The Influence of Dietary Fat on Liver Fat Accumulation



Abstract
:1. Introduction
2. Quantifying Hepatic Steatosis
3. Liver Fatty Acid Metabolism: Transitioning from the Fasted to Fed State
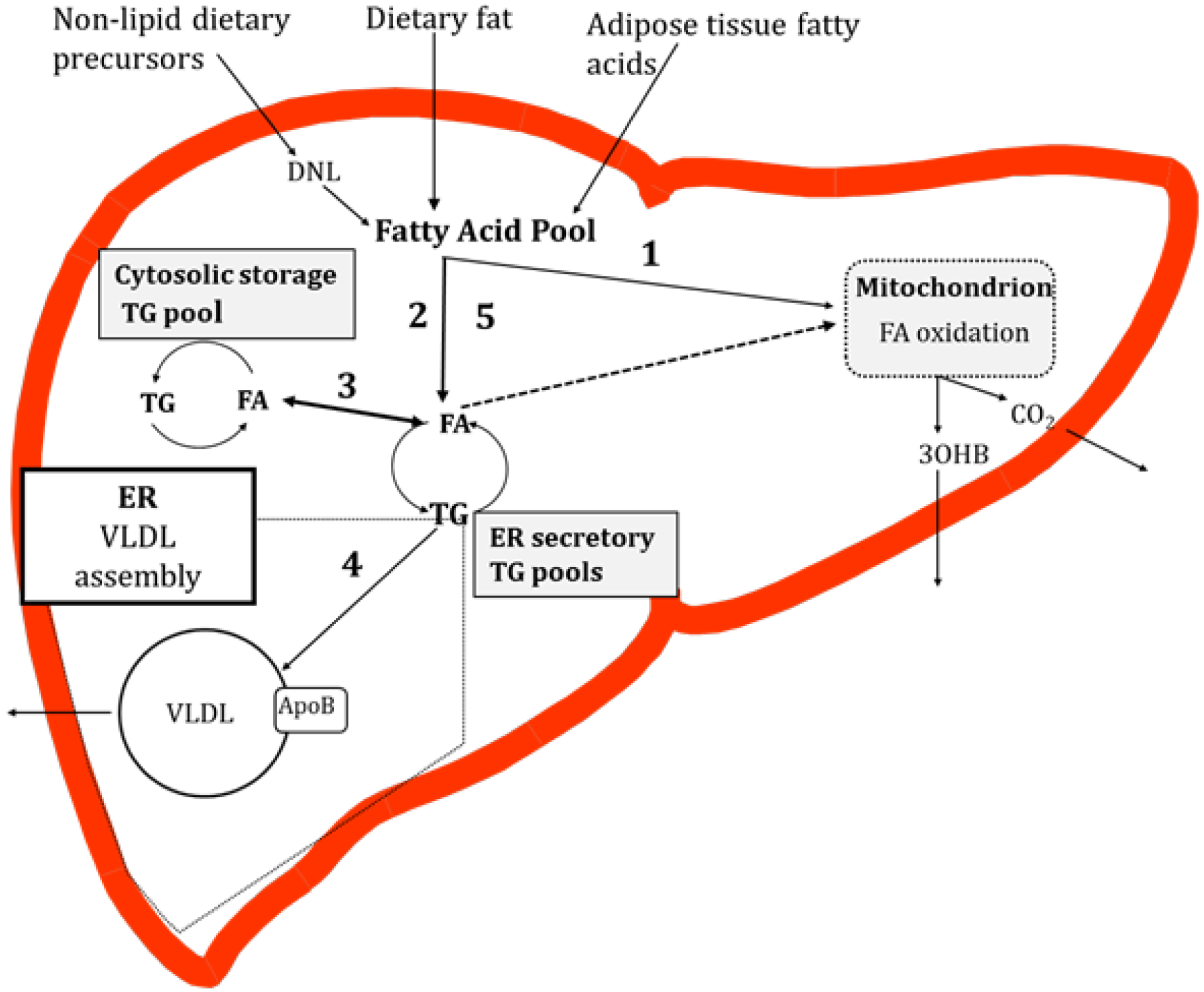
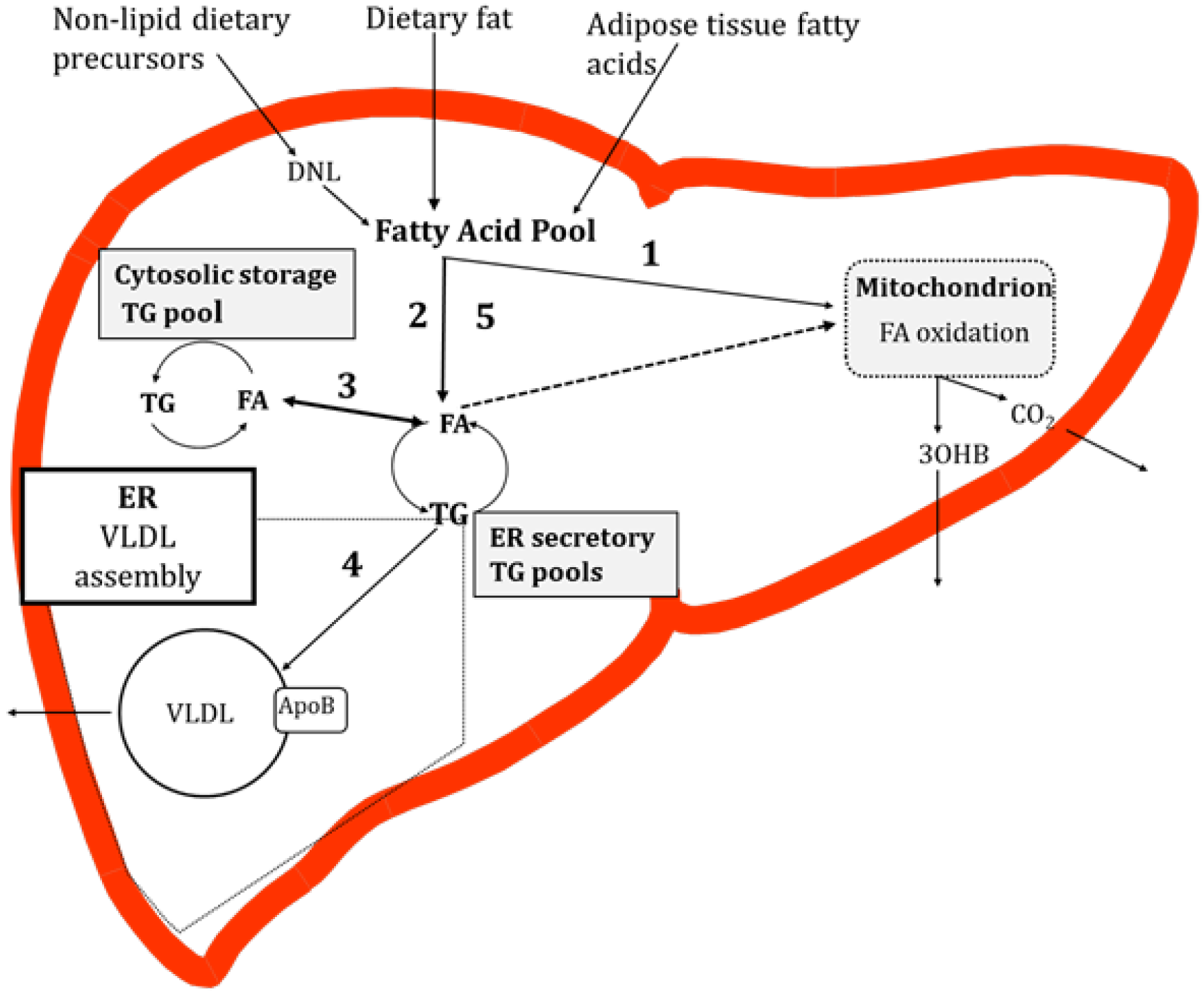
4. Potential Causes of Hepatic TG Accumulation
4.1. Contribution of Specific Fatty Acids to Liver TG
4.2. Trafficking of Dietary Fatty Acids to Liver TG
5. Associations between Dietary Fat and Liver TG
6. Intervention Studies: Evidence for Dietary Fat Altering Liver Fat Content

{kind=link}
| Ref | Subjects | Dsn | Lngt | Eng | Diet | Fat (%TE) | Measure Liver Fat | Baseline Liver Fat (%) | Change Liver Fat (%) |
|---|---|---|---|---|---|---|---|---|---|
| [70] | 10 F | X | 2 wk | Iso | LF | 16% Tot | MRS | 10 | ↓20 |
| BMI 33 | HF | 56% Tot | ↑35 | ||||||
| Age 43 | |||||||||
| [67] | 7 M/13 F | P | 4 wk | Iso | LF, low SFA, low GI | 23% Tot | MRS | 2.2 † | ↓0.44 |
| BMI 26.9 | 7% SFA | ||||||||
| Age 69 | |||||||||
| 6 M/9 F | HF, high SFA, high GI | 43% Tot | 1.2 † | ↑0.001 | |||||
| BMI 28.1 | 24% SFA | ||||||||
| Age 69 | |||||||||
| [69] | 20 M | P | 3 wk | Iso | LF vs. HF | 20% Tot | MRS | 2.2 | ↓13 |
| BMI 29 | 55% Tot | ↑17 | |||||||
| Age 34 | |||||||||
| [61] | 37M/8F | P | 8 wk | Iso | MUFA −ex | 42% Tot; 7% SFA; 5% PUFA; 27% MUFA −ex | MRS | 7.4 | ↓30 |
| T2D | |||||||||
| BMI 30 | MUFA +ex | 42% Tot; 7% SFA; 5% PUFA; 16% MUFA +ex | 11.6 | ↓22 | |||||
| Age 35–70 ‡ | |||||||||
| [60] | 67M/F | P | 10 wk | Iso | SFA vs. n-6 PUFA | ~42% Tot, ~20% SFA, ~4% PUFA | MRS | 3.2 | ↑8 |
| BMI 30.5 | ~39% Tot, ~10% SFA, ~13% PUFA | ↓26 | |||||||
| Age 30–65 y ‡ | |||||||||
| [62] | 5 M/13 F | P | 2 wk | Hypo | LC | 34% Tot | MRS | 19 | ↓26 |
| BMI 35 | LCHO | 59% Tot | 22 | ↓55 | |||||
| Age 45 y | |||||||||
| [63] | 35 M/135 F | P | 6 m | Hypo | LCHO | 30% Tot | MRS | 7.6 | ↓47 |
| BMI 32 | LF | ≤20% Tot | 9.6 | ↓42 | |||||
| Age 45 y | |||||||||
| [64] | 4 M/18 F | P | 11 wk | Hypo | LF vs. HF | 20% Tot | MRS | 11.2 | ↓>45 § |
| BMI 37 | 75% Tot | 12.4 | ↓>35 § | ||||||
| Age 44 y | |||||||||
| [71] | 9 M/17 F | I | 7 m | Hypo | LC | 30% Tot | MRS | 10.8 | ↓28 |
| BMI 32.4 | 10% SFA | ||||||||
| 10% MUFA | |||||||||
| Age 52y | 10% PUFA | ||||||||
| [66] | 39 M | P | 4 d | Hyper | HF | 60% Tot, 28% SFA | MRS | ~11 § | ↑86 |
| BMI 23 | HF/HFrc | 60% Tot, 3.5g Frc/kg FFM | ~12 § | ↑133 | |||||
| Age 24 y | |||||||||
| [68] | 15 M | I | 3 d | Hyper | HEHF | 69% Tot | MRS | 2.01 | ↑112 |
| BMI 23.4 | |||||||||
| Age 25 y | |||||||||
| [65] | 41 M/F | P | 7 wk | Hyper | SFA | 37% Tot, 17% SFA, 5% PUFA | MRI | 0.96 | ↑58 |
| BMI 18–27 ‡ | n-6 PUFA | 40% Tot, SFA 11%, PUFA 13% | 0.75 | ↑5 | |||||
| Age 20–38 y ‡ |
6.1. Iso-Caloric Diets
6.2. Hypo-Caloric Diets
6.3. Hyper-Caloric Diets
6.4. Supplementation Studies
7. Dietary Fat Alters Liver Fat: Potential Mechanisms
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frayn, K.N. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002, 45, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Yki-Jarvinen, H. Liver fat in the pathogenesis of insulin resistance and type 2 diabetes. Dig. Dis. 2010, 28, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Babin, P.J.; Gibbons, G.F. The evolution of plasma cholesterol: Direct utility or a “spandrel” of hepatic lipid metabolism? Prog. Lipid Res. 2009, 48, 73–91. [Google Scholar] [CrossRef]
- Diraison, F.; Beylot, M. Role of human liver lipogenesis and reesterification in triglycerides secretion and in ffa reesterification. Am. J. Physiol. 1998, 274, E321–E327. [Google Scholar]
- Sidossis, L.S.; Mittendorfer, B.; Walser, E.; Chinkes, D.; Wolfe, R.R. Hyperglycemia-induced inhibition of splanchnic fatty acid oxidation increases hepatic triacylglycerol secretion. Am. J. Physiol. 1998, 275, E798–E805. [Google Scholar] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.S.; He, Q. A current update on the rule of alternative and complementary medicine in the treatment of liver diseases. Evid. Based Complement. Altern. Med. 2013, 2013, 321234. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the AMERICAN Gastroenterological association. Am. J. Gastroenterol. 2012, 107, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Pais, R.; Bellentani, S.; Day, C.P.; Ratziu, V.; Loria, P.; Lonardo, A. From nafld in clinical practice to answers from guidelines. J. Hepatol. 2013, 59, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D. Dorothy hodgkin lecture 2012: Non-alcoholic fatty liver disease, insulin resistance and ectopic fat: A new problem in diabetes management. Diabet. Med. 2012, 29, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.M.; Brunt, E.M. Pathological features of fatty liver disease. Gastroenterology 2014, 147, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.B. Non-alcoholic fatty liver disease: The hepatic consequence of obesity and the metabolic syndrome. Proc. Nutr. Soc. 2010, 69, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Olufadi, R.; Bruce, K.D.; Cagampang, F.R.; Ahmed, M.H. Metabolic disturbances in non-alcoholic fatty liver disease. Clin. Sci. (Lond.) 2009, 116, 539–564. [Google Scholar] [CrossRef]
- Lonardo, A.; Sookoian, S.; Chonchol, M.; Loria, P.; Targher, G. Cardiovascular and systemic risk in nonalcoholic fatty liver disease—Atherosclerosis as a major player in the natural course of nafld. Curr. Pharm. Des. 2013, 19, 5177–5192. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Conte, C.; Magkos, F. Methods for assessing intrahepatic fat content and steatosis. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Galle, P.R. Animal models of non-alcoholic steatohepatitis: of Mice and man. Dig. Dis. 2010, 28, 247–254. [Google Scholar] [CrossRef] [PubMed]
- El-Badry, A.M.; Breitenstein, S.; Jochum, W.; Washington, K.; Paradis, V.; Rubbia-Brandt, L.; Puhan, M.A.; Slankamenac, K.; Graf, R.; Clavien, P.A.; et al. Assessment of hepatic steatosis by expert pathologists: The end of a gold standard. Ann. Surg. 2009, 250, 691–697. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- El-Badry, A.M.; Moritz, W.; Contaldo, C.; Tian, Y.; Graf, R.; Clavien, P.A. Prevention of reperfusion injury and microcirculatory failure in macrosteatotic mouse liver by omega-3 fatty acids. Hepatology 2007, 45, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Reeder, S.B.; Cruite, I.; Hamilton, G.; Sirlin, C.B. Quantitative assessment of liver fat with magnetic resonance imaging and spectroscopy. J. Magn. Reson. Imaging 2011, 34, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Pournik, O.; Alavian, S.M.; Ghalichi, L.; Seifizarei, B.; Mehrnoush, L.; Aslani, A.; Anjarani, S.; Eslami, S. Inter-observer and intra-observer agreement in pathological evaluation of non-alcoholic fatty liver disease suspected liver biopsies. Hepat. Mon. 2014, 14, e15167. [Google Scholar] [CrossRef] [PubMed]
- Vuppalanchi, R.; Unalp, A.; van Natta, M.L.; Cummings, O.W.; Sandrasegaran, K.E.; Hameed, T.; Tonascia, J.; Chalasani, N. Effects of liver biopsy sample length and number of readings on sampling variability in nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2009, 7, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Havel, R.J.; Kane, J.P.; Balasse, E.O.; Segel, N.; Basso, L.V. Splanchnic metabolism of free fatty acids and production of triglycerides of very low density lipoproteins in normotriglyceridemic and hypertriglyceridemic humans. J. Clin. Investig. 1970, 49, 2017–2035. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Fielding, B.A. Trafficking and partitioning of fatty acids: The transition from fasted to fed state. Clin. Lipidol. 2010, 5, 131–144. [Google Scholar] [CrossRef]
- Gibbons, G.F.; Bartlett, S.M.; Sparks, C.E.; Sparks, J.D. Extracellular fatty acids are not utilized directly for the synthesis of very-low-density lipoprotein in primary cultures of rat hepatocytes. Biochem. J. 1992, 287((Pt. 3)), 749–753. [Google Scholar] [PubMed]
- Gibbons, G.F.; Wiggins, D. Intracellular triacylglycerol lipase: Its role in the assembly of hepatic very-low-density lipoprotein (vldl). Adv. Enzym. Regul. 1995, 35, 179–198. [Google Scholar] [CrossRef]
- Gibbons, G.F. Assembly and secretion of hepatic very-low-density lipoprotein. Biochem. J. 1990, 268, 1–13. [Google Scholar] [PubMed]
- Zammit, V.A. Role of insulin in hepatic fatty acid partitioning: Emerging concepts. Biochem. J. 1996, 314((Pt. 1)), 1–14. [Google Scholar] [PubMed]
- Gibbons, G.F.; Islam, K.; Pease, R.J. Mobilisation of triacylglycerol stores. Biochim. Biophys. Acta 2000, 1483, 37–57. [Google Scholar] [CrossRef] [PubMed]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar]
- Ruge, T.; Hodson, L.; Cheeseman, J.; Dennis, A.L.; Fielding, B.A.; Humphreys, S.M.; Frayn, K.N.; Karpe, F. Fasted to fed trafficking of fatty acids in human adipose tissue reveals a novel regulatory step for enhanced fat storage. J. Clin. Endocrinol. Metab. 2009, 94, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Pot, G.K.; Prynne, C.J.; Roberts, C.; Olson, A.; Nicholson, S.K.; Whitton, C.; Teucher, B.; Bates, B.; Henderson, H.; Pigott, S.; et al. National diet and nutrition survey: Fat and fatty acid intake from the first year of the rolling programme and comparison with previous surveys. Br. J. Nutr. 2012, 107, 405–415. [Google Scholar] [CrossRef]
- Bergouignan, A.; Rudwill, F.; Simon, C.; Blanc, S. Physical inactivity as the culprit of metabolic inflexibility: Evidence from bed-rest studies. J. Appl. Physiol. 2011, 111, 1201–1210. [Google Scholar] [CrossRef]
- Enjoji, M.; Nakamuta, M. Is the control of dietary cholesterol intake sufficiently effective to ameliorate nonalcoholic fatty liver disease? World J. Gastroenterol. 2010, 16, 800–803. [Google Scholar]
- Papandreou, D.; Karabouta, Z.; Rousso, I. Are dietary cholesterol intake and serum cholesterol levels related to nonalcoholic fatty liver disease in obese children? Cholesterol 2012, 2012, 572820. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the united states population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Senturk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc. Nutr. Soc. 2013, 72, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Frayn, K.N. Hepatic fatty acid partitioning. Curr. Opin. Lipidol. 2011, 22, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (nafld). Prog. Lipid Res. 2009, 48, 1–26. [Google Scholar] [CrossRef]
- Scorletti, E.; Byrne, C.D. Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2013, 33, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Does fructose consumption contribute to non-alcoholic fatty liver disease? Clin. Res. Hepatol. Gastroenterol. 2012, 36, 554–560. [Google Scholar] [CrossRef]
- Yasutake, K.; Kohjima, M.; Kotoh, K.; Nakashima, M.; Nakamuta, M.; Enjoji, M. Dietary habits and behaviors associated with nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Barrows, B.R.; Parks, E.J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Bickerton, A.S.; McQuaid, S.E.; Roberts, R.; Karpe, F.; Frayn, K.N.; Fielding, B.A. The contribution of splanchnic fat to vldl triglyceride is greater in insulin-resistant than insulin-sensitive men and women: Studies in the postprandial state. Diabetes 2007, 56, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Vedala, A.; Wang, W.; Neese, R.A.; Christiansen, M.P.; Hellerstein, M.K. Delayed secretory pathway contributions to vldl-triglycerides from plasma nefa, diet, and de novo lipogenesis in humans. J. Lipid Res. 2006, 47, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Carey, P.E.; Snaar, J.E.; Deelchand, D.K.; Cook, D.B.; Neely, R.D.; English, P.T.; Firbank, M.J.; Morris, P.G.; Taylor, R.; et al. Real-time assessment of postprandial fat storage in liver and skeletal muscle in health and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E789–E797. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Borggrefe, J.; Barbaresko, J.; Groth, G.; Jacobs, G.; Siegert, S.; Lieb, W.; Muller, M.J.; Bosy-Westphal, A.; Heller, M.; et al. Dietary patterns associated with magnetic resonance imaging-determined liver fat content in a general population study. Am. J. Clin. Nutr. 2014, 99, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Kuk, J.L.; Davidson, L.E.; Hudson, R.; Kilpatrick, K.; Bacskai, K.; Ross, R. Association between dietary fat intake, liver fat, and insulin sensitivity in sedentary, abdominally obese, older men. Appl. Physiol. Nutr. Metab. 2008, 33, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Mollard, R.C.; Senechal, M.; MacIntosh, A.C.; Hay, J.; Wicklow, B.A.; Wittmeier, K.D.; Sellers, E.A.; Dean, H.J.; Ryner, L.; Berard, L.; et al. Dietary determinants of hepatic steatosis and visceral adiposity in overweight and obese youth at risk of type 2 diabetes. Am. J. Clin. Nutr. 2014, 99, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Sathiaraj, E.; Chutke, M.; Reddy, M.Y.; Pratap, N.; Rao, P.N.; Reddy, D.N.; Raghunath, M. A case-control study on nutritional risk factors in non-alcoholic fatty liver disease in indian population. Eur. J. Clin. Nutr. 2011, 65, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Bingham, S.A. The dietary assessment of individuals; methods, accuracy, new techniques and recommendations. Nutr. Abstr. Rev. (Ser. A) 1987, 57, 705–742. [Google Scholar]
- Biro, G.; Hulshof, K.F.; Ovesen, L.; Amorim Cruz, J.A. Selection of methodology to assess food intake. Eur. J. Clin. Nutr. 2002, 56 (Suppl. 2), S25–S32. [Google Scholar] [CrossRef]
- Hodson, L.; Skeaff, C.M.; Fielding, B.A. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res. 2008, 47, 348–380. [Google Scholar] [CrossRef] [PubMed]
- Bjermo, H.; Iggman, D.; Kullberg, J.; Dahlman, I.; Johansson, L.; Persson, L.; Berglund, J.; Pulkki, K.; Basu, S.; Uusitupa, M.; et al. Effects of n-6 pufas compared with sfas on liver fat, lipoproteins, and inflammation in abdominal obesity: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 95, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Bozzetto, L.; Prinster, A.; Annuzzi, G.; Costagliola, L.; Mangione, A.; Vitelli, A.; Mazzarella, R.; Longobardo, M.; Mancini, M.; Vigorito, C.; et al. Liver fat is reduced by an isoenergetic mufa diet in a controlled randomized study in type 2 diabetic patients. Diabetes Care 2012, 35, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Baker, J.A.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-term weight loss and hepatic triglyceride reduction: Evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr. 2011, 93, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Haufe, S.; Engeli, S.; Kast, P.; Bohnke, J.; Utz, W.; Haas, V.; Hermsdorf, M.; Mahler, A.; Wiesner, S.; Birkenfeld, A.L.; et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011, 53, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.; Reeds, D.N.; Finck, B.N.; Mayurranjan, S.M.; Patterson, B.W.; Klein, S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology 2009, 136, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Rosqvist, F.; Iggman, D.; Kullberg, J.; Cedernaes, J.; Johansson, H.E.; Larsson, A.; Johansson, L.; Ahlstrom, H.; Arner, P.; Dahlman, I.; et al. Overfeeding polyunsaturated and saturated fat causes distinct effects on liver and visceral fat accumulation in humans. Diabetes 2014, 63, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Sobrecases, H.; Le, K.A.; Bortolotti, M.; Schneiter, P.; Ith, M.; Kreis, R.; Boesch, C.; Tappy, L. Effects of short-term overfeeding with fructose, fat and fructose plus fat on plasma and hepatic lipids in healthy men. Diabetes Metab. 2010, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Bayer-Carter, J.L.; Arbuckle, M.D.; Tidwell, J.M.; Richards, T.L.; Craft, S. Beneficial effect of a weight-stable, low-fat/low-saturated fat/low-glycaemic index diet to reduce liver fat in older subjects. Br. J. Nutr. 2013, 109, 1096–1104. [Google Scholar] [CrossRef]
- Van der Meer, R.W.; Hammer, S.; Lamb, H.J.; Frolich, M.; Diamant, M.; Rijzewijk, L.J.; de Roos, A.; Romijn, J.A.; Smit, J.W. Effects of short-term high-fat, high-energy diet on hepatic and myocardial triglyceride content in healthy men. J. Clin. Endocrinol. Metab. 2008, 93, 2702–2708. [Google Scholar] [CrossRef] [PubMed]
- Van Herpen, N.A.; Schrauwen-Hinderling, V.B.; Schaart, G.; Mensink, R.P.; Schrauwen, P. Three weeks on a high-fat diet increases intrahepatic lipid accumulation and decreases metabolic flexibility in healthy overweight men. J. Clin. Endocrinol. Metab. 2011, 96, E691–E695. [Google Scholar] [CrossRef] [PubMed]
- Westerbacka, J.; Lammi, K.; Hakkinen, A.M.; Rissanen, A.; Salminen, I.; Aro, A.; Yki-Jarvinen, H. Dietary fat content modifies liver fat in overweight nondiabetic subjects. J. Clin. Endocrinol. Metab. 2005, 90, 2804–2809. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Hakkarainen, A.; Lundbom, N.; Yki-Jarvinen, H. Effects of dietary interventions on liver volume in humans. Obesity (Silver Spring) 2014, 22, 989–995. [Google Scholar] [CrossRef]
- Hodson, L.; McQuaid, S.E.; Karpe, F.; Frayn, K.N.; Fielding, B.A. Differences in partitioning of meal fatty acids into blood lipid fractions: A comparison of linoleate, oleate, and palmitate. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E64–E71. [Google Scholar] [CrossRef] [PubMed]
- Cussons, A.J.; Watts, G.F.; Mori, T.A.; Stuckey, B.G. Omega-3 fatty acid supplementation decreases liver fat content in polycystic ovary syndrome: A randomized controlled trial employing proton magnetic resonance spectroscopy. J. Clin. Endocrinol. Metab. 2009, 94, 3842–3848. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Bhatia, L.; McCormick, K.G.; Clough, G.F.; Nash, K.; Hodson, L.; Moyses, H.E.; Calder, P.C.; Byrne, C.D.; On Behalf of the W.S.I. Effects of purified eicosapentaenoic and docosahexaenoic acids in non-alcoholic fatty liver disease: Results from the *WELCOME study. Hepatology 2014, 60, 1211–1221. [Google Scholar] [CrossRef]
- Vega, G.L.; Chandalia, M.; Szczepaniak, L.S.; Grundy, S.M. Effects of n-3 fatty acids on hepatic triglyceride content in humans. J. Investig. Med. 2008, 56, 780–785. [Google Scholar] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Tariq, Z.; Green, C.J.; Hodson, L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (nash)? Liver Int. 2014, 34, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Fielding, B.A. Stearoyl-coa desaturase: Rogue or innocent bystander? Prog. Lipid Res. 2013, 52, 15–42. [Google Scholar] [CrossRef]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Karpe, F.; Hodson, L. Caution on the interpretation of plasma fatty acid composition as a proxy marker for scd1 activity: Particular implications for using the 16:1/16:0 ratio in qtl studies involving hyperlipidemic patients. Arterioscler. Thromb. Vasc. Biol. 2008, 28, e152. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S.; et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446.e2. [Google Scholar] [CrossRef] [PubMed]
- Koopman, K.E.; Caan, M.W.; Nederveen, A.J.; Pels, A.; Ackermans, M.T.; Fliers, E.; la Fleur, S.E.; Serlie, M.J. Hypercaloric diets with increased meal frequency, but not meal size, increase intrahepatic triglycerides: A randomized controlled trial. Hepatology 2014, 60, 545–553. [Google Scholar] [CrossRef] [PubMed]
- DeLany, J.P.; Windhauser, M.M.; Champagne, C.M.; Bray, G.A. Differential oxidation of individual dietary fatty acids in humans. Am. J. Clin. Nutr. 2000, 72, 905–911. [Google Scholar] [PubMed]
- Moussavi, N.; Gavino, V.; Receveur, O. Could the quality of dietary fat, and not just its quantity, be related to risk of obesity? Obesity (Silver Spring) 2008, 16, 7–15. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Green, C.J.; Hodson, L. The Influence of Dietary Fat on Liver Fat Accumulation. Nutrients 2014, 6, 5018-5033. https://doi.org/10.3390/nu6115018
Green CJ, Hodson L. The Influence of Dietary Fat on Liver Fat Accumulation. Nutrients. 2014; 6(11):5018-5033. https://doi.org/10.3390/nu6115018
Chicago/Turabian StyleGreen, Charlotte J., and Leanne Hodson. 2014. "The Influence of Dietary Fat on Liver Fat Accumulation" Nutrients 6, no. 11: 5018-5033. https://doi.org/10.3390/nu6115018
APA StyleGreen, C. J., & Hodson, L. (2014). The Influence of Dietary Fat on Liver Fat Accumulation. Nutrients, 6(11), 5018-5033. https://doi.org/10.3390/nu6115018