L. Swedish obese subjects (SOS): An obesity intervention study with a nutritional perspective. Eur. J. Clin. Nutr. 1998, 52, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Del Mar Bibiloni, M.; Maffeis, C.; Llompart, I.; Pons, A.; Tur, J.A. Dietary factors associated with subclinical inflammation among girls. Eur. J. Clin. Nutr. 2013, 67, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Rose, F.J.; Webster, J.; Barry, J.B.; Phillips, L.K.; Richards, A.A.; Whitehead, J.P. Synergistic effects of ascorbic acid and thiazolidinedione on secretion of high molecular weight adiponectin from human adipocytes. Diabetes Obes. Metab. 2010, 12, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Afkhami-Ardekani, M.; Shojaoddiny-Ardekani, A. Effect of vitamin C on blood glucose, serum lipids & serum insulin in type 2 diabetes patients. Indian J. Med. Res. 2007, 126, 471–474. [Google Scholar] [PubMed]
- Soares, A.F.; Guichardant, M.; Cozzone, D.; Bernoud-Hubac, N.; Bouzaidi-Tiali, N.; Lagarde, M.; Geloen, A. Effects of oxidative stress on adiponectin secretion and lactate production in 3T3-L1 adipocytes. Free Radic. Biol. Med. 2005, 38, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the american gastroenterological association, american association for the study of liver diseases, and american college of gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Leone, N.; Vanni, E.; Marchesini, G.; Brunello, F.; Carucci, P.; Musso, A.; de Paolis, P.; Capussotti, L.; Salizzoni, M.; et al. Expanding the natural history of nonalcoholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002, 123, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Angelico, F.; del Ben, M.; Conti, R.; Francioso, S.; Feole, K.; Fiorello, S.; Cavallo, M.G.; Zalunardo, B.; Lirussi, F.; Alessandri, C.; et al. Insulin resistance, the metabolic syndrome, and nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2005, 90, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, F.H.; Desaive, C.; Thiry, A.; Dewe, W.; Scheen, A.J.; Gielen, J.E.; Lefebvre, P.J. Liver abnormalities in severely obese subjects: Effect of drastic weight loss after gastroplasty. Int. J. Obes. Relat. Metab. Disord. 1998, 22, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.; Farooq, S.; Deepa, M.; Ravikumar, R.; Pitchumoni, C.S. Prevalence of non-alcoholic fatty liver disease in urban south indians in relation to different grades of glucose intolerance and metabolic syndrome. Diabetes Res. Clin. Pract. 2009, 84, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Diehl, A.M. Nonalcoholic fatty liver disease as a complication of insulin resistance. Med. Clin. N. Am. 2007, 91, 1125–1149. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; deFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Bardini, G.; Rotella, C.M.; Giannini, S. Dyslipidemia and diabetes: Reciprocal impact of impaired lipid metabolism and beta-cell dysfunction on micro- and macrovascular complications. Rev. Diabet. Stud. 2012, 9, 82–93. [Google Scholar] [CrossRef] [PubMed]
- McRae, M.P. Vitamin C supplementation lowers serum low-density lipoprotein cholesterol and triglycerides: A meta-analysis of 13 randomized controlled trials. J. Chiropr. Med. 2008, 7, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Molnar, C.; Geiger, S.; Graziadei, I.; Ebenbichler, C.F.; Weiss, H.; Kaser, S.; Kaser, A.; Tilg, H. Anti-inflammatory effects of excessive weight loss: Potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut 2010, 59, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Tamura, S.; Kiso, S.; Matsumoto, H.; Saji, Y.; Yoshida, Y.; Fukui, K.; Maeda, N.; Nishizawa, H.; Nagaretani, H.; et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 2003, 125, 1796–1807. [Google Scholar] [CrossRef] [PubMed]
- Finelli, C.; Tarantino, G. What is the role of adiponectin in obesity related non-alcoholic fatty liver disease? World J. Gastroenterol. 2013, 19, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing Il-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Does fructose consumption contribute to non-alcoholic fatty liver disease? Clin. Res. Hepatol. Gastroenterol. 2012, 36, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Podszun, M.C.; Grebenstein, N.; Spruss, A.; Schlueter, T.; Kremoser, C.; Bergheim, I.; Frank, J. Dietary α-tocopherol and atorvastatin reduce high-fat-induced lipid accumulation and down-regulate cd36 protein in the liver of guinea pigs. J. Nutr. Biochem. 2014, 25, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Cheah, I.K.; Halliwell, B. A high-fat and cholesterol diet causes fatty liver in guinea pigs. The role of iron and oxidative damage. Free Radic. Res. 2013, 47, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Coban, J.; Evran, B.; Ozkan, F.; Cevik, A.; Dogru-Abbasoglu, S.; Uysal, M. Effect of blueberry feeding on lipids and oxidative stress in the serum, liver and aorta of guinea pigs fed on a high-cholesterol diet. Biosci. Biotechnol. Biochem. 2013, 77, 389–391. [Google Scholar] [PubMed]
- Kim, J.E.; Clark, R.M.; Park, Y.; Lee, J.; Fernandez, M.L. Lutein decreases oxidative stress and inflammation in liver and eyes of guinea pigs fed a hypercholesterolemic diet. Nutr. Res. Pract. 2012, 6, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and FAS-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Senturk, H. From fatty liver to fibrosis: A tale of “second hit”. World J. Gastroenterol. 2013, 19, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Larter, C.Z.; Yeh, M.M.; Haigh, W.G.; Teoh, N.; Farrell, G.C. Ezetimibe and atorvastatin ameliorate liver injury in foz/foz mice with NASH. J. Gastroenterol. Hepatol. 2011, 26, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Kawada, N. Inflammation and fibrogenesis in steatohepatitis. J. Gastroenterol. 2012, 47, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.; Nicco, C.; Tran van Nhieu, J.; Borderie, D.; Chereau, C.; Conti, F.; Jaffray, P.; Soubrane, O.; Calmus, Y.; Weill, B.; et al. Pivotal role of superoxide anion and beneficial effect of antioxidant molecules in murine steatohepatitis. Hepatology 2004, 39, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. Nash is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Gokulakrishnan, K.; Sampathkumar, R.; Farooq, S.; Ravikumar, R.; Mohan, V.; Balasubramanyam, M. Oxidative stress is independently associated with non-alcoholic fatty liver disease (NAFLD) in subjects with and without type 2 diabetes. Clin. Biochem. 2010, 43, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Valdecantos, M.P.; Perez-Matute, P.; Quintero, P.; Martinez, J.A. Vitamin C, resveratrol and lipoic acid actions on isolated rat liver mitochondria: All antioxidants but different. Redox Rep. 2010, 15, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; de Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Faga, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Ferolla, S.M.; Ferrari, T.C.; Lima, M.L.; Reis, T.O.; Tavares, W.C., Jr.; Couto, O.F.; Vidigal, P.V.; Fausto, M.A.; Couto, C.A. Dietary patterns in brazilian patients with nonalcoholic fatty liver disease: A cross-sectional study. Clinics 2013, 68, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar] [PubMed]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quinones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, H.; Lykkesfeldt, J. Role of marginal vitamin C deficiency in atherogenesis: In vivo models and clinical studies. Basic Clin. Pharmacol. Toxicol. 2009, 104, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.W. Association between hyperlipidemia and hepatic cytochrome p-450 in guinea pigs. Res. Commun. Mol. Pathol. Pharmacol. 1997, 97, 139–150. [Google Scholar] [PubMed]
- Montano, C.E.; Fernandez, M.L.; McNamara, D.J. Regulation of apolipoprotein B-containing lipoproteins by vitamin C level and dietary fat saturation in guinea pigs. Metabolism 1998, 47, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Ginter, E.; Cerven, J.; Nemec, R.; Mikus, L. Lowered cholesterol catabolism in guinea pigs with chronic ascorbic acid deficiency. Am. J. Clin. Nutr. 1971, 24, 1238–1245. [Google Scholar] [PubMed]
- Ginter, E. Ascorbic acid in cholesterol and bile acid metabolism. Ann. N. Y. Acad. Sci. 1975, 258, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Pramod, J.; Sharma, P.K.; Sapra, M.; Manorma; Kothari, L.K. Effect of vitamin C deficiency and excess on the liver: A histopathological and biochemical study in guinea pigs fed normal or high cholesterol diet. Indian J. Pathol. Microbiol. 1990, 33, 307–313. [Google Scholar] [PubMed]
- Frikke-Schmidt, H.; Tveden-Nyborg, P.; Birck, M.M.; Lykkesfeldt, J. High dietary fat and cholesterol exacerbates chronic vitamin C deficiency in guinea pigs. Br. J. Nutr. 2011, 105, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Horio, F.; Ozaki, K.; Oda, H.; Makino, S.; Hayashi, Y.; Yoshida, A. Effect of dietary ascorbic acid, cholesterol and PCB on cholesterol concentrations in serum and liver in a rat mutant unable to synthesize ascorbic acid. J. Nutr. 1987, 117, 1036–1044. [Google Scholar] [PubMed]
- Uchida, K.; Nomura, Y.; Takase, H.; Tasaki, T.; Seo, S.; Hayashi, Y.; Takeuchi, N. Effect of vitamin C depletion on serum cholesterol and lipoprotein levels in ODS (od/od) rats unable to synthesize ascorbic acid. J. Nutr. 1990, 120, 1140–1147. [Google Scholar] [PubMed]
- Fernandez, M.L. Guinea pigs as models for cholesterol and lipoprotein metabolism. J. Nutr. 2001, 131, 10–20. [Google Scholar] [PubMed]
- Yang, R.; Guo, P.; Song, X.; Liu, F.; Gao, N. Hyperlipidemic guinea pig model: Mechanisms of triglyceride metabolism disorder and comparison to rat. Biol. Pharm. Bull. 2011, 34, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Barja, G.; Lopez-Torres, M.; Perez-Campo, R.; Rojas, C.; Cadenas, S.; Prat, J.; Pamplona, R. Dietary vitamin C decreases endogenous protein oxidative damage, malondialdehyde, and lipid peroxidation and maintains fatty acid unsaturation in the guinea pig liver. Free Radic. Biol. Med. 1994, 17, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kitamura, R.; Imaoka, S.; Funae, Y.; Kitada, M.; Kamataki, T. Examination for lipid peroxidation in liver microsomes of guinea pigs as a causal factor in the decrease in the content of cytochrome p-450 due to ascorbic acid deficiency. Res. Commun. Chem. Pathol. Pharmacol. 1992, 75, 209–219. [Google Scholar] [PubMed]
- Amano, A.; Sato, Y.; Kishimoto, Y.; Takahashi, K.; Handa, S.; Aigaki, T.; Maruyama, N.; Ishigami, A. Effects of ascorbic acid deficiency on protein and lipid oxidation in livers from SMP30/GNL knockout mice. J. Nutr. Sci. Vitaminol. 2013, 59, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.R.; Diasio, D.L.; Rodrigues, L.M.; Harrison, F.E.; May, J.M. Combined vitamin C and E deficiency induces motor defects in gulo(−/−)/SVCT2(+/−) mice. Nutr. Neurosci. 2013, 16, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo(−/−) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Horio, F.; Kiyama, K.; Kobayashi, M.; Kawai, K.; Tsuda, T. Ascorbic acid deficiency stimulates hepatic expression of inflammatory chemokine, cytokine-induced neutrophil chemoattractant-1, in scurvy-prone ODS rats. J. Nutr. Sci. Vitaminol. 2006, 52, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, A.; Yazdanparast, R.; Molaei, M. Amelioration of diet-induced nonalcoholic steatohepatitis in rats by Mn-salen complexes via reduction of oxidative stress. J. Biomed. Sci. 2012, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, A.; Yazdanparast, R. Prevention of nonalcoholic steatohepatitis in rats by two manganese-salen complexes. Iran. Biomed. J. 2014, 18, 41–48. [Google Scholar] [PubMed]
- Oliveira, C.P.; Gayotto, L.C.; Tatai, C.; Della Nina, B.I.; Lima, E.S.; Abdalla, D.S.; Lopasso, F.P.; Laurindo, F.R.; Carrilho, F.J. Vitamin C and vitamin E in prevention of nonalcoholic fatty liver disease (NAFLD) in choline deficient diet fed rats. Nutr. J. 2003, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Nambisan, B.; Kurup, P.A. Ascorbic acid and glycosaminoglycan and lipid metabolism in guinea pigs fed normal and atherogenic diets. Atherosclerosis 1975, 22, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Hathcock, J.N.; Azzi, A.; Blumberg, J.; Bray, T.; Dickinson, A.; Frei, B.; Jialal, I.; Johnston, C.S.; Kelly, F.J.; Kraemer, K.; et al. Vitamins E and C are safe across a broad range of intakes. Am. J. Clin. Nutr. 2005, 81, 736–745. [Google Scholar] [PubMed]
- The National Academies; Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids; The National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Monsen, E.R. Dietary reference intakes for the antioxidant nutrients: Vitamin C, vitamin E, selenium, and carotenoids. J. Am. Diet. Assoc. 2000, 100, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.K. Dietary intake—How do we measure what people are really eating? Obes. Res. 2002, 10, 63s–68s. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jo, A.N.; Lee, S.M.; Bae, H.S.; Jun, D.W.; Cho, Y.K.; Suk, K.T.; Yoon, J.H.; Ahn, S.B.; Cho, Y.J.; et al. Associations between intakes of individual nutrients or whole food groups and non-alcoholic fatty liver disease among korean adults. J. Gastroenterol. Hepatol. 2014, 29, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Colvin, R.; Belt, P.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Schwimmer, J.B.; Tonascia, J.; Unalp, A.; Lavine, J.E. Correlation of vitamin E, uric acid, and diet composition with histologic features of pediatric NAFLD. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.R.; Patterson, C.; So, S.; Rogenstein, C.D.; Wykes, L.J.; Roberts, E.A. Dietary and physical activity patterns in children with fatty liver. Eur. J. Clin. Nutr. 2010, 64, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, H.E.; Arendt, B.M.; Noureldin, S.A.; Therapondos, G.; Guindi, M.; Allard, J.P. A cross-sectional study assessing dietary intake and physical activity in canadian patients with nonalcoholic fatty liver disease vs. healthy controls. J. Acad. Nutr. Diet. 2014, 114, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Madan, K.; Bhardwaj, P.; Thareja, S.; Gupta, S.D.; Saraya, A. Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD). J. Clin. Gastroenterol. 2006, 40, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Canbakan, B.; Senturk, H.; Tahan, V.; Hatemi, I.; Balci, H.; Toptas, T.; Sonsuz, A.; Velet, M.; Aydin, S.; Dirican, A.; et al. Clinical, biochemical and histological correlations in a group of non-drinker subjects with non-alcoholic fatty liver disease. Acta Gastro-Enterol. Belgica 2007, 70, 277–284. [Google Scholar]
- Dehghan, M.; Akhtar-Danesh, N.; McMillan, C.R.; Thabane, L. Is plasma vitamin C an appropriate biomarker of vitamin C intake? A systematic review and meta-analysis. Nutr. J. 2007, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Sharp, S.J.; Luben, R.; Wareham, N.J.; Khaw, K.T. Plasma vitamin C predicts incident heart failure in men and women in european prospective investigation into cancer and nutrition-norfolk prospective study. Am. Heart J. 2011, 162, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Manco, M.; Devito, R.; Ciampalini, P.; Piemonte, F.; Marcellini, M. Effect of vitamin E on aminotransferase levels and insulin resistance in children with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2006, 24, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Manco, M.; Devito, R.; di Ciommo, V.; Comparcola, D.; Sartorelli, M.R.; Piemonte, F.; Marcellini, M.; Angulo, P. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: A randomized, controlled trial. Hepatology 2008, 48, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.; Ward, J.; Schenker, S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2003, 98, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Angulo, P. Vitamins E and C for the treatment of NASH: Duplication of results but lack of demonstration of efficacy. Am. J. Gastroenterol. 2003, 98, 2348–2350. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro-Carrero, V.M.; Piñeiro, E.O. Liver. Pediatrics 2004, 113, 1097–1106. [Google Scholar] [PubMed]
- Papandreou, D.; Rousso, I.; Mavromichalis, I. Update on non-alcoholic fatty liver disease in children. Clin. Nutr. 2007, 26, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Kawanaka, M.; Nishino, K.; Nakamura, J.; Suehiro, M.; Goto, D.; Urata, N.; Oka, T.; Kawamoto, H.; Nakamura, H.; Yodoi, J.; et al. Treatment of nonalcoholic steatohepatitis with vitamins E and C: A pilot study. Hepat. Med. 2013, 5, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Ersöz, G.; Gunsar, F.; Karasu, Z.; Akay, S.; Batur, Y.; Akarca, U.S. Management of fatty liver disease with vitamin E and C compared to ursodeoxycholic acid treatment. Turkish J. Gastroenterol. 2005, 16, 124–128. [Google Scholar]
- Foster, T.; Budoff, M.J.; Saab, S.; Ahmadi, N.; Gordon, C.; Guerci, A.D. Atorvastatin and antioxidants for the treatment of nonalcoholic fatty liver disease: The St. Francis Heart Study randomized clinical trial. Am. J. Gastroenterol. 2011, 106, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Rodella, S.; Lippi, G.; Franchini, M.; Zoppini, G.; Muggeo, M.; Day, C.P. Nash predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity 2008, 16, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; Meigs, J.B.; Sahani, D.V.; Hirschhorn, J.N.; O’Donnell, C.J.; Fox, C.S. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: The framingham heart study. Hepatology 2010, 51, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Hatzitolios, A.; Savopoulos, C.; Lazaraki, G.; Sidiropoulos, I.; Haritanti, P.; Lefkopoulos, A.; Karagiannopoulou, G.; Tzioufa, V.; Dimitrios, K. Efficacy of omega-3 fatty acids, atorvastatin and orlistat in non-alcoholic fatty liver disease with dyslipidemia. Indian J. Gastroenterol. 2004, 23, 131–134. [Google Scholar] [PubMed]
- Saadeh, S.; Younossi, Z.M.; Remer, E.M.; Gramlich, T.; Ong, J.P.; Hurley, M.; Mullen, K.D.; Cooper, J.N.; Sheridan, M.J. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 745–750. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ipsen, D.H.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does Vitamin C Deficiency Promote Fatty Liver Disease Development? Nutrients 2014, 6, 5473-5499. https://doi.org/10.3390/nu6125473
Ipsen DH, Tveden-Nyborg P, Lykkesfeldt J. Does Vitamin C Deficiency Promote Fatty Liver Disease Development? Nutrients. 2014; 6(12):5473-5499. https://doi.org/10.3390/nu6125473
Chicago/Turabian StyleIpsen, David Højland, Pernille Tveden-Nyborg, and Jens Lykkesfeldt. 2014. "Does Vitamin C Deficiency Promote Fatty Liver Disease Development?" Nutrients 6, no. 12: 5473-5499. https://doi.org/10.3390/nu6125473
APA StyleIpsen, D. H., Tveden-Nyborg, P., & Lykkesfeldt, J. (2014). Does Vitamin C Deficiency Promote Fatty Liver Disease Development? Nutrients, 6(12), 5473-5499. https://doi.org/10.3390/nu6125473