
Mechanisms Linking Obesity, Insulin Resistance, and Alzheimer’s Disease: Effects of Polyphenols and Omega-3 Polyunsaturated Fatty Acids

 , , and
, , and 
Abstract

1. Introduction
1.1. Obesity, Chronic Inflammation, and AD

{kind=link}
{kind=link}
{kind=link}
| Study | Subjects | Findings | Ref |
|---|---|---|---|
| Launceston study Case–control study | 50 elderly patients with AD, male and female | Obesity (BMI > 30 kg/m2), abdominal obesity (waist-to-hip ratio (WHR): >0.8 (women), >0.9 (men)) were associated with AD | [50] |
| New York longitudinal study | Male and female, without dementia at baseline, 145 AD patients at the end | WHR was correlated with higher risk of AD | [51] |
| The HUNT Study Longitudinal study | 654 male and female patients with AD | Significant associations between AD and obesity | [52] |
| AD Neuroimaging Initiative and the Cardiovascular Health Study-Cognition Study. Observational study | 700 male patients with MCI or AD | A higher BMI was associated with brain volume deficits in frontal, temporal, parietal, and occipital lobes measured by MRI | [53] |
| AD Model | Intervention | Findings | Ref |
|---|---|---|---|
| APP/PS1 male and female | 45% HFD 8 and 12 months | HFD resulted in increased food intake, body weight and glucose intolerance, inflammation, and tau and Aβ in the cortex | [54] |
| Tg2576 AD male | 60% HFD 7 months | HFD resulted in IR, higher BW, higher Aβ plaques, and GSK | [55] |
| Tg2576 AD female | 42% HFD 4–16 weeks | Increased food intake, body weight, and Aβ content in the brain | [56] |
| APP/PS1 TG male | High-sucrose diet 10 to 20 or 28 weeks | Higher BW and fat mass, leptin resistance, hyperinsulinemia | [57] |
| 3xTg-CD AD male and female | 60% HFD 2 months | HFD aggravated brain atrophy and memory impairments, impaired brain structural integrity | [58] |
| AppNL/NL knock-in male | 60% HFD 10 months | HFD impaired hippocampal potentiation | [59] |
| APP/PS1 male and female | 42% HFD 17 months | Poorer memory performance, impaired social interactions, and increases in Aβ monomers and plaques with HFD | [60] |
| AppNL−F/NL−F male mice | 40% HFD 14 months | Decreased the expression of the Aβ-binding protein transthyretin and Aβ deposition in hippocampus | [61] |
1.2. Diabetes and AD
| Study | Subjects | Findings | Reference |
|---|---|---|---|
| The Rotterdam Study Prospective population-based cohort study | n = 6370 male and female 2.1 years follow-up | T2DM increased the risk of dementia and AD in elderly individuals; insulin-treated patients had the highest risk and T2DM-attributable risk of dementia by 8.8% | [75] |
| Observational study | n = 1262 male and female 4.3 years follow-up | Diabetes was associated with AD and cognitive impairment without dementia | [76] |
| Longitudinal cohort study | n = 824 male and female 5.5 years follow-up | T2DM was associated with developing AD, and the risk of incidence of AD was 65% higher in those with diabetes | [77] |
| Honolulu–Asia Aging Study Prospective cohort study | n = 2574 male | T2DM is a risk factor for AD, with the association being stronger among APOEε4 carriers | [78] |
| Israeli Ischemic Heart Disease Study Observational, longitudinal study | n = 1892 male | Confirmed diabetes as a risk factor for dementia | [79] |
| AD Model/Sex/Age | Intervention | Findings | Ref |
|---|---|---|---|
| B6-STZ/male/8–12 weeks | STZ-induced diabetic | Downregulation of LRP1 expression and its function of transporting Aβ across the BBB in diabetic mice | [80] |
| APP-STZ/male/8 months | STZ-induced diabetic | Mice with both APP overexpression and diabetes showed insulin receptor reduction and more phosphorylated tau and Aβ plaques | [81] |
| 5xFAD-STZ/-/5 months | STZ-induced diabetic | STZ-induced insulin-deficient diabetes exacerbated Aβ accumulation by elevating expression levels of the β-secretase enzyme BACE1 and its substrate APP in the 5xFAD mouse model of AD | [82] |
| SAMP8/-/11 months | Metformin 20 mg/kgor 200 mg/kg subcutaneously (8 weeks) | Metformin improved learning and memory, decreased Aβ and tau proteins | [83] |
| 3xTg Psen1tm1Mpm/male/12 months | - | Proteomics data showed altered O-GlcNAcylation levels in TG mice | [84] |
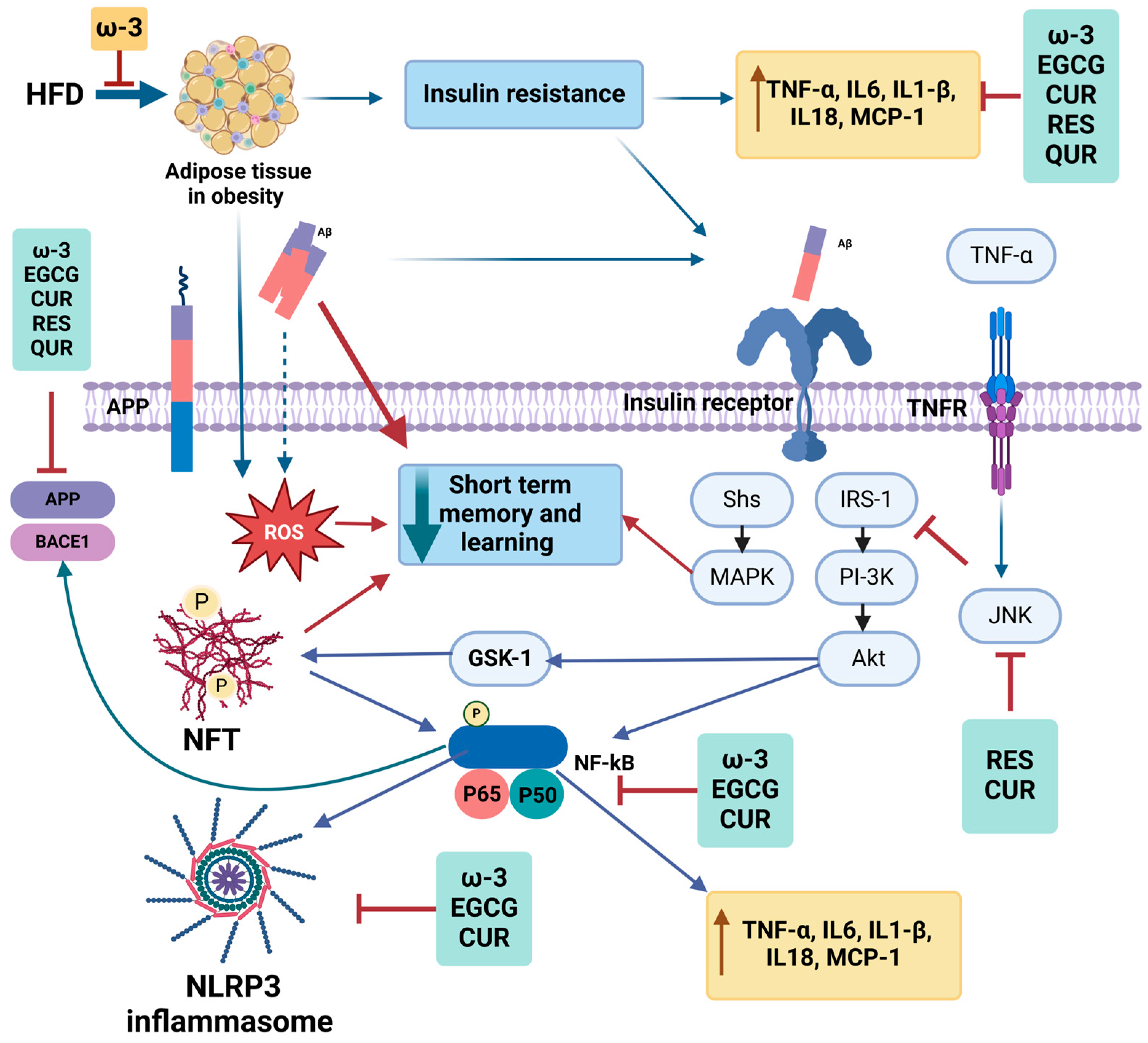
1.3. Insulin Signaling in the Brain in AD
1.4. Adipokines and AD
1.5. Role of Bioactive Food Compounds in AD
1.6. Resveratrol
1.7. Epigallocatechin-3-Gallate (EGCG)
1.8. Curcumin
1.9. Quercetin
1.10. Omega-3 Polyunsaturated Fatty Acids
| AD Model/Sex/Age | Intervention | Findings | Ref |
|---|---|---|---|
| Polyphenols | |||
| Wistar rats- STZ/-/8–10 months | Aβ hippocampal injection -Resveratrol: 25 mg/kg BW | ↑ SIRT1 expression, MDA, ↓ Memory impairment, GSH, SOD, IL-1β, IL-16 | [182] |
| B6/male/8 weeks | Resveratrol: 100 mg/kg BW and Metformin 250 mg/kg BW | ↑ AMPK phosphorylation, mTOR activation by resveratrol and combo ↓ BDNF | [183] |
| Tg2576 male AD mice and hUC-MSCs transplanted mice/-/5 months | Resveratrol/200 mg/kg BW | Shower synergistic effects in neuroprotection ↓ SIRT1, neural apoptosis ↑ p53, p21, neurogenesis, cognition | [184] |
| Tg19959/ male/4 months | Resveratrol: 300 mg/kg BW | ↓ Reduction in plaque counts and plaque burden in medial cortex, GSH | [105] |
| 3xTg-AD/male/7 months | Resveratrol: 481 mg/kg BW and exercise training | ↓ NF-κB, GFAP, PARP, Aβ, BACE1 by RES or combo ↑ BDNF, NGF, synaptophysin, PSD-95 RES or combo | [185] |
| Tg6799/male/6 months | Resveratrol: 60 mg/kg BW | ↓ Cognitive impairment, Aβ42, BACE1, APP No effect on SIRT1 | [186] |
| 5xFAD/male/10 months | Trans-resveratrol 1 g/kg BW and HFD | ↓ Cognitive impairment, APP, tau, BACE1, and Aβ plaques compared to HFD | [187] |
| Tg2576/male and female/14 months | EGCG: 50 mg/kg BW | ↓ Aβ pathology, cognitive impairments | [188] |
| SAMP8/-/- | EGCG: 5 and 15 mg/kg BW | ↓ Aβ1–42 accumulations, tau phosphorylation, and BACE-1 | [189] |
| Sprague-Dawley (SD) rats injected with Aβ/male/- | EGCG: 100, 250 and 625 mg/kg BW | ↓ Aβ, tau phosphorylation, BACE-1 ↑ Learning and memory | [190] |
| APP/PS1/-/6 months | EGCG: 50 mg/kg BW | ↓ Aβ, Iba ↑ synapsin-1, synaptophysin, PSD93 and GluR1, IL-10 and IL-13 | [191] |
| Swiss Albino injected with Aβ/female/18–22 months | Free curcumin: 50 mg/kg BW, Lipid core nano capsuled curcumin: 10 mg/kg | ↓ Aβ, NF-κB, TNFα, IL-1β, IL-6, IFN-γ Similar results in the high dose and nanocapsule low dose | [192] |
| 3xTg-AD/-/16 months | Quercetin 100 mg/kg BW | ↓ Aβ, and tau hyperphosphorylation ↑ cognitive function | [193] |
| SAMP8 mice/male/7 months | Quercetin-loaded nanoparticles: 25 mg/kg BW | ↑ cognition and memory impairments | [154] |
| 3xTg/-/21–24 months | Quercetin 25 mg/kg BW | ↓ Aβ, tau level, GFAP ↑ spinal learning | [194] |
| ω-3 fatty acids | |||
| SAMP8/male/12 months | EPA and DHA 10 g/kg BW | DHA and EPA ↓ PS1 and BACE1, soluble Aβ40 DHA ↓ cognitive impairment | [195] |
| B6 /male/2.5 months | Lysophosphatidylcholine-EPA for 2 weeks | ↑ the amount of EPA and DHA in brain ↓ BDNF, CREB, and 5-HT1A, TNFα ↑ phosphorylation of CREB | [196] |
| SAMP8/male/9 months | 200 mg/kg BW of DHA, 200 mg/kg BW of EPA Oral gavage | ↓ p-JNK, PHF-1 by DHA | [197] |
| APP/PS1/male and female/18 months | HFD supplemented with 36 g/kg BW EPA | ↓ Aβ-40 compared to HFD in the serum of the male group, ↓ serum leptin and ↑ serum adiponectin | [168] |
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AKT | Protein kinase B |
| APP | Amyloid precursor protein |
| Aβ | Amyloid-beta |
| BACE1 | Beta-secretase 1 |
| BBB | Blood–brain barrier |
| CUR | Curcumin |
| DIO | Diet-induced obese |
| EGCG | Epigallocatechin-3-gallate |
| FDA | Food and Drug Administration |
| GSK3 | Glycogen synthase kinase 3 |
| HFD | High-fat diet |
| IDE | Insulin-degrading enzyme |
| IL18 | Interleukin 18 |
| IL1β | Interleukin 1 beta |
| IL6 | Interleukin 6 |
| IR | Insulin resistance |
| IRS-1 | Insulin receptor substrate |
| JNK | c-Jun N-terminal kinase |
| LRP | Low-density lipoprotein receptor-related protein 1 |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| NFT | Neurofibrillary tangle |
| NF-kB | Nuclear factor kappa |
| NLRP3 | NOD-, LRR-, and pyrin domain-containing protein 3 |
| PI3K | Phosphatidylinositol 3-kinase |
| PUFAs | Polyunsaturated fatty acids |
| QUR | Quercetin |
| RES | Resveratrol |
| ROS | Reactive oxygen species |
| SIRT1 | Silent information regulator 1 |
| T2DM | Type two diabetes mellitus |
| TNFα | Tumor necrosis factor-alpha |
| TNFR | Tumor necrosis factor receptor |
| ω-3 | Omega-3 fatty acid |
References
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-onset Alzheimer’s disease: What is missing in research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar]
- Tan, J.Z.A.; Gleeson, P.A. The role of membrane trafficking in the processing of amyloid precursor protein and production of amyloid peptides in Alzheimer’s disease. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2019, 1861, 697–712. [Google Scholar]
- de la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 2020, 9, 35–66. [Google Scholar]
- Felstead, C.; Perkins, L.; Stott, J.; Hui, E.K.; Spector, A. A systematic literature review of group-based training interventions for informal carers: Impact on the behavioural and psychological symptoms of dementia (BPSD). Aging Ment. Health 2023, 27, 1246–1255. [Google Scholar]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar]
- Audronyte, E.; Sutnikiene, V.; Pakulaite-Kazliene, G.; Kaubrys, G. Brief Test of Olfactory Dysfunction Based on Diagnostic Features of Specific Odors in Early-Stage Alzheimer Disease. Med. Sci. Monit. 2023, 29, e940363. [Google Scholar] [CrossRef]
- Akhgarjand, C.; Hashemi, R.; Amini, M.; Rasekhi, H.; Farazandeh, D.; Etesam, F.; Rasooli, A.; Houjaghani, H.; Faezi, S.; Vahabi, Z. The relationship between micronutrients and cognitive ability in an elderly population with mild cognitive impairment and Alzheimer’s disease: A cross-sectional study. BMC Neurol. 2024, 24, 416. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s disease. Neurotherapeutics 2021, 19, 173–185. [Google Scholar]
- Selkoe, D.J. Deciphering the genesis and fate of amyloid β-protein yields novel therapies for Alzheimer disease. J. Clin. Investig. 2002, 110, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Seubert, P.; Oltersdorf, T.; Lee, M.G.; Barbour, R.; Blomquist, C.; Davis, D.L.; Bryant, K.; Fritz, L.C.; Galasko, D.; Thal, L.J. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature 1993, 361, 260–263. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease β-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Paolicelli, R.C. Microglia-mediated synapse loss in Alzheimer’s disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef]
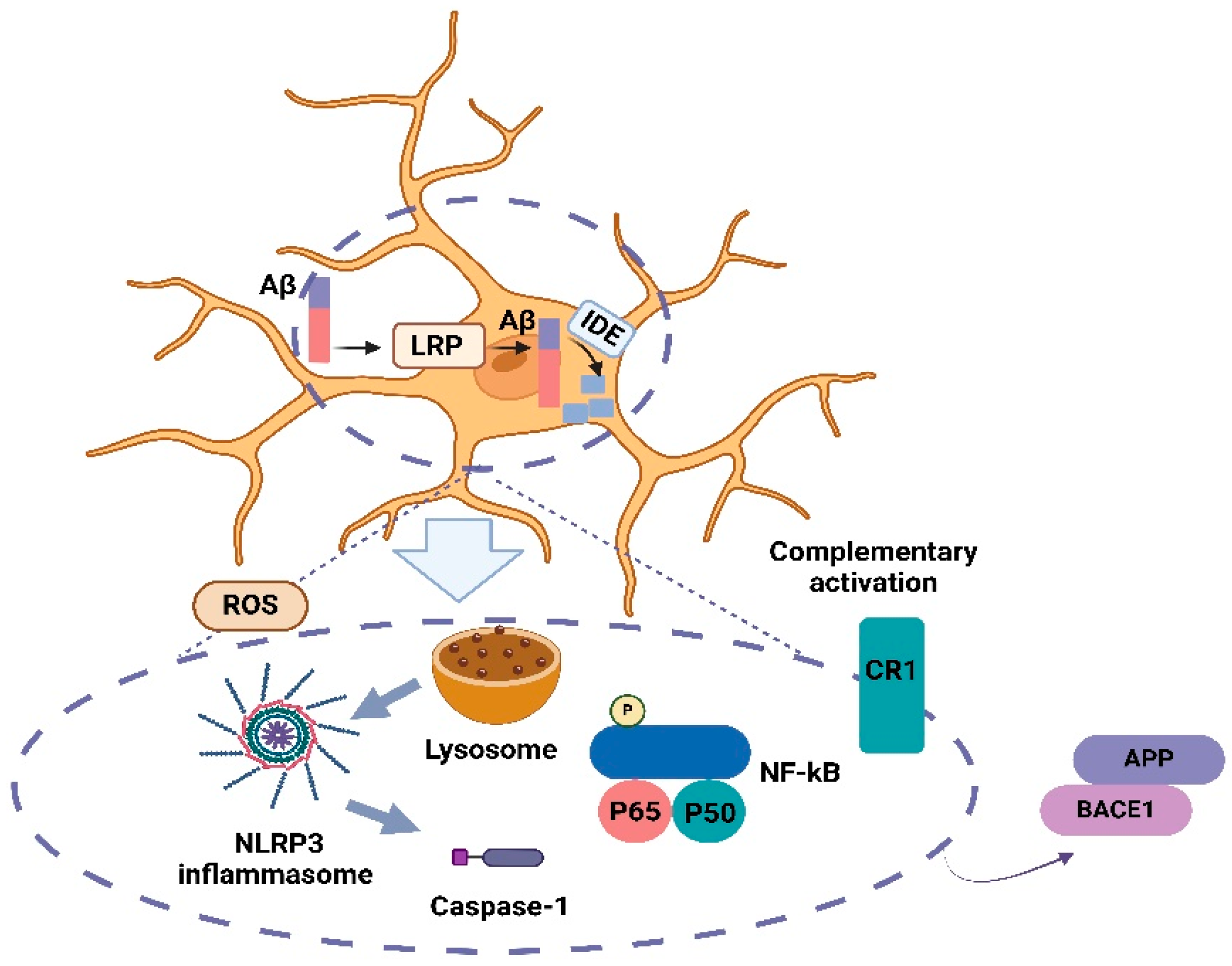
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, B.; Storck, S.E.; Reekmans, S.M.; Lechat, B.; Gordts, P.L.S.M.; Pradier, L.; Pietrzik, C.U.; Roebroek, A.J.M. LRP1 Has a Predominant Role in Production over Clearance of Aβ in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7234–7245. [Google Scholar] [CrossRef] [PubMed]
- Tanokashira, D.; Motoki, K.; Minegishi, S.; Hosaka, A.; Mamada, N.; Tamaoka, A.; Okada, T.; Lakshmana, M.K.; Araki, W. LRP1 Downregulates the Alzheimer’s β-Secretase BACE1 by Modulating Its Intraneuronal Trafficking. eNeuro 2015, 2, ENEURO.0006-15.2015. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflammation 2014, 11, 48. [Google Scholar] [CrossRef]
- Rogers, J.; Lue, L.-F. Microglial chemotaxis, activation, and phagocytosis of amyloid β-peptide as linked phenomena in Alzheimer’s disease. Neurochem. Int. 2001, 39, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Edison, P. Cardiometabolic risk factors and neurodegeneration: A review of the mechanisms underlying diabetes, obesity and hypertension in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2024, 95, 581. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Olloquequi, J.; Ettcheto, M.; Busquets, O.; Sánchez-López, E.; Cano, A.; Espinosa-Jiménez, T.; García, M.L.; Beas-Zarate, C.; Casadesús, G.; et al. The Involvement of Peripheral and Brain Insulin Resistance in Late Onset Alzheimer’s Dementia. Front. Aging Neurosci. 2019, 11, 236. [Google Scholar] [CrossRef]
- Götz, J.; Bodea, L.-G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef] [PubMed]
- McKean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 3168. [Google Scholar] [CrossRef]
- Qian, Z.; Li, Y.; Ye, K. Advancements and challenges in mouse models of Alzheimer’s disease. Trends Mol. Med. 2024, 30, 1152–1164. [Google Scholar] [CrossRef]
- Martin-Jiménez, C.A.; Gaitán-Vaca, D.M.; Echeverria, V.; González, J.; Barreto, G.E. Relationship Between Obesity, Alzheimer’s Disease, and Parkinson’s Disease: An Astrocentric View. Mol. Neurobiol. 2017, 54, 7096–7115. [Google Scholar] [CrossRef]
- Fröhlich, M.; Imhof, A.; Berg, G.; Hutchinson, W.L.; Pepys, M.B.; Boeing, H.; Muche, R.; Brenner, H.; Koenig, W. Association between C-reactive protein and features of the metabolic syndrome: A population-based study. Diabetes Care 2000, 23, 1835–1839. [Google Scholar]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar]
- Hotamisligil, G.S. Foundations of immunometabolism and implications for metabolic health and disease. Immunity 2017, 47, 406–420. [Google Scholar]
- Heilbronn, L.K.; Campbell, L.V. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr. Pharm. Des. 2008, 14, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [PubMed]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases–an update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar]
- Whitmer, R.; Gustafson, D.; Barrett-Connor, E.; Haan, M.; Gunderson, E.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar]
- Zhuang, Q.-S.; Zheng, H.; Gu, X.-D.; Shen, L.; Ji, H.-F. Detecting the genetic link between Alzheimer’s disease and obesity using bioinformatics analysis of GWAS data. Oncotarget 2017, 8, 55915. [Google Scholar]
- Zhuang, Q.-S.; Meng, L.; Wang, Z.; Shen, L.; Ji, H.-F. Associations Between Obesity and Alzheimer’s Disease: Multiple Bioinformatic Analyses. J. Alzheimer’s Dis. 2021, 80, 271–281. [Google Scholar] [CrossRef]
- Nuzzo, D.; Galizzi, G.; Amato, A.; Terzo, S.; Picone, P.; Cristaldi, L.; Mulè, F.; Di Carlo, M. Regular intake of pistachio mitigates the deleterious effects of a high fat-diet in the brain of obese mice. Antioxidants 2020, 9, 317. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Marino Gammazza, A.; Cappello, F.; Mulè, F.; Di Carlo, M. Insulin resistance as common molecular denominator linking obesity to Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [PubMed]
- Parimisetty, A.; Dorsemans, A.-C.; Awada, R.; Ravanan, P.; Diotel, N.; Lefebvre d’Hellencourt, C. Secret talk between adipose tissue and central nervous system via secreted factors—An emerging frontier in the neurodegenerative research. J. Neuroinflammation 2016, 13, 1–13. [Google Scholar]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. Embo J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Barber, A.J.; Del Genio, C.L.; Swain, A.B.; Pizzi, E.M.; Watson, S.C.; Tapiavala, V.N.; Zanazzi, G.J.; Gaur, A.B. Age, sex and Alzheimer’s disease: A longitudinal study of 3xTg-AD mice reveals sex-specific disease trajectories and inflammatory responses mirrored in postmortem brains from Alzheimer’s patients. Alzheimers Res. Ther. 2024, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.J.; Ma, Y.H.; Bi, Y.L.; Shen, X.N.; Hou, X.H.; Cao, X.P.; Ou, Y.N.; Zhao, B.; Dong, Q.; Tan, L.; et al. Metabolically healthy obesity and lipids may be protective factors for pathological changes of Alzheimer’s disease in cognitively normal adults. J. Neurochem. 2021, 157, 834–845. [Google Scholar] [CrossRef]
- Lee, J.Y.; Han, K.; Han, E.; Kim, G.; Cho, H.; Kim, K.J.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Brayne, C.; et al. Risk of Incident Dementia According to Metabolic Health and Obesity Status in Late Life: A Population-Based Cohort Study. J. Clin. Endocrinol. Metab. 2019, 104, 2942–2952. [Google Scholar] [CrossRef]
- Al-Delaimy, W.K.; von Muhlen, D.; Barrett-Connor, E. Insulinlike growth factor-1, insulinlike growth factor binding protein-1, and cognitive function in older men and women. J. Am. Geriatr. Soc. 2009, 57, 1441–1446. [Google Scholar] [CrossRef]
- Lee, E.B. Obesity, leptin, and Alzheimer’s disease. Ann. New York Acad. Sci. 2011, 1243, 15–29. [Google Scholar] [CrossRef]
- Nourhashémi, F.; Andrieu, S.; Gillette-Guyonnet, S.; Reynish, E.; Albarède, J.L.; Grandjean, H.; Vellas, B. Is there a relationship between fat-free soft tissue mass and low cognitive function? Results from a study of 7,105 women. J. Am. Geriatr. Soc. 2002, 50, 1796–1801. [Google Scholar] [CrossRef]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. Obesity, Abdominal Obesity and Alzheimer Disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 173–176. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Cheng, D.; Tang, M.X.; Schupf, N.; Mayeux, R. Central Obesity in the Elderly is Related to Late-onset Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 101–105. [Google Scholar] [CrossRef]
- Rasmussen Eid, H.; Rosness, T.A.; Bosnes, O.; Salvesen, Ø.; Knutli, M.; Stordal, E. Smoking and Obesity as Risk Factors in Frontotemporal Dementia and Alzheimer’s Disease: The HUNT Study. Dement. Geriatr. Cogn. Disord. Extra 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Ho, A.J.; Raji, C.A.; Becker, J.T.; Lopez, O.L.; Kuller, L.H.; Hua, X.; Lee, S.; Hibar, D.; Dinov, I.D.; Stein, J.L. Obesity is linked with lower brain volume in 700 AD and MCI patients. Neurobiol. Aging 2010, 31, 1326–1339. [Google Scholar] [PubMed]
- Walker, J.M.; Dixit, S.; Saulsberry, A.C.; May, J.M.; Harrison, F.E. Reversal of high fat diet-induced obesity improves glucose tolerance, inflammatory response, β-amyloid accumulation and cognitive decline in the APP/PSEN1 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2017, 100, 87–98. [Google Scholar] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar]
- Kohjima, M.; Sun, Y.; Chan, L. Increased food intake leads to obesity and insulin resistance in the tg2576 Alzheimer’s disease mouse model. Endocrinology 2010, 151, 1532–1540. [Google Scholar] [PubMed]
- Yeh, S.H.-H.; Shie, F.-S.; Liu, H.-K.; Yao, H.-H.; Kao, P.-C.; Lee, Y.-H.; Chen, L.-M.; Hsu, S.-M.; Chao, L.-J.; Wu, K.-W. A high-sucrose diet aggravates Alzheimer’s disease pathology, attenuates hypothalamic leptin signaling, and impairs food-anticipatory activity in APPswe/PS1dE9 mice. Neurobiol. Aging 2020, 90, 60–74. [Google Scholar]
- Rollins, C.P.E.; Gallino, D.; Kong, V.; Ayranci, G.; Devenyi, G.A.; Germann, J.; Chakravarty, M.M. Contributions of a high-fat diet to Alzheimer’s disease-related decline: A longitudinal behavioural and structural neuroimaging study in mouse models. NeuroImage Clin. 2019, 21, 101606. [Google Scholar] [CrossRef]
- Salas, I.H.; Weerasekera, A.; Ahmed, T.; Callaerts-Vegh, Z.; Himmelreich, U.; D’Hooge, R.; Balschun, D.; Saido, T.C.; De Strooper, B.; Dotti, C.G. High fat diet treatment impairs hippocampal long-term potentiation without alterations of the core neuropathological features of Alzheimer disease. Neurobiol. Dis. 2018, 113, 82–96. [Google Scholar] [CrossRef]
- Bracko, O.; Vinarcsik, L.K.; Cruz Hernández, J.C.; Ruiz-Uribe, N.E.; Haft-Javaherian, M.; Falkenhain, K.; Ramanauskaite, E.M.; Ali, M.; Mohapatra, A.; Swallow, M.A.; et al. High fat diet worsens Alzheimer’s disease-related behavioral abnormalities and neuropathology in APP/PS1 mice, but not by synergistically decreasing cerebral blood flow. Sci. Rep. 2020, 10, 9884. [Google Scholar] [CrossRef]
- Mazzei, G.; Ikegami, R.; Abolhassani, N.; Haruyama, N.; Sakumi, K.; Saito, T.; Saido, T.C.; Nakabeppu, Y. A high-fat diet exacerbates the Alzheimer’s disease pathology in the hippocampus of the AppNL−F/NL−F knock-in mouse model. Aging Cell 2021, 20, e13429. [Google Scholar] [CrossRef]
- Akter, K.; Lanza, E.A.; Martin, S.A.; Myronyuk, N.; Rua, M.; Raffa, R.B. Diabetes mellitus and Alzheimer’s disease: Shared pathology and treatment? Br. J. Clin. Pharmacol. 2011, 71, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kroner, Z. The Relationship between Alzheimer’s Disease and Diabetes: Type 3 Diabetes. Altern. Med. Rev. 2009, 14, 373. [Google Scholar] [PubMed]
- Huang, C.C.; Chung, C.M.; Leu, H.B.; Lin, L.Y.; Chiu, C.C.; Hsu, C.Y.; Chiang, C.H.; Huang, P.H.; Chen, T.J.; Lin, S.J.; et al. Diabetes mellitus and the risk of Alzheimer’s disease: A nationwide population-based study. PLoS ONE 2014, 9, e87095. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef]
- Murray, R.K.; Granner, D.K.; Mayes, P.A.; Rodwell, V.W. Harper’s Illustrated Biochemistry; Mcgraw-Hill: New York, NY, USA, 2014. [Google Scholar]
- Haque, R.; Nazir, A. Insulin-degrading enzyme: A link between Alzheimer’s and type 2 diabetes mellitus. CNS Neurol. Disord. -Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2014, 13, 259–264. [Google Scholar]
- Ebrahimpour, S.; Zakeri, M.; Esmaeili, A. Crosstalk between obesity, diabetes, and Alzheimer’s disease: Introducing quercetin as an effective triple herbal medicine. Ageing Res. Rev. 2020, 62, 101095. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, Y.; Ho, G.; Koike, W.; Sugama, S.; Takenouchi, T.; Waragai, M.; Wei, J.; Sekiyama, K.; Hashimoto, M. Combined immunotherapy with “anti-insulin resistance” therapy as a novel therapeutic strategy against neurodegenerative diseases. NPJ Park. Dis. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Rad, S.K.; Arya, A.; Karimian, H.; Madhavan, P.; Rizwan, F.; Koshy, S.; Prabhu, G. Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: Link between type 2 diabetes and Alzheimer’s disease. Drug Des. Devel. Ther. 2018, 12, 3999–4021. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, N.-Q.; Yan, F.; Jin, H.; Zhou, S.-Y.; Shi, J.-S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Yamaguchi, K.; Matsui, K.; Sano, T.; Kubota, T.; Hashimoto, T.; Mano, A.; Yamada, K.; Matsuo, Y.; Kubota, N.; et al. Differential effects of diet- and genetically-induced brain insulin resistance on amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Tran, A.; Ishimwe, E.; Denner, L.; Dave, N.; Oddo, S.; Dineley, K.T. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 1–13. [Google Scholar] [CrossRef]
- Schwartz, S.S.; Herman, M.E.; Tun, M.T.H.; Barone, E.; Butterfield, D.A. The double life of glucose metabolism: Brain health, glycemic homeostasis, and your patients with type 2 diabetes. BMC Med. 2024, 22, 582. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.; van Harskamp, F.; Pols, H.; Hofman, A.; Breteler, M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar] [PubMed]
- Luchsinger, J.A.; Tang, M.-X.; Stern, Y.; Shea, S.; Mayeux, R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am. J. Epidemiol. 2001, 154, 635–641. [Google Scholar]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [PubMed]
- Beeri, M.S.; Goldbourt, U.; Silverman, J.M.; Noy, S.; Schmeidler, J.; Ravona-Springer, R.; Sverdlick, A.; Davidson, M. Diabetes mellitus in midlife and the risk of dementia three decades later. Neurology 2004, 63, 1902–1907. [Google Scholar]
- Hong, H.; Liu, L.P.; Liao, J.M.; Wang, T.S.; Ye, F.Y.; Wu, J.; Wang, Y.Y.; Wang, Y.; Li, Y.Q.; Long, Y.; et al. Downregulation of LPR1 at the blood–brain barrier in streptozotocin-induced diabetic mice. Neuropharmacology 2009, 56, 1054–1059. [Google Scholar] [CrossRef]
- Jolivalt, C.G.; Hurford, R.; Lee, C.A.; Dumaop, W.; Rockenstein, E.; Masliah, E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp. Neurol. 2010, 223, 422–431. [Google Scholar] [CrossRef]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer’s disease. PLoS ONE 2012, 7, e32792. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Roesler, E.; Niehoff, M.L.; Roby, D.A.; McKee, A.; Morley, J.E. Metformin improves learning and memory in the SAMP8 mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 68, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Sharma, N.; Barone, E.; Lanzillotta, C.; Castellani, A.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Butterfield, D.A.; Gaetani, S.; et al. Proteomic identification of altered protein O-GlcNAcylation in a triple transgenic mouse model of Alzheimer’s disease. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3309–3321. [Google Scholar] [CrossRef]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xiang, Z.; Haroutunian, V.; Buxbaum, J.D.; Stetka, B.; Pasinetti, G.M. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiol. Aging 2007, 28, 824–830. [Google Scholar] [CrossRef]
- Miller, B.W.; Willett, K.C.; Desilets, A.R. Rosiglitazone and pioglitazone for the treatment of Alzheimer’s disease. Ann. Pharmacother. 2011, 45, 1416–1424. [Google Scholar] [CrossRef]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARγ agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Flynn, E.R. Insulin resistance contributes to aberrant stress responses in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2004, 17, 500–506. [Google Scholar] [CrossRef]
- Forny-Germano, L.; De Felice, F.G.; Vieira, M.N.d.N. The role of leptin and adiponectin in obesity-associated cognitive decline and Alzheimer’s disease. Front. Neurosci. 2019, 12, 1027. [Google Scholar]
- Mejido, D.C.; Andrade, J.; Vieira, M.N.; Ferreira, S.T.; De Felice, F.G. Insulin and leptin as potential cognitive enhancers in metabolic disorders and Alzheimer’s disease. Neuropharmacology 2020, 171, 108115. [Google Scholar] [PubMed]
- Ma, J.; Zhang, W.; Wang, H.-F.; Wang, Z.-X.; Jiang, T.; Tan, M.-S.; Yu, J.-T.; Tan, L. Peripheral blood adipokines and insulin levels in patients with Alzheimer’s disease: A replication study and meta-analysis. Curr. Alzheimer Res. 2016, 13, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Elias, C.F.; Fukuda, M.; Williams, K.W.; Berglund, E.D.; Holland, W.L.; Cho, Y.-R.; Chuang, J.-C.; Xu, Y.; Choi, M.; et al. Direct Insulin and Leptin Action on Pro-opiomelanocortin Neurons Is Required for Normal Glucose Homeostasis and Fertility. Cell Metab. 2010, 11, 286–297. [Google Scholar] [CrossRef]
- Li, X.L.; Aou, S.; Oomura, Y.; Hori, N.; Fukunaga, K.; Hori, T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 2002, 113, 607–615. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Hsu, H.-C.; Kao, P.-C.; Shiao, Y.-J.; Yeh, S.H.-H.; Shie, F.-S.; Hsu, S.-M.; Yeh, C.-W.; Liu, H.-K.; Yang, S.-B. Augmented insulin and leptin resistance of high fat diet-fed APPswe/PS1dE9 transgenic mice exacerbate obesity and glycemic dysregulation. Int. J. Mol. Sci. 2018, 19, 2333. [Google Scholar] [CrossRef]
- Elfeky, M.; Yoneshiro, T.; Okamatsu-Ogura, Y.; Kimura, K. Adiponectin suppression of late inflammatory mediator, HMGB1-induced cytokine expression in RAW264 macrophage cells. J. Biochem. 2017, 163, 143–153. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, H.; Xie, Y.; Ding, Y.; Kong, D.; Yu, H. Research Progress on Alzheimer’s Disease and Resveratrol. Neurochem. Res. 2020, 45, 989–1006. [Google Scholar] [CrossRef]
- Le, K.; Daliv, E.C.; Wu, S.; Qian, F.; Ali, A.I.; Yu, D.; Guo, Y. SIRT1-regulated HMGB1 release is partially involved in TLR4 signal transduction: A possible anti-neuroinflammatory mechanism of resveratrol in neonatal hypoxic-ischemic brain injury. Int. Immunopharmacol. 2019, 75, 105779. [Google Scholar] [CrossRef]
- Huang, J.; Huang, N.; Xu, S.; Luo, Y.; Li, Y.; Jin, H.; Yu, C.; Shi, J.; Jin, F. Signaling mechanisms underlying inhibition of neuroinflammation by resveratrol in neurodegenerative diseases. J. Nutr. Biochem. 2021, 88, 108552. [Google Scholar]
- Hou, Y.; Zhang, Y.; Mi, Y.; Wang, J.; Zhang, H.; Xu, J.; Yang, Y.; Liu, J.; Ding, L.; Yang, J. A Novel Quinolyl-Substituted Analogue of Resveratrol Inhibits LPS-Induced Inflammatory Responses in Microglial Cells by Blocking the NF-κB/MAPK Signaling Pathways. Mol. Nutr. Food Res. 2019, 63, 1801380. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Wang, X.; Wang, J.; Ye, Y.; Zhang, M.; Lang, Y.; Ding, S. Dihydro-resveratrol ameliorates NLRP3 inflammasome-mediated neuroinflammation via Bnip3-dependent mitophagy in Alzheimer’s disease. Br. J. Pharmacol. 2025, 182, 1005–1024. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.-L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflammation 2017, 14, 1. [Google Scholar]
- Zhu, C.W.; Grossman, H.; Neugroschl, J.; Parker, S.; Burden, A.; Luo, X.; Sano, M. A randomized, double-blind, placebo-controlled trial of resveratrol with glucose and malate (RGM) to slow the progression of Alzheimer’s disease: A pilot study. Alzheimers Dement 2018, 4, 609–616. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Cottart, C.H.; Nivet-Antoine, V.; Laguillier-Morizot, C.; Beaudeux, J.L. Resveratrol bioavailability and toxicity in humans. Mol. Nutr. Food Res. 2010, 54, 7–16. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Bartra, C.; Yuan, Y.; Vuraić, K.; Valdés-Quiroz, H.; Garcia-Baucells, P.; Slevin, M.; Pastorello, Y.; Suñol, C.; Sanfeliu, C. Resveratrol Activates Antioxidant Protective Mechanisms in Cellular Models of Alzheimer’s Disease Inflammation. Antioxidants 2024, 13, 177. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, W.; Wang, Z.; Wang, Z.; Jin, X.; Xu, J.; Bai, L.; Li, Y.; Cui, J.; Cai, L. Resveratrol attenuates testicular apoptosis in type 1 diabetic mice: Role of Akt-mediated Nrf2 activation and p62-dependent Keap1 degradation. Redox Biol. 2018, 14, 609–617. [Google Scholar]
- Ozpak, L.; Bağca, B.G. Neuroprotective effects of resveratrol through modulation of PI3K/Akt/GSK-3β pathway and metalloproteases. IUBMB Life 2024, 76, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [PubMed]
- Rashet, A.; Abdi, A.; Barari, A. Synergistic Role of Aerobic Training and Resveratrol on AMPK/PGC1-α/SIRT1 Pathway in the Hippocampus of Rats with Alzheimer's Disease. J. Arch. Mil. Med. 2024, 12, e144281. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Yazdi, H.S.; Samarghandian, S. The protective effects of green tea catechins in the management of neurodegenerative diseases: A review. Curr. Drug Discov. Technol. 2019, 16, 57–65. [Google Scholar]
- de la Torre, R.; de Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar]
- Loftis, J.M.; Wilhelm, C.J.; Huckans, M. Effect of epigallocatechin gallate supplementation in schizophrenia and bipolar disorder: An 8-week, randomized, double-blind, placebo-controlled study. Ther. Adv. Psychopharmacol. 2013, 3, 21–27. [Google Scholar] [CrossRef]
- Valverde-Salazar, V.; Ruiz-Gabarre, D.; García-Escudero, V. Alzheimer’s Disease and Green Tea: Epigallocatechin-3-Gallate as a Modulator of Inflammation and Oxidative Stress. Antioxidants 2023, 12, 1460. [Google Scholar] [CrossRef]
- Khalifa, M.K.A.; Abdel-Sattar, S.A.; Amin, O.M.; Kohaf, N.A.; Zaky, H.S.; Abd El-Fattah, M.A.; Mohammed, K.H.A.; Badawi, N.M.; Mansoor, I.; Eassa, H.A. Effectiveness of epigallocatechin gallate nanoparticles on the in-vivo treatment of Alzheimer’s disease in a rat/mouse model: A systematic review. Daru 2024, 32, 319–337. [Google Scholar] [CrossRef]
- Lv, L.; Yang, F.; Li, H.; Yuan, J. Brain-targeted co-delivery of β-amyloid converting enzyme 1 shRNA and epigallocatechin-3-gallate by multifunctional nanocarriers for Alzheimer’s disease treatment. IUBMB Life 2020, 72, 1819–1829. [Google Scholar] [CrossRef]
- Cheng-Chung Wei, J.; Huang, H.-C.; Chen, W.-J.; Huang, C.-N.; Peng, C.-H.; Lin, C.-L. Epigallocatechin gallate attenuates amyloid β-induced inflammation and neurotoxicity in EOC 13.31 microglia. Eur. J. Pharmacol. 2016, 770, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J. Neurosci. 2005, 25, 8807–8814. [Google Scholar]
- He, M.; Liu, M.-Y.; Wang, S.; Tang, Q.-S.; Yao, W.-F.; Zhao, H.-S.; Wei, M.-J. Research on EGCG improving the degenerative changes of the brain in AD model mice induced with chemical drugs. Zhong Yao Cai = Zhongyaocai = J. Chin. Med. Mater. 2012, 35, 1641–1644. [Google Scholar]
- Lin, C.-L.; Chen, T.-F.; Chiu, M.-J.; Way, T.-D.; Lin, J.-K. Epigallocatechin gallate (EGCG) suppresses β-amyloid-induced neurotoxicity through inhibiting c-Abl/FE65 nuclear translocation and GSK3β activation. Neurobiol. Aging 2009, 30, 81–92. [Google Scholar]
- Babaei, F.G.; Saburi, E.; Forouzanfar, F.; Asgari, M.; Keshavarzi, Z.; Hajali, V. Effect of epigallocatechin-3-gallate (EGCG) on cognitive functioning and the expression of APP and BDNF in the hippocampus of rats with streptozotocin -induced Alzheimer-like disease. Biochem. Biophys. Rep. 2025, 41, 101930. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Abdollahi, E.; Nikfar, B.; Chaichian, S.; Ekhlasi-Hundrieser, M. Curcumin as a potential modulator of M1 and M2 macrophages: New insights in atherosclerosis therapy. Heart Fail. Rev. 2019, 24, 399–409. [Google Scholar] [PubMed]
- Meng, N.; Gong, Y.; Zhang, J.; Mu, X.; Song, Z.; Feng, R.; Zhang, H. A novel curcumin-loaded nanoparticle restricts atherosclerosis development and promotes plaques stability in apolipoprotein E deficient mice. J. Biomater. Appl. 2019, 33, 946–954. [Google Scholar] [CrossRef]
- Zhang, S.; Zou, J.; Li, P.; Zheng, X.; Feng, D. Curcumin Protects against Atherosclerosis in Apolipoprotein E-Knockout Mice by Inhibiting Toll-like Receptor 4 Expression. J. Agric. Food Chem. 2018, 66, 449–456. [Google Scholar] [CrossRef]
- Edwards, R.L.; Luis, P.B.; Varuzza, P.V.; Joseph, A.I.; Presley, S.H.; Chaturvedi, R.; Schneider, C. The anti-inflammatory activity of curcumin is mediated by its oxidative metabolites. J. Biol. Chem. 2017, 292, 21243–21252. [Google Scholar]
- Sundar Dhilip Kumar, S.; Houreld, N.N.; Abrahamse, H. Therapeutic potential and recent advances of curcumin in the treatment of aging-associated diseases. Molecules 2018, 23, 835. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, N.; Takahashi, Y.; Amakusa, Y.; Tanno, Y.; Tuji, Y.; Niwa, H.; Murakami, N.; Krishna, U. Effects of turmeric on Alzheimer’s disease with behavioral and psychological symptoms of dementia. Ayu 2012, 33, 499. [Google Scholar] [CrossRef]
- Rainey-Smith, S.R.; Brown, B.M.; Sohrabi, H.R.; Shah, T.; Goozee, K.G.; Gupta, V.B.; Martins, R.N. Curcumin and cognition: A randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br. J. Nutr. 2016, 115, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Siddarth, P.; Li, Z.; Miller, K.J.; Ercoli, L.; Emerson, N.D.; Martinez, J.; Wong, K.P.; Liu, J.; Merrill, D.A.; et al. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am. J. Geriatr. Psychiatry 2018, 26, 266–277. [Google Scholar] [CrossRef]
- DiSilvestro, R.A.; Joseph, E.; Zhao, S.; Bomser, J. Diverse effects of a low dose supplement of lipidated curcumin in healthy middle aged people. Nutr. J. 2012, 11, 79. [Google Scholar] [CrossRef]
- Zheng, K.; Dai, X.; Wu, X.; Wei, Z.; Fang, W.; Zhu, Y.; Zhang, J.; Chen, X. Curcumin ameliorates memory decline via inhibiting BACE1 expression and β-Amyloid pathology in 5× FAD transgenic mice. Mol. Neurobiol. 2017, 54, 1967–1977. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; De Simone, A.; Andrisano, V.; Bisignano, P.; Bisi, A.; Gobbi, S.; Rampa, A.; Fato, R.; Bergamini, C.; Perez, D.I.; et al. Versatility of the Curcumin Scaffold: Discovery of Potent and Balanced Dual BACE-1 and GSK-3β Inhibitors. J. Med. Chem. 2016, 59, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, X.; Wang, C.; Teng, Z.; Li, Y. Curcumin Decreases Hyperphosphorylation of Tau by Down-Regulating Caveolin-1/GSK-3β in N2a/APP695swe Cells and APP/PS1 Double Transgenic Alzheimer’s Disease Mice. Am. J. Chin. Med. 2017, 45, 1667–1682. [Google Scholar] [CrossRef]
- Feng, H.-L.; Fan, H.; Dang, H.-Z.; Chen, X.-P.; Ren, Y.; Yang, J.-D.; Wang, P.-W. Neuroprotective effect of curcumin to Aβ of double transgenic mice with Alzheimer’s disease. Zhongguo Zhong Yao Za Zhi = Zhongguo Zhongyao Zazhi = China J. Chin. Mater. Medica 2014, 39, 3846–3849. [Google Scholar]
- Wang, P.; Su, C.; Feng, H.; Chen, X.; Dong, Y.; Rao, Y.; Ren, Y.; Yang, J.; Shi, J.; Tian, J. Curcumin regulates insulin pathways and glucose metabolism in the brains of APPswe/PS1dE9 mice. Int. J. Immunopathol. Pharmacol. 2017, 30, 25–43. [Google Scholar] [CrossRef]
- Wang, H.-M.; Zhao, Y.-X.; Zhang, S.; Liu, G.-D.; Kang, W.-Y.; Tang, H.-D.; Ding, J.-Q.; Chen, S.-D. PPARγ agonist curcumin reduces the amyloid-β-stimulated inflammatory responses in primary astrocytes. J. Alzheimer’s Dis. 2010, 20, 1189–1199. [Google Scholar]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [PubMed]
- Garcia-Alloza, M.; Robbins, E.M.; Zhang-Nunes, S.X.; Purcell, S.M.; Betensky, R.A.; Raju, S.; Prada, C.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 2006, 24, 516–524. [Google Scholar]
- Das, T.K.; Chakrabarti, S.K.; Zulkipli, I.N.; Abdul Hamid, M.R.W. Curcumin Ameliorates the Impaired Insulin Signaling Involved in the Pathogenesis of Alzheimer’s Disease in Rats. J. Alzheimer’s Dis. Rep. 2019, 3, 59–70. [Google Scholar] [CrossRef]
- Zaplatic, E.; Bule, M.; Shah, S.Z.A.; Uddin, M.S.; Niaz, K. Molecular mechanisms underlying protective role of quercetin in attenuating Alzheimer’s disease. Life Sci. 2019, 224, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Luo, G.; Tang, Y.; Yao, P. Quercetin and iron metabolism: What we know and what we need to know. Food Chem. Toxicol. 2018, 114, 190–203. [Google Scholar]
- Matsuzaki, K.; Noguch, T.; Wakabayashi, M.; Ikeda, K.; Okada, T.; Ohashi, Y.; Hoshino, M.; Naiki, H. Inhibitors of amyloid β-protein aggregation mediated by GM1-containing raft-like membranes. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2007, 1768, 122–130. [Google Scholar]
- Sharma, V.; Mishra, M.; Ghosh, S.; Tewari, R.; Basu, A.; Seth, P.; Sen, E. Modulation of interleukin-1β mediated inflammatory response in human astrocytes by flavonoids: Implications in neuroprotection. Brain Res. Bull. 2007, 73, 55–63. [Google Scholar]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2019, 10, 59. [Google Scholar] [CrossRef]
- Srinivasan, P.; Vijayakumar, S.; Kothandaraman, S.; Palani, M. Anti-diabetic activity of quercetin extracted from Phyllanthus emblica L. fruit: In silico and in vivo approaches. J. Pharm. Anal. 2018, 8, 109–118. [Google Scholar] [CrossRef]
- Yang, D.K.; Kang, H.-S. Anti-diabetic effect of cotreatment with quercetin and resveratrol in streptozotocin-induced diabetic rats. Biomol. Ther. 2018, 26, 130. [Google Scholar]
- Ho, L.; Ferruzzi, M.G.; Janle, E.M.; Wang, J.; Gong, B.; Chen, T.Y.; Lobo, J.; Cooper, B.; Wu, Q.L.; Talcott, S.T. Identification of brain-targeted bioactive dietary quercetin-3-O-glucuronide as a novel intervention for Alzheimer’s disease. FASEB J. 2013, 27, 769–781. [Google Scholar]
- Moreno, L.C.G.e.I.; Puerta, E.; Suárez-Santiago, J.E.; Santos-Magalhães, N.S.; Ramirez, M.J.; Irache, J.M. Effect of the oral administration of nanoencapsulated quercetin on a mouse model of Alzheimer’s disease. Int. J. Pharm. 2017, 517, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, J.; Zhong, L.; Wang, N.; Yang, L.; Liu, C.-C.; Li, H.; Wang, X.; Zhou, Y.; Zhang, Y.; et al. Quercetin stabilizes apolipoprotein E and reduces brain Aβ levels in amyloid model mice. Neuropharmacology 2016, 108, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Aliaga, K.; Bermejo-Bescós, P.; Benedí, J.; Martín-Aragón, S. Quercetin and rutin exhibit antiamyloidogenic and fibril-disaggregating effects in vitro and potent antioxidant activity in APPswe cells. Life Sci. 2011, 89, 939–945. [Google Scholar]
- Hu, T.; Lu, X.-Y.; Shi, J.-J.; Liu, X.-Q.; Chen, Q.-B.; Wang, Q.; Chen, Y.-B.; Zhang, S.-J. Quercetin protects against diabetic encephalopathy via SIRT1/NLRP3 pathway in db/db mice. J. Cell. Mol. Med. 2020, 24, 3449–3459. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Kiliaan, A.J. Fatty acids, lipid metabolism and Alzheimer pathology. Eur. J. Pharmacol. 2008, 585, 176–196. [Google Scholar] [PubMed]
- Jicha, G.A.; Markesbery, W.R. Omega-3 fatty acids: Potential role in the management of early Alzheimer’s disease. Clin. Interv. Aging 2010, 5, 45–61. [Google Scholar] [CrossRef]
- Cleland, L.G.; James, M.J.; Proudman, S.M. Fish oil: What the prescriber needs to know. Arthritis Res. Ther. 2006, 8, 202. [Google Scholar] [CrossRef]
- Stillwell, W.; Wassall, S.R. Docosahexaenoic acid: Membrane properties of a unique fatty acid. Chem. Phys. Lipids 2003, 126, 1–27. [Google Scholar]
- Bouyanfif, A.; Jayarathne, S.; Koboziev, I.; Moustaid-Moussa, N. The Nematode Caenorhabditis elegans as a Model Organism to Study Metabolic Effects of ω-3 Polyunsaturated Fatty Acids in Obesity. Adv. Nutr. 2019, 10, 165–178. [Google Scholar] [PubMed]
- Spencer, M.; Finlin, B.S.; Unal, R.; Zhu, B.; Morris, A.J.; Shipp, L.R.; Lee, J.; Walton, R.G.; Adu, A.; Erfani, R. Omega-3 fatty acids reduce adipose tissue macrophages in human subjects with insulin resistance. Diabetes 2013, 62, 1709–1717. [Google Scholar]
- Thota, R.N.; Acharya, S.H.; Garg, M.L. Curcumin and/or omega-3 polyunsaturated fatty acids supplementation reduces insulin resistance and blood lipids in individuals with high risk of type 2 diabetes: A randomised controlled trial. Lipids Health Dis. 2019, 18, 31. [Google Scholar]
- Lim, G.P.; Calon, F.; Morihara, T.; Yang, F.; Teter, B.; Ubeda, O.; Salem, N.; Frautschy, S.A.; Cole, G.M. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005, 25, 3032–3040. [Google Scholar]
- Pahlavani, M.; Razafimanjato, F.; Ramalingam, L.; Kalupahana, N.S.; Moussa, H.; Scoggin, S.; Moustaid-Moussa, N. Eicosapentaenoic acid regulates brown adipose tissue metabolism in high-fat-fed mice and in clonal brown adipocytes. J. Nutr. Biochem. 2017, 39, 101–109. [Google Scholar]
- Kalupahana, N.S.; Claycombe, K.; Newman, S.J.; Stewart, T.; Siriwardhana, N.; Matthan, N.; Lichtenstein, A.H.; Moustaid-Moussa, N. Eicosapentaenoic acid prevents and reverses insulin resistance in high-fat diet-induced obese mice via modulation of adipose tissue inflammation. J. Nutr. 2010, 140, 1915–1922. [Google Scholar] [PubMed]
- Yavari, M.; Ramalingam, L.; Harris, B.N.; Kahathuduwa, C.N.; Chavira, A.; Biltz, C.; Mounce, L.; Maldonado, K.A.; Scoggin, S.; Zu, Y.; et al. Eicosapentaenoic Acid Protects against Metabolic Impairments in the APPswe/PS1dE9 Alzheimer’s Disease Mouse Model. J. Nutr. 2023, 153, 1038–1051. [Google Scholar] [CrossRef]
- Fotuhi, M.; Mohassel, P.; Yaffe, K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: A complex association. Nat. Rev. Neurol. 2009, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Conquer, J.A.; Tierney, M.C.; Zecevic, J.; Bettger, W.J.; Fisher, R.H. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids 2000, 35, 1305–1312. [Google Scholar] [CrossRef]
- Kyle, D.; Schaefer, E.; Patton, G.; Beiser, A. Low serum docosahexaenoic acid is a significant risk factor for Alzheimer’s dementia. Lipids 1999, 34, S245. [Google Scholar] [CrossRef]
- Heude, B.; Ducimetière, P.; Berr, C. Cognitive decline and fatty acid composition of erythrocyte membranes—The EVA Study. Am. J. Clin. Nutr. 2003, 77, 803–808. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Wilson, R.S. Fish Consumption and Cognitive Decline With Age in a Large Community Study. Arch. Neurol. 2005, 62, 1849–1853. [Google Scholar] [CrossRef] [PubMed]
- van Gelder, B.M.; Tijhuis, M.; Kalmijn, S.; Kromhout, D. Fish consumption, n−3 fatty acids, and subsequent 5-y cognitive decline in elderly men: The Zutphen Elderly Study. Am. J. Clin. Nutr. 2007, 85, 1142–1147. [Google Scholar] [CrossRef]
- Freund Levi, Y.; Vedin, I.; Cederholm, T.; Basun, H.; Faxen Irving, G.; Eriksdotter, M.; Hjorth, E.; Schultzberg, M.; Vessby, B.; Wahlund, L.O. Transfer of omega-3 fatty acids across the blood–brain barrier after dietary supplementation with a docosahexaenoic acid-rich omega-3 fatty acid preparation in patients with A lzheimer’s disease: The O meg AD study. J. Intern. Med. 2014, 275, 428–436. [Google Scholar] [CrossRef]
- Koivisto, H.; Grimm, M.O.; Rothhaar, T.L.; Berkecz, R.; Lütjohann, D.; Giniatullina, R.; Takalo, M.; Miettinen, P.O.; Lahtinen, H.-M.; Giniatullin, R.; et al. Special lipid-based diets alleviate cognitive deficits in the APPswe/PS1dE9 transgenic mouse model of Alzheimer’s disease independent of brain amyloid deposition. J. Nutr. Biochem. 2014, 25, 157–169. [Google Scholar] [CrossRef]
- Freund-Levi, Y.; Hjorth, E.; Lindberg, C.; Cederholm, T.; Faxen-Irving, G.; Vedin, I.; Palmblad, J.; Wahlund, L.O.; Schultzberg, M.; Basun, H.; et al. Effects of Omega-3 Fatty Acids on Inflammatory Markers in Cerebrospinal Fluid and Plasma in Alzheimer’s Disease: The OmegAD Study. Dement. Geriatr. Cogn. Disord. 2009, 27, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Hjorth, E.; Zhu, M.; Toro, V.C.; Vedin, I.; Palmblad, J.; Cederholm, T.; Freund-Levi, Y.; Faxen-Irving, G.; Wahlund, L.-O.; Basun, H. Omega-3 fatty acids enhance phagocytosis of alzheimer’s disease-related amyloid-β 42 by human microglia and decrease inflammatory markers. J. Alzheimer’s Dis. 2013, 35, 697–713. [Google Scholar]
- Zhao, Y.; Calon, F.; Julien, C.; Winkler, J.W.; Petasis, N.A.; Lukiw, W.J.; Bazan, N.G. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase-and PPARγ-mediated mechanisms in Alzheimer’s disease models. PLoS ONE 2011, 6, e15816. [Google Scholar]
- Ma, Q.-L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y. β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar]
- Nisbet, R.M.; Polanco, J.-C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef]
- Ma, X.; Sun, Z.; Han, X.; Li, S.; Jiang, X.; Chen, S.; Zhang, J.; Lu, H. Neuroprotective effect of resveratrol via activation of Sirt1 signaling in a rat model of combined diabetes and Alzheimer’s disease. Front. Neurosci. 2020, 13, 1400. [Google Scholar]
- Yang, A.J.; Frendo-Cumbo, S.; MacPherson, R.E. Resveratrol and metformin recover prefrontal cortex AMPK activation in diet-induced obese mice but reduce BDNF and synaptophysin protein content. J. Alzheimer’s Dis. 2019, 71, 945–956. [Google Scholar]
- Wang, X.; Ma, S.; Yang, B.; Huang, T.; Meng, N.; Xu, L.; Xing, Q.; Zhang, Y.; Zhang, K.; Li, Q.; et al. Resveratrol promotes hUC-MSCs engraftment and neural repair in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2018, 339, 297–304. [Google Scholar] [CrossRef]
- Broderick, T.L.; Rasool, S.; Li, R.; Zhang, Y.; Anderson, M.; Al-Nakkash, L.; Plochocki, J.H.; Geetha, T.; Babu, J.R. Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7337. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, G.W.; Liang, Z.M.; Sheng, S.Y.; Shi, Y.S.; Peng, L.; Wang, Y.P.; Wang, F.; Zhang, X.M. Resveratrol improves cognition and decreases amyloid plaque formation in Tg6799 mice. Mol. Med. Rep. 2019, 19, 3783–3790. [Google Scholar]
- Sarroca, S.; Gatius, A.; Rodríguez-Farré, E.; Vilchez, D.; Pallàs, M.; Griñán-Ferré, C.; Sanfeliu, C.; Corpas, R. Resveratrol confers neuroprotection against high-fat diet in a mouse model of Alzheimer’s disease via modulation of proteolytic mechanisms. J. Nutr. Biochem. 2021, 89, 108569. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces β-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008, 1214, 177–187. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, Y.; Nan, Y.; Wang, X.; Chen, Y.; Wang, S. (−)-Epigallocatechin-3-gallate ameliorates memory impairment and rescues the abnormal synaptic protein levels in the frontal cortex and hippocampus in a mouse model of Alzheimer’s disease. NeuroReport 2017, 28, 590–597. [Google Scholar]
- Nan, S.; Wang, P.; Zhang, Y.; Fan, J. Epigallocatechin-3-Gallate Provides Protection Against Alzheimer’s Disease-Induced Learning and Memory Impairments in Rats. Drug Des. Devel Ther. 2021, 15, 2013–2024. [Google Scholar] [CrossRef]
- Bao, J.; Liu, W.; Zhou, H.-Y.; Gui, Y.-R.; Yang, Y.-H.; Wu, M.-J.; Xiao, Y.-F.; Shang, J.-T.; Long, G.-F.; Shu, X.-J. Epigallocatechin-3-gallate Alleviates Cognitive Deficits in APP/PS1 Mice. Curr. Med. Sci. 2020, 40, 18–27. [Google Scholar] [CrossRef]
- Giacomeli, R.; Izoton, J.C.; dos Santos, R.B.; Boeira, S.P.; Jesse, C.R.; Haas, S.E. Neuroprotective effects of curcumin lipid-core nanocapsules in a model Alzheimer’s disease induced by β-amyloid 1-42 peptide in aged female mice. Brain Res. 2019, 1721, 146325. [Google Scholar] [CrossRef] [PubMed]
- Paula, P.-C.; Angelica Maria, S.-G.; Luis, C.-H.; Gloria Patricia, C.-G. Preventive Effect of Quercetin in a Triple Transgenic Alzheimer’s Disease Mice Model. Molecules 2019, 24, 2287. [Google Scholar] [CrossRef] [PubMed]
- Sabogal-Guáqueta, A.M.; Muñoz-Manco, J.I.; Ramírez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gómez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Zhou, M.; Zhang, T.; Zhang, L.; Ding, L.; Yanagita, T.; Xu, J.; Xue, C.; Wang, Y. Comparative study of the effects of phosphatidylcholine rich in DHA and EPA on Alzheimer’s disease and the possible mechanisms in CHO-APP/PS1 cells and SAMP8 mice. Food Funct. 2018, 9, 643–654. [Google Scholar]
- Yalagala, P.R.; Sugasini, D.; Dasarathi, S.; Pahan, K.; Subbaiah, P.V. Dietary lysophosphatidylcholine-EPA enriches both EPA and DHA in the brain: Potential treatment for depression[S]. J. Lipid Res. 2019, 60, 566–578. [Google Scholar] [CrossRef]
- Vela, S.; Sainz, N.; Moreno-Aliaga, M.J.; Solas, M.; Ramirez, M.J. DHA Selectively Protects SAMP-8-Associated Cognitive Deficits Through Inhibition of JNK. Mol. Neurobiol. 2019, 56, 1618–1627. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yavari, M.; Kalupahana, N.S.; Harris, B.N.; Ramalingam, L.; Zu, Y.; Kahathuduwa, C.N.; Moustaid-Moussa, N. Mechanisms Linking Obesity, Insulin Resistance, and Alzheimer’s Disease: Effects of Polyphenols and Omega-3 Polyunsaturated Fatty Acids. Nutrients 2025, 17, 1203. https://doi.org/10.3390/nu17071203
Yavari M, Kalupahana NS, Harris BN, Ramalingam L, Zu Y, Kahathuduwa CN, Moustaid-Moussa N. Mechanisms Linking Obesity, Insulin Resistance, and Alzheimer’s Disease: Effects of Polyphenols and Omega-3 Polyunsaturated Fatty Acids. Nutrients. 2025; 17(7):1203. https://doi.org/10.3390/nu17071203
Chicago/Turabian StyleYavari, Mahsa, Nishan Sudheera Kalupahana, Breanna N. Harris, Latha Ramalingam, Yujiao Zu, Chanaka Nadeeshan Kahathuduwa, and Naima Moustaid-Moussa. 2025. "Mechanisms Linking Obesity, Insulin Resistance, and Alzheimer’s Disease: Effects of Polyphenols and Omega-3 Polyunsaturated Fatty Acids" Nutrients 17, no. 7: 1203. https://doi.org/10.3390/nu17071203
APA StyleYavari, M., Kalupahana, N. S., Harris, B. N., Ramalingam, L., Zu, Y., Kahathuduwa, C. N., & Moustaid-Moussa, N. (2025). Mechanisms Linking Obesity, Insulin Resistance, and Alzheimer’s Disease: Effects of Polyphenols and Omega-3 Polyunsaturated Fatty Acids. Nutrients, 17(7), 1203. https://doi.org/10.3390/nu17071203