
Lifestyle Interventions to Tackle Cardiovascular Risk in Thyroid Hormone Signaling Disorders


Abstract
:1. Introduction
2. Materials and Methods
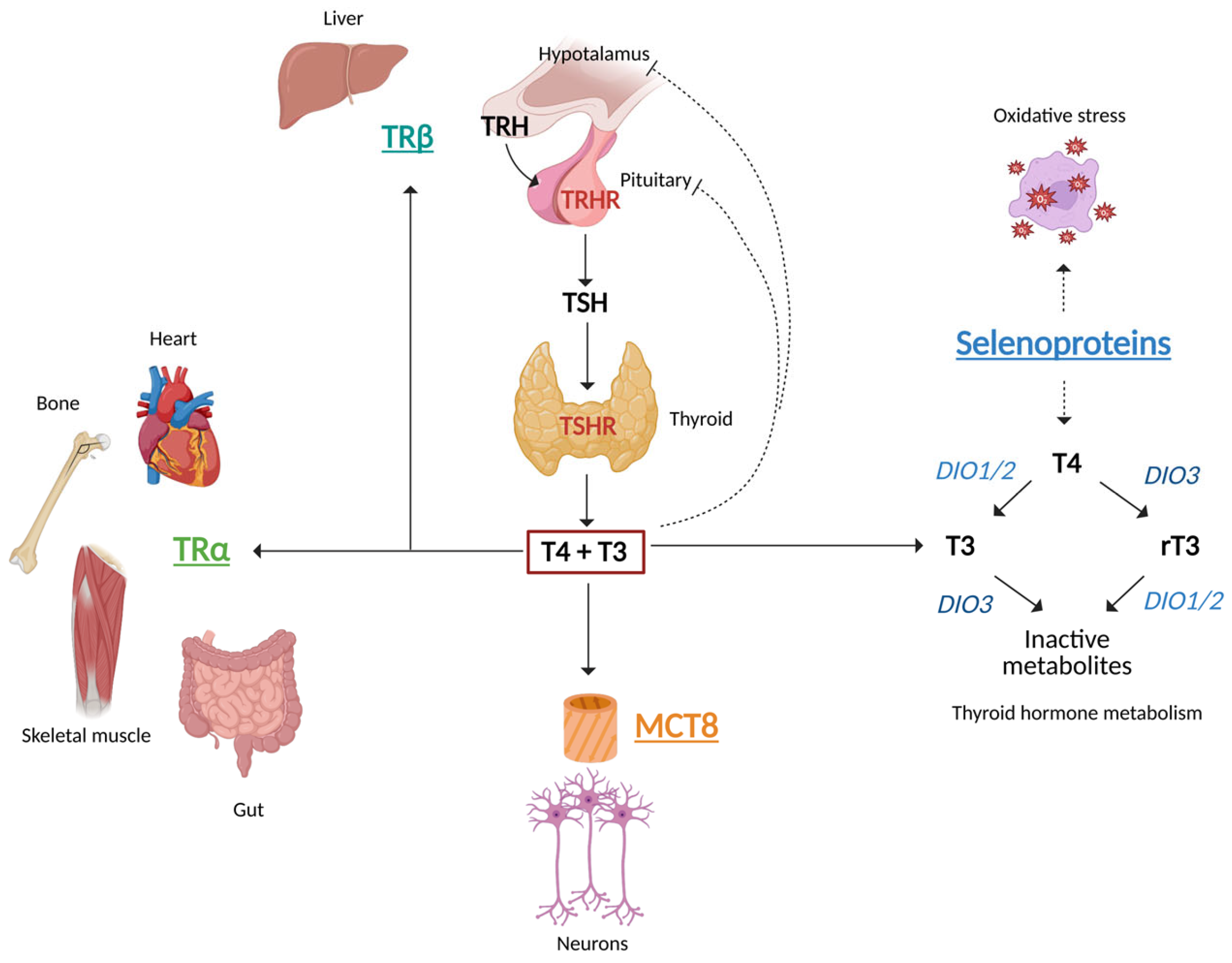
3. Hypothalamus–Pituitary–Thyroid Axis
4. Thyroid Hormone Synthesis, Metabolism and Action
5. Primary Thyroid Dysfunction and the CV System
6. Thyroid Dysfunction and Muscles
7. Thyroid Dysfunction and the Liver
8. Thyroid Dysfunction and Adipose Tissue
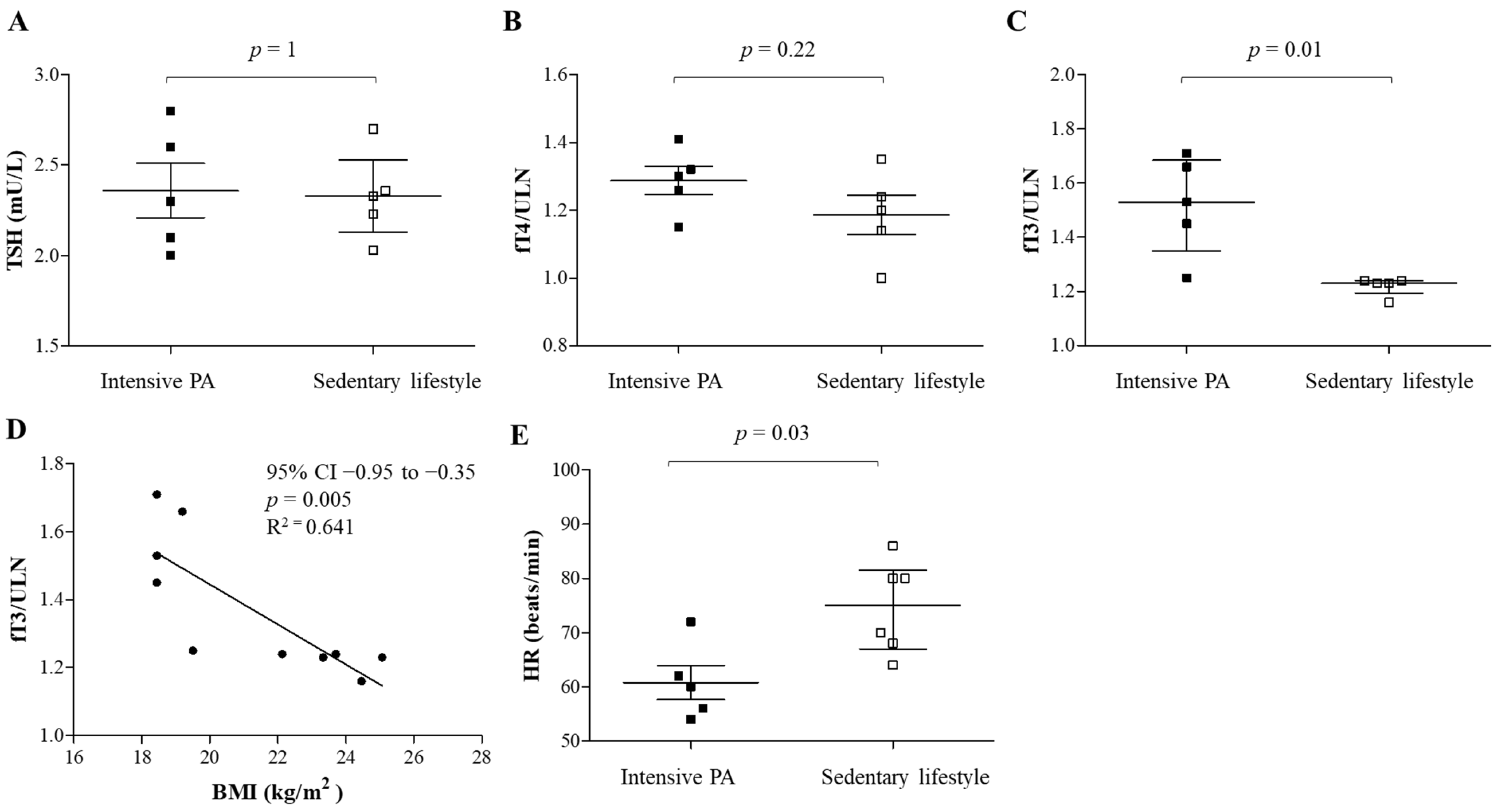
9. Impact of Physical Exercise on CV Risk and Thyroid Function
10. Thyroid Dysfunction and Food Intake
11. Resistance to Thyroid Hormone β (RTHβ)
Role of Lifestyle Modifications in RTHβ
12. Resistance to Thyroid Hormone α (RTHα)
Role of Lifestyle Modifications in RTHα
13. Monocarboxylate Transporter 8 (MCT8) Defects
Practical Considerations for Diet and Physical Exercise
14. Selenoprotein Deficiency
Role of Lifestyle Modifications
15. Limitations of the Study
16. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AF | Atrial fibrillation |
| AgRP | Agouti-related protein |
| BAT | Brown adipose tissue |
| BP | Blood pressure |
| CV | Cardiovascular |
| D1 | Type 1 deiodinase |
| D2 | Type 2 deiodinase |
| D3 | Type 3 deiodinase |
| DIOs | Iodothyronine deiodinases |
| 3, 5-DIT | Diiodotyrosine |
| HF | Heart failure |
| HPTA | Hypothalamus–pituitary–thyroid axis |
| HR | Heart rate |
| IRD | Inner-ring deiodination |
| LT-4 | Levothyroxine |
| MACEs | Major CV events |
| MASLD | Metabolic Dysfunction-Associated Steatotic Liver Disease |
| MCT8 | Monocarboxylate Transporter 8 |
| 3′-MIT | Monoiodotyrosine |
| NIS | Sodium/iodide symporter |
| NPY | Neuropeptide Y |
| ORD | Outer-ring deiodination |
| PA | Physical activity |
| POMC | Proopiomelanocortin |
| PVN | paraventricular nucleus |
| RAAS | Renin–angiotensin–aldosterone system |
| REE | Resting energy expenditure |
| RTHα | Resistance to thyroid hormone α |
| RTHβ | Resistance to thyroid hormone β |
| SECISBP2 | Selenocysteine Insertion Sequence-Binding Protein 2 |
| SELENON | Seloprotein N |
| SLC16A2 | Solute Carrier Family 16 Member 2 |
| T3 | Triiodothyronine |
| T4 | Thyroxine |
| THs | Thyroid hormones |
| TPO | Thyroid peroxidase |
| TRH | Thyrotropin-releasing hormone |
| TRHR | Thyrotropin-releasing hormone receptor |
| TRIAC | 3,3′,5-Triiodothyroacetic Acid |
| TRs | Thyroid hormone receptors |
| TRα | Thyroid hormone receptor α |
| TRβ | Thyroid hormone receptor β |
| TSH | Thyroid stimulating hormone |
| UV | Ultraviolet |
| VMH | Ventromedial nucleus of the hypothalamus |
| WAT | White adipose tissue |
References
- Persani, L.; Rodien, P.; Moran, C.; Visser, W.E.; Groeneweg, S.; Peeters, R.; Refetoff, S.; Gurnell, M.; Beck-Peccoz, P.; Chatterjee, K. 2024 European Thyroid Association Guidelines on diagnosis and management of genetic disorders of thyroid hormone transport, metabolism and action. Eur. Thyroid, J. 2024, 13, e240125. [Google Scholar] [CrossRef]
- Feldt-Rasmussen, U.; Effraimidis, G.; Klose, M. The hypothalamus-pituitary-thyroid (HPT)-axis and its role in physiology and pathophysiology of other hypothalamus-pituitary functions. Mol. Cell. Endocrinol. 2021, 525, 111173. [Google Scholar] [CrossRef]
- Melmed, S.; Auchus, R.J.; Goldfine, A.B.; Koenig, R.J.; Rosen, C.J. Williams Textbook of Endocrinology, 14th ed.; Elsevier: Philadelphia, PA, USA, 2020; pp. xiv, 1777. [Google Scholar]
- Refetoff, S.; Weiss, R.E.; Usala, S.J. The syndromes of resistance to thyroid hormone. Endocr. Rev. 1993, 14, 348–399. [Google Scholar] [CrossRef]
- Gardner, D.G.; Shoback, D.M. Greenspan’s Basic & Clinical Endocrinology, 10th ed.; McGraw-Hill Education: New York, NY, USA, 2018; pp. xxi, 916. [Google Scholar]
- Groeneweg, S.; van Geest, F.S.; Abacı, A.; Alcantud, A.; Ambegaonkar, G.P.; Armour, C.M.; Bakhtiani, P.; Barca, D.; Bertini, E.S.; van Beynum, I.M.; et al. Disease characteristics of MCT8 deficiency: An international, retrospective, multicentre cohort study. Lancet Diabetes Endocrinol. 2020, 8, 594–605. [Google Scholar] [CrossRef]
- Bianco, A.C.; Larsen, P.R. Cellular and structural biology of the deiodinases. Thyroid 2005, 15, 777–786. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Chatterjee, K. Human Genetic Disorders Resulting in Systemic Selenoprotein Deficiency. Int. J. Mol. Sci. 2021, 22, 12927. [Google Scholar] [CrossRef]
- Dumitrescu, A.M.; Liao, X.H.; Abdullah, M.S.; Lado-Abeal, J.; Majed, F.A.; Moeller, L.C.; Boran, G.; Schomburg, L.; Weiss, R.E.; Refetoff, S. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat. Genet. 2005, 37, 1247–1252. [Google Scholar] [CrossRef]
- Vargas-Uricoechea, H.; Bonelo-Perdomo, A.; Sierra-Torres, C.H. Effects of thyroid hormones on the heart. Clin. Investig. Arterioscler. 2014, 26, 296–309. [Google Scholar] [CrossRef]
- Vargas, F.; Rodríguez-Gómez, I.; Vargas-Tendero, P.; Jimenez, E.; Montiel, M. The renin-angiotensin system in thyroid disorders and its role in cardiovascular and renal manifestations. J. Endocrinol. 2012, 213, 25–36. [Google Scholar] [CrossRef]
- Klein, I.; Danzi, S. Thyroid Disease and the Heart. Curr. Probl. Cardiol. 2016, 41, 65–92. [Google Scholar] [CrossRef]
- Klein, I.; Ojamaa, K. Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 2001, 344, 501–509. [Google Scholar] [CrossRef]
- Biondi, B. Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur. J. Endocrinol. 2012, 167, 609–618. [Google Scholar] [CrossRef]
- Frost, L.; Vestergaard, P.; Mosekilde, L. Hyperthyroidism and risk of atrial fibrillation or flutter: A population-based study. Arch. Intern. Med. 2004, 164, 1675–1678. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Mikhailidis, D.P.; Rysz, J.; Banach, M. The mechanisms of atrial fibrillation in hyperthyroidism. Thyroid Res. 2009, 2, 4. [Google Scholar] [CrossRef]
- Selmer, C.; Olesen, J.B.; Hansen, M.L.; von Kappelgaard, L.M.; Madsen, J.C.; Hansen, P.R.; Pedersen, O.D.; Faber, J.; Torp-Pedersen, C.; Gislason, G.H. Subclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events: A large population study. J. Clin. Endocrinol. Metab. 2014, 99, 2372–2382. [Google Scholar] [CrossRef]
- Groothof, D.; Flores-Guerrero, J.L.; Nolte, I.M.; Bouma, H.R.; Gruppen, E.G.; Bano, A.; Post, A.; Kootstra-Ros, J.E.; Hak, E.; Bos, J.H.J.; et al. Thyroid function and risk of all-cause and cardiovascular mortality: A prospective population-based cohort study. Endocrine 2021, 71, 385–396. [Google Scholar] [CrossRef]
- Bano, A.; Chaker, L.; Mattace-Raso, F.U.S.; Terzikhan, N.; Kavousi, M.; Ikram, M.A.; Peeters, R.P.; Franco, O.H. Thyroid function and life expectancy with and without noncommunicable diseases: A population-based study. PLoS Med. 2019, 16, e1002957. [Google Scholar] [CrossRef]
- Chaker, L.; van den Berg, M.E.; Niemeijer, M.N.; Franco, O.H.; Dehghan, A.; Hofman, A.; Rijnbeek, P.R.; Deckers, J.W.; Eijgelsheim, M.; Stricker, B.H.; et al. Thyroid Function and Sudden Cardiac Death: A Prospective Population-Based Cohort Study. Circulation 2016, 134, 713–722. [Google Scholar] [CrossRef]
- Ochs, N.; Auer, R.; Bauer, D.C.; Nanchen, D.; Gussekloo, J.; Cornuz, J.; Rodondi, N. Meta-analysis: Subclinical thyroid dysfunction and the risk for coronary heart disease and mortality. Ann. Intern. Med. 2008, 148, 832–845. [Google Scholar] [CrossRef]
- Biondi, B.; Cooper, D.S. The clinical significance of subclinical thyroid dysfunction. Endocr. Rev. 2008, 29, 76–131. [Google Scholar] [CrossRef]
- Baumgartner, C.; da Costa, B.R.; Collet, T.H.; Feller, M.; Floriani, C.; Bauer, D.C.; Cappola, A.R.; Heckbert, S.R.; Ceresini, G.; Gussekloo, J.; et al. Thyroid Function within the Normal Range, Subclinical Hypothyroidism, and the Risk of Atrial Fibrillation. Circulation 2017, 136, 2100–2116. [Google Scholar] [CrossRef]
- Xu, Y.; Derakhshan, A.; Hysaj, O.; Wildisen, L.; Ittermann, T.; Pingitore, A.; Abolhassani, N.; Medici, M.; Kiemeney, L.A.L.M.; Riksen, N.P.; et al. The optimal healthy ranges of thyroid function defined by the risk of cardiovascular disease and mortality: Systematic review and individual participant data meta-analysis. Lancet Diabetes Endocrinol. 2023, 11, 743–754. [Google Scholar] [CrossRef]
- Klein, I.; Danzi, S. Thyroid disease and the heart. Circulation 2007, 116, 1725–1735. [Google Scholar] [CrossRef]
- Kannan, L.; Shaw, P.A.; Morley, M.P.; Brandimarto, J.; Fang, J.C.; Sweitzer, N.K.; Cappola, T.P.; Cappola, A.R. Thyroid Dysfunction in Heart Failure and Cardiovascular Outcomes. Circ. Heart Fail. 2018, 11, e005266. [Google Scholar] [CrossRef]
- Stamatouli, A.; Bedoya, P.; Yavuz, S. Hypothyroidism: Cardiovascular Endpoints of Thyroid Hormone Replacement. Front. Endocrinol 2019, 10, 888. [Google Scholar] [CrossRef]
- Biondi, B.; Klein, I. Hypothyroidism as a risk factor for cardiovascular disease. Endocrine 2004, 24, 1–13. [Google Scholar] [CrossRef]
- Stabouli, S.; Papakatsika, S.; Kotsis, V. Hypothyroidism and hypertension. Expert. Rev. Cardiovasc. Ther. 2010, 8, 1559–1565. [Google Scholar] [CrossRef]
- Ripoli, A.; Pingitore, A.; Favilli, B.; Bottoni, A.; Turchi, S.; Osman, N.F.; De Marchi, D.; Lombardi, M.; L’Abbate, A.; Iervasi, G. Does subclinical hypothyroidism affect cardiac pump performance? Evidence from a magnetic resonance imaging study. J. Am. Coll. Cardiol. 2005, 45, 439–445. [Google Scholar] [CrossRef]
- Brenta, G.; Mutti, L.A.; Schnitman, M.; Fretes, O.; Perrone, A.; Matute, M.L. Assessment of left ventricular diastolic function by radionuclide ventriculography at rest and exercise in subclinical hypothyroidism, and its response to L-thyroxine therapy. Am. J. Cardiol. 2003, 91, 1327–1330. [Google Scholar] [CrossRef]
- Cappola, A.R.; Fried, L.P.; Arnold, A.M.; Danese, M.D.; Kuller, L.H.; Burke, G.L.; Tracy, R.P.; Ladenson, P.W. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA 2006, 295, 1033–1041. [Google Scholar] [CrossRef]
- Selmer, C.; Olesen, J.B.; Hansen, M.L.; Lindhardsen, J.; Olsen, A.M.; Madsen, J.C.; Faber, J.; Hansen, P.R.; Pedersen, O.D.; Torp-Pedersen, C.; et al. The spectrum of thyroid disease and risk of new onset atrial fibrillation: A large population cohort study. BMJ 2012, 345, e7895. [Google Scholar] [CrossRef]
- Cooper, D.S.; Biondi, B. Subclinical thyroid disease. Lancet 2012, 379, 1142–1154. [Google Scholar] [CrossRef]
- Inoue, K.; Ritz, B.; Brent, G.A.; Ebrahimi, R.; Rhee, C.M.; Leung, A.M. Association of Subclinical Hypothyroidism and Cardiovascular Disease with Mortality. JAMA Netw. Open 2020, 3, e1920745. [Google Scholar] [CrossRef]
- Hyland, K.A.; Arnold, A.M.; Lee, J.S.; Cappola, A.R. Persistent subclinical hypothyroidism and cardiovascular risk in the elderly: The cardiovascular health study. J. Clin. Endocrinol. Metab. 2013, 98, 533–540. [Google Scholar] [CrossRef]
- Salvatore, D.; Simonides, W.S.; Dentice, M.; Zavacki, A.M.; Larsen, P.R. Thyroid hormones and skeletal muscle–New insights and potential implications. Nat. Rev. Endocrinol. 2014, 10, 206–214. [Google Scholar] [CrossRef]
- Brix, T.H.; Kyvik, K.O.; Hegedüs, L. Validity of self-reported hyperthyroidism and hypothyroidism: Comparison of self-reported questionnaire data with medical record review. Thyroid 2001, 11, 769–773. [Google Scholar] [CrossRef]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef]
- Simonides, W.S.; van Hardeveld, C. Thyroid hormone as a determinant of metabolic and contractile phenotype of skeletal muscle. Thyroid 2008, 18, 205–216. [Google Scholar] [CrossRef]
- Brennan, M.D.; Powell, C.; Kaufman, K.R.; Sun, P.C.; Bahn, R.S.; Nair, K.S. The impact of overt and subclinical hyperthyroidism on skeletal muscle. Thyroid 2006, 16, 375–380. [Google Scholar] [CrossRef]
- Skovgaard, C.; Brandt, N.; Pilegaard, H.; Bangsbo, J. Combined speed endurance and endurance exercise amplify the exercise-induced PGC-1α and PDK4 mRNA response in trained human muscle. Physiol. Rep. 2016, 4, e12864. [Google Scholar] [CrossRef]
- Bloise, F.F.; Cordeiro, A.; Ortiga-Carvalho, T.M. Role of thyroid hormone in skeletal muscle physiology. J. Endocrinol. 2018, 236, R57–R68. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Olmo, J.L.; Mastaglia, F.L. Changes in histochemical profile of rat respiratory muscles in hypo- and hyperthyroidism. Q. J. Exp. Physiol. 1983, 68, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, S.L.; Inder, W.J.; Stowasser, M.; Stanton, T. Hyperdynamic Right Heart Function in Graves’ Hyperthyroidism Measured by Echocardiography Normalises on Restoration of Euthyroidism. Heart Lung Circ. 2017, 26, 580–585. [Google Scholar] [CrossRef]
- Fu, S.E.; Liang, X.H.; Huang, L.L.; Xian, J.; Wu, X.Z.; Pan, J.; Chen, X.L.; Kuang, Y.Q.; Wu, C.J.; Li, Q.L.; et al. Chronic thyrotoxic myopathy development is associated with thyroid hormone sensitivity index, predicted by lower-limb fatigue and the squat-up test. Sci. Rep. 2024, 14, 24364. [Google Scholar] [CrossRef]
- Sindoni, A.; Rodolico, C.; Pappalardo, M.A.; Portaro, S.; Benvenga, S. Hypothyroid myopathy: A peculiar clinical presentation of thyroid failure. Review of the literature. Rev. Endocr. Metab. Disord. 2016, 17, 499–519. [Google Scholar] [CrossRef]
- Horak, H.A.; Pourmand, R. Endocrine myopathies. Neurol. Clin. 2000, 18, 203–213. [Google Scholar] [CrossRef]
- Rodolico, C.; Bonanno, C.; Pugliese, A.; Nicocia, G.; Benvenga, S.; Toscano, A. Endocrine myopathies: Clinical and histopathological features of the major forms. Acta Myol. 2020, 39, 130–135. [Google Scholar] [CrossRef]
- Flores-Morales, A.; Gullberg, H.; Fernandez, L.; Ståhlberg, N.; Lee, N.H.; Vennström, B.; Norstedt, G. Patterns of liver gene expression governed by TRbeta. Mol. Endocrinol. 2002, 16, 1257–1268. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Vallejo, C.G.; Seguido, A.M.; Testillano, P.S.; Risueño, M.C. Thyroid hormone regulates tubulin expression in mammalian liver. Effects of deleting thyroid hormone receptor-alpha or -beta. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E87–E94. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Peng, H.; Chen, X.; Wu, X.; Wang, B. Hyperlipidemia and hypothyroidism. Clin. Chim. Acta 2022, 527, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Mavromati, M.; Jornayvaz, F.R. Hypothyroidism-Associated Dyslipidemia: Potential Molecular Mechanisms Leading to NAFLD. Int. J. Mol. Sci. 2021, 22, 12797. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Gusdon, A.M.; Qu, S. Cross-talk between the thyroid and liver: A new target for nonalcoholic fatty liver disease treatment. World J. Gastroenterol. 2013, 19, 8238–8246. [Google Scholar] [CrossRef]
- Mazo, D.F.; Lima, V.M.; Stefano, J.T.; Rabelo, F.; Faintuch, J.; Oliveira, C.P. Gluco-lipidic indices in treated hypothyroidism associated with nonalcoholic fatty liver disease. Arq. Gastroenterol. 2011, 48, 186–189. [Google Scholar] [CrossRef]
- Perra, A.; Simbula, G.; Simbula, M.; Pibiri, M.; Kowalik, M.A.; Sulas, P.; Cocco, M.T.; Ledda-Columbano, G.M.; Columbano, A. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. 2008, 22, 2981–2989. [Google Scholar] [CrossRef]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef]
- Yau, W.W.; Yen, P.M. Thermogenesis in Adipose Tissue Activated by Thyroid Hormone. Int. J. Mol. Sci. 2020, 21, 3020. [Google Scholar] [CrossRef]
- Cioffi, F.; Gentile, A.; Silvestri, E.; Goglia, F.; Lombardi, A. Effect of Iodothyronines on Thermogenesis: Focus on Brown Adipose Tissue. Front. Endocrinol. 2018, 9, 254. [Google Scholar] [CrossRef]
- Johann, K.; Cremer, A.L.; Fischer, A.W.; Heine, M.; Pensado, E.R.; Resch, J.; Nock, S.; Virtue, S.; Harder, L.; Oelkrug, R.; et al. Thyroid-Hormone-Induced Browning of White Adipose Tissue Does Not Contribute to Thermogenesis and Glucose Consumption. Cell Rep. 2019, 27, 3385–3400.e3383. [Google Scholar] [CrossRef]
- Weiner, J.; Hankir, M.; Heiker, J.T.; Fenske, W.; Krause, K. Thyroid hormones and browning of adipose tissue. Mol. Cell. Endocrinol. 2017, 458, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Klieverik, L.P.; Janssen, S.F.; van Riel, A.; Foppen, E.; Bisschop, P.H.; Serlie, M.J.; Boelen, A.; Ackermans, M.T.; Sauerwein, H.P.; Fliers, E.; et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc. Natl. Acad. Sci. USA 2009, 106, 5966–5971. [Google Scholar] [CrossRef] [PubMed]
- Zybek-Kocik, A.; Sawicka-Gutaj, N.; Szczepanek-Parulska, E.; Andrusiewicz, M.; Waligórska-Stachura, J.; Białas, P.; Krauze, T.; Guzik, P.; Skrobisz, J.; Ruchała, M. The association between irisin and muscle metabolism in different thyroid disorders. Clin. Endocrinol. 2018, 88, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, G.X.; Ma, S.L.; Jung, D.Y.; Ha, H.; Altamimi, T.; Zhao, X.Y.; Guo, L.; Zhang, P.; Hu, C.R.; et al. Nrg4 promotes fuel oxidation and a healthy adipokine profile to ameliorate diet-induced metabolic disorders. Mol. Metab. 2017, 6, 863–872. [Google Scholar] [CrossRef]
- Li, M.; Chen, Y.; Jiang, J.; Lu, Y.; Song, Z.; Zhang, S.; Sun, C.; Ying, H.; Fan, X.; Song, Y.; et al. Elevated serum neuregulin 4 levels in patients with hyperthyroidism. Endocr. Connect. 2019, 8, 728–735. [Google Scholar] [CrossRef]
- Bakiner, O.; Bozkirli, E.; Ertugrul, D.; Sezgin, N.; Ertorer, E. Plasma fetuin-A levels are reduced in patients with hypothyroidism. Eur. J. Endocrinol. 2014, 170, 411–418. [Google Scholar] [CrossRef]
- Felske, D.; Gagnon, A.; Sorisky, A. Interacting Effects of TSH and Insulin on Human Differentiated Adipocytes. Horm. Metab. Res. 2015, 47, 681–685. [Google Scholar] [CrossRef]
- Bull, F.C.; Al-Ansari, S.S.; Biddle, S.; Borodulin, K.; Buman, M.P.; Cardon, G.; Carty, C.; Chaput, J.P.; Chastin, S.; Chou, R.; et al. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br. J. Sports Med. 2020, 54, 1451–1462. [Google Scholar] [CrossRef]
- Noone, J.; Mucinski, J.M.; DeLany, J.P.; Sparks, L.M.; Goodpaster, B.H. Understanding the variation in exercise responses to guide personalized physical activity prescriptions. Cell Metab. 2024, 36, 702–724. [Google Scholar] [CrossRef]
- Fogarasi, A.; Gonzalez, K.; Dalamaga, M.; Magkos, F. The Impact of the Rate of Weight Loss on Body Composition and Metabolism. Curr. Obes. Rep. 2022, 11, 33–44. [Google Scholar] [CrossRef]
- Kerksick, C.M.; Wismann-Bunn, J.; Fogt, D.; Thomas, A.R.; Taylor, L.; Campbell, B.I.; Wilborn, C.D.; Harvey, T.; Roberts, M.D.; La Bounty, P.; et al. Changes in weight loss, body composition and cardiovascular disease risk after altering macronutrient distributions during a regular exercise program in obese women. Nutr. J. 2010, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Lepe, M.A.; Miranda-Gil, M.I.; Valbuena-Gregorio, E.; Olivas-Aguirre, F.J. Exercise Programs Combined with Diet Supplementation Improve Body Composition and Physical Function in Older Adults with Sarcopenia: A Systematic Review. Nutrients 2023, 15, 1998. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Standley, R.A.; Yi, F.; Carnero, E.A.; Sparks, L.M.; Goodpaster, B.H. Individual Response Variation in the Effects of Weight Loss and Exercise on Insulin Sensitivity and Cardiometabolic Risk in Older Adults. Front. Endocrinol. 2020, 11, 632. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.J.; Ryan, A.S.; Stevenson, T.G.; Goldberg, A.P. Metabolic inflexibility during submaximal aerobic exercise is associated with glucose intolerance in obese older adults. Obesity 2014, 22, 451–457. [Google Scholar] [CrossRef]
- Ryan, A.S.; Katzel, L.I.; Prior, S.J.; McLenithan, J.C.; Goldberg, A.P.; Ortmeyer, H.K. Aerobic exercise plus weight loss improves insulin sensitivity and increases skeletal muscle glycogen synthase activity in older men. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 790–798. [Google Scholar] [CrossRef]
- Boulé, N.G.; Weisnagel, S.J.; Lakka, T.A.; Tremblay, A.; Bergman, R.N.; Rankinen, T.; Leon, A.S.; Skinner, J.S.; Wilmore, J.H.; Rao, D.C.; et al. Effects of exercise training on glucose homeostasis: The HERITAGE Family Study. Diabetes Care 2005, 28, 108–114. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Mu, W.J.; Zhu, J.Y.; Chen, M.; Guo, L. Exercise-Mediated Browning of White Adipose Tissue: Its Significance, Mechanism and Effectiveness. Int. J. Mol. Sci. 2021, 22, 11512. [Google Scholar] [CrossRef]
- Facioli, T.P.; Philbois, S.V.; Gastaldi, A.C.; Almeida, D.S.; Maida, K.D.; Rodrigues, J.A.L.; Sánchez-Delgado, J.C.; Souza, H.C.D. Study of heart rate recovery and cardiovascular autonomic modulation in healthy participants after submaximal exercise. Sci. Rep. 2021, 11, 3620. [Google Scholar] [CrossRef]
- Souza, H.C.D.; Philbois, S.V.; Veiga, A.C.; Aguilar, B.A. Heart Rate Variability and Cardiovascular Fitness: What We Know so Far. Vasc. Health Risk Manag. 2021, 17, 701–711. [Google Scholar] [CrossRef]
- Ylli, D.; Wartofsky, L. Can We Link Thyroid Status, Energy Expenditure, and Body Composition to Management of Subclinical Thyroid Dysfunction? J. Clin. Endocrinol. Metab. 2019, 104, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Yu, M.D.; Lee, M.S.; Cheng, C.Y.; Yang, S.P.; Chin, H.M.; Wu, S.Y. Effect of treadmill exercise on circulating thyroid hormone measurements. Med. Princ. Pract. 2004, 13, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Benso, A.; Broglio, F.; Aimaretti, G.; Lucatello, B.; Lanfranco, F.; Ghigo, E.; Grottoli, S. Endocrine and metabolic responses to extreme altitude and physical exercise in climbers. Eur. J. Endocrinol. 2007, 157, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Altaye, K.Z.; Mondal, S.; Legesse, K.; Abdulkedir, M. Effects of aerobic exercise on thyroid hormonal change responses among adolescents with intellectual disabilities. BMJ Open Sport Exerc. Med. 2019, 5, e000524. [Google Scholar] [CrossRef]
- Ciloglu, F.; Peker, I.; Pehlivan, A.; Karacabey, K.; Ilhan, N.; Saygin, O.; Ozmerdivenli, R. Exercise intensity and its effects on thyroid hormones. Neuro Endocrinol. Lett. 2005, 26, 830–834. [Google Scholar]
- Hanke, L.; Poeten, P.; Spanke, L.; Britz, S.; Diel, P. The Influence of Levothyroxine on Body Composition and Physical Performance in Subclinical Hypothyroidism. Horm. Metab. Res. 2023, 55, 51–58. [Google Scholar] [CrossRef]
- Klasson, C.L.; Sadhir, S.; Pontzer, H. Daily physical activity is negatively associated with thyroid hormone levels, inflammation, and immune system markers among men and women in the NHANES dataset. PLoS ONE 2022, 17, e0270221. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Matthews, C.H.; Mohr-Kahaly, S.; Richards, C.A.; Chatterjee, V.K. Cardiac involvement in thyroid hormone resistance. J. Clin. Endocrinol. Metab. 2002, 87, 204–212. [Google Scholar] [CrossRef]
- Ylli, D.; Klubo-Gwiezdzinska, J.; Wartofsky, L. Thyroid emergencies. Pol. Arch. Intern. Med. 2019, 129, 526–534. [Google Scholar] [CrossRef]
- Coker, R.H.; Hays, N.P.; Williams, R.H.; Brown, A.D.; Freeling, S.A.; Kortebein, P.M.; Sullivan, D.H.; Starling, R.D.; Evans, W.J. Exercise-induced changes in insulin action and glycogen metabolism in elderly adults. Med. Sci. Sports Exerc. 2006, 38, 433–438. [Google Scholar] [CrossRef]
- McAllister, R.M.; Delp, M.D.; Laughlin, M.H. Thyroid status and exercise tolerance. Cardiovascular and metabolic considerations. Sports Med. 1995, 20, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Duñabeitia, I.; González-Devesa, D.; Varela-Martínez, S.; Diz-Gómez, J.C.; Ayán-Pérez, C. Effect of physical exercise in people with hypothyroidism: Systematic review and meta-analysis. Scand. J. Clin. Lab. Investig. 2023, 83, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Piercy, K.L.; Troiano, R.P.; Ballard, R.M.; Carlson, S.A.; Fulton, J.E.; Galuska, D.A.; George, S.M.; Olson, R.D. The Physical Activity Guidelines for Americans. JAMA 2018, 320, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Biondi, B.; Cooper, D.S. Thyroid hormone therapy for hypothyroidism. Endocrine 2019, 66, 18–26. [Google Scholar] [CrossRef]
- Lankhaar, J.A.; de Vries, W.R.; Jansen, J.A.; Zelissen, P.M.; Backx, F.J. Impact of overt and subclinical hypothyroidism on exercise tolerance: A systematic review. Res. Q. Exerc. Sport 2014, 85, 365–389. [Google Scholar] [CrossRef]
- Medici, B.R.; Nygaard, B.; la Cour, J.L.; Krakauer, M.; Brønden, A.; Sonne, M.P.; Holst, J.J.; Rehfeld, J.F.; Vilsbøll, T.; Faber, J.; et al. Effects of levothyroxine substitution therapy on hunger and food intake in individuals with hypothyroidism. Endocr. Connect. 2023, 12, e230314. [Google Scholar] [CrossRef]
- Pijl, H.; de Meijer, P.H.; Langius, J.; Coenegracht, C.I.; van den Berk, A.H.; Chandie Shaw, P.K.; Boom, H.; Schoemaker, R.C.; Cohen, A.F.; Burggraaf, J.; et al. Food choice in hyperthyroidism: Potential influence of the autonomic nervous system and brain serotonin precursor availability. J. Clin. Endocrinol. Metab. 2001, 86, 5848–5853. [Google Scholar] [CrossRef]
- Amin, A.; Dhillo, W.S.; Murphy, K.G. The central effects of thyroid hormones on appetite. J. Thyroid Res. 2011, 2011, 306510. [Google Scholar] [CrossRef]
- Villicev, C.M.; Freitas, F.R.; Aoki, M.S.; Taffarel, C.; Scanlan, T.S.; Moriscot, A.S.; Ribeiro, M.O.; Bianco, A.C.; Gouveia, C.H. Thyroid hormone receptor beta-specific agonist GC-1 increases energy expenditure and prevents fat-mass accumulation in rats. J. Endocrinol. 2007, 193, 21–29. [Google Scholar] [CrossRef]
- Hameed, S.; Patterson, M.; Dhillo, W.S.; Rahman, S.A.; Ma, Y.; Holton, C.; Gogakos, A.; Yeo, G.S.H.; Lam, B.Y.H.; Polex-Wolf, J.; et al. Thyroid Hormone Receptor Beta in the Ventromedial Hypothalamus Is Essential for the Physiological Regulation of Food Intake and Body Weight. Cell Rep. 2017, 19, 2202–2209. [Google Scholar] [CrossRef]
- Ortiga-Carvalho, T.M.; Sidhaye, A.R.; Wondisford, F.E. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat. Rev. Endocrinol. 2014, 10, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Staníková, D.; Krajčovičová, L.; Demková, L.; Forišek-Paulová, P.; Slobodová, L.; Vitariušová, E.; Tichá, L.; Ukropcová, B.; Staník, J.; Ukropec, J. Food preferences and thyroid hormones in children and adolescents with obesity. Front. Psychiatry 2022, 13, 962949. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, A.N. The role of the thyrotropin-releasing hormone (TRH) neuron as a metabolic sensor. Thyroid 2008, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Miro, C.; Cicatiello, A.G.; Nappi, A.; Sagliocchi, S.; Acampora, L.; Restolfer, F.; Cuomo, O.; de Alteris, G.; Pugliese, G.; Torabinejad, S.; et al. Leptin enhances the intracellular thyroid hormone activation in skeletal muscle to boost energy balance. Cell Metab. 2025, 37, 936–953.e937. [Google Scholar] [CrossRef]
- McConnell, R.J.; Menendez, C.E.; Smith, F.R.; Henkin, R.I.; Rivlin, R.S. Defects of taste and smell in patients with hypothyroidism. Am. J. Med. 1975, 59, 354–364. [Google Scholar] [CrossRef]
- Deniz, F.; Ay, S.A.; Salihoglu, M.; Kurt, O.; Baskoy, K.; Altundag, A.; Tekeli, H.; Yonem, A.; Hummel, T. Thyroid Hormone Replacement Therapy Improves Olfaction and Taste Sensitivity in Primary Hypothyroid Patients: A Prospective Randomised Clinical Trial. Exp. Clin. Endocrinol. Diabetes 2016, 124, 562–567. [Google Scholar] [CrossRef]
- Clark, A.A.; Dotson, C.D.; Elson, A.E.; Voigt, A.; Boehm, U.; Meyerhof, W.; Steinle, N.I.; Munger, S.D. TAS2R bitter taste receptors regulate thyroid function. FASEB J. 2015, 29, 164–172. [Google Scholar] [CrossRef]
- Agostini, M.; Schoenmakers, E.; Syanda, A.; Romartinez-Alonso, B.; Cacciottolo, T.; Rashid, T.; Schwabe, J.; Chatterjee, K. Human resistance to thyroid hormone beta operates via a mechanism requiring receptor binding to DNA. Endocr. Abstr. 2023, 92, PS2-20-05. [Google Scholar] [CrossRef]
- Dumitrescu, A.M.; Refetoff, S. The syndromes of reduced sensitivity to thyroid hormone. Biochim. Biophys. Acta 2013, 1830, 3987–4003. [Google Scholar] [CrossRef]
- Moran, C.; Schoenmakers, N.; Visser, W.E.; Schoenmakers, E.; Agostini, M.; Chatterjee, K. Genetic disorders of thyroid development, hormone biosynthesis and signalling. Clin. Endocrinol. 2022, 97, 502–514. [Google Scholar] [CrossRef]
- Pulcrano, M.; Palmieri, E.A.; Mannavola, D.; Ciulla, M.; Campi, I.; Covelli, D.; Lombardi, G.; Biondi, B.; Beck-Peccoz, P. Impact of resistance to thyroid hormone on the cardiovascular system in adults. J. Clin. Endocrinol. Metab. 2009, 94, 2812–2816. [Google Scholar] [CrossRef] [PubMed]
- Brucker-Davis, F.; Skarulis, M.C.; Grace, M.B.; Benichou, J.; Hauser, P.; Wiggs, E.; Weintraub, B.D. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone. The National Institutes of Health Prospective Study. Ann. Intern. Med. 1995, 123, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Hauser, P.; Zametkin, A.J.; Martinez, P.; Vitiello, B.; Matochik, J.A.; Mixson, A.J.; Weintraub, B.D. Attention deficit-hyperactivity disorder in people with generalized resistance to thyroid hormone. N. Engl. J. Med. 1993, 328, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Uter, J.; Heldmann, M.; Rogge, B.; Obst, M.; Steinhardt, J.; Brabant, G.; Moran, C.; Chatterjee, K.; Münte, T.F. Patients with mutations of the Thyroid hormone beta-receptor show an ADHD-like phenotype for performance monitoring: An electrophysiological study. NeuroImage Clin. 2020, 26, 102250. [Google Scholar] [CrossRef]
- Mixson, A.J.; Parrilla, R.; Ransom, S.C.; Wiggs, E.A.; McClaskey, J.H.; Hauser, P.; Weintraub, B.D. Correlations of language abnormalities with localization of mutations in the beta-thyroid hormone receptor in 13 kindreds with generalized resistance to thyroid hormone: Identification of four new mutations. J. Clin. Endocrinol. Metab. 1992, 75, 1039–1045. [Google Scholar] [CrossRef]
- Illouz, F.; Briet, C.; Mirebeau-Prunier, D.; Bouhours-Nouet, N.; Coutant, R.; Sibilia, P.; Rodien, P. Cardiac complications of thyroid hormone resistance syndromes. Ann. Endocrinol. 2021, 82, 167–169. [Google Scholar] [CrossRef]
- Okosieme, O.E.; Usman, D.; Taylor, P.N.; Dayan, C.M.; Lyons, G.; Moran, C.; Chatterjee, K.; Rees, D.A. Cardiovascular morbidity and mortality in patients in Wales, UK with resistance to thyroid hormone β (RTHβ): A linked-record cohort study. Lancet Diabetes Endocrinol. 2023, 11, 657–666. [Google Scholar] [CrossRef]
- Campi, I.; Censi, S.; Prodam, F.; Petrone, L.; Brigante, G.; Porcelli, T.; Ruggeri, R.M.; Vigone, M.C.; Rurale, G.; Lio, S.; et al. Increased cardiovascular morbidity and reduced life expectancy in a large Italian cohort of patients with resistance to thyroid hormone β (RTHβ). Eur. J. Endocrinol. 2024, 191, 407–415. [Google Scholar] [CrossRef]
- Davis, T.M.E.; Davis, W.A.; Moran, C.; Lyons, G.; Bryden, E.; Chatterjee, K. Cardiovascular Risk and Plasma N-terminal Pro-B-type Natriuretic Peptide in Adults with Resistance to Thyroid Hormone β. J. Endocr. Soc. 2025, 9, bvaf023. [Google Scholar] [CrossRef]
- Owen, P.J.; Chatterjee, V.K.; John, R.; Halsall, D.; Lazarus, J.H. Augmentation index in resistance to thyroid hormone (RTH). Clin. Endocrinol. 2009, 70, 650–654. [Google Scholar] [CrossRef]
- Moran, C.; McEniery, C.M.; Schoenmakers, N.; Mitchell, C.; Sleigh, A.; Watson, L.; Lyons, G.; Burling, K.; Barker, P.; Chatterjee, K. Dyslipidemia, Insulin Resistance, Ectopic Lipid Accumulation, and Vascular Function in Resistance to Thyroid Hormone β. J. Clin. Endocrinol. Metab. 2021, 106, e2005–e2014. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.; Bruinstroop, E.; Refetoff, S.; Yen, P.M.; Anselmo, J. Increased Hepatic Fat Content in Patients with Resistance to Thyroid Hormone Beta. Thyroid 2021, 31, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Domingues-Hajj, P.M.S.; Gomes, P.M.; Magalhães, P.K.R.; Maciel, L.M.Z. Assessment of Cardiometabolic Risk Factors and Insulin Sensitivity by Hyperinsulinemic-Euglycemic Clamp in Resistance to Thyroid Hormone β Syndrome. Thyroid 2024, 34, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.S.; Savage, D.B.; Dufour, S.; Schoenmakers, N.; Murgatroyd, P.; Befroy, D.; Halsall, D.; Northcott, S.; Raymond-Barker, P.; Curran, S.; et al. Resistance to thyroid hormone is associated with raised energy expenditure, muscle mitochondrial uncoupling, and hyperphagia. J. Clin. Investig. 2010, 120, 1345–1354. [Google Scholar] [CrossRef]
- Harington, C.R.; Pitt-Rivers, R. Note on the synthesis of the acetic acid analogue of thyroxine. Biochem. J. 1952, 50, 438–439. [Google Scholar] [CrossRef]
- Braverman, L.E.; Ingbar, S.H.; Sterling, K. Conversion of thyroxine (T4) to triiodothyronine (T3) in athyreotic human subjects. J. Clin. Investig. 1970, 49, 855–864. [Google Scholar] [CrossRef]
- Beck-Peccoz, P.; Piscitelli, G.; Cattaneo, M.G.; Faglia, G. Successful treatment of hyperthyroidism due to nonneoplastic pituitary TSH hypersecretion with 3,5,3′-triiodothyroacetic acid (TRIAC). J. Endocrinol. Investig. 1983, 6, 217–223. [Google Scholar] [CrossRef]
- Usala, S.J.; Bale, A.E.; Gesundheit, N.; Weinberger, C.; Lash, R.W.; Wondisford, F.E.; McBride, O.W.; Weintraub, B.D. Tight linkage between the syndrome of generalized thyroid hormone resistance and the human c-erbA beta gene. Mol. Endocrinol. 1988, 2, 1217–1220. [Google Scholar] [CrossRef]
- Manka, P.; Bechmann, L.; Best, J.; Sydor, S.; Claridge, L.C.; Coombes, J.D.; Canbay, A.; Moeller, L.; Gerken, G.; Wedemeyer, H.; et al. Low Free Triiodothyronine Is Associated with Advanced Fibrosis in Patients at High Risk for Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2019, 64, 2351–2358. [Google Scholar] [CrossRef]
- Chung, M.K.; Eckhardt, L.L.; Chen, L.Y.; Ahmed, H.M.; Gopinathannair, R.; Joglar, J.A.; Noseworthy, P.A.; Pack, Q.R.; Sanders, P.; Trulock, K.M. Lifestyle and Risk Factor Modification for Reduction of Atrial Fibrillation: A Scientific Statement From the American Heart Association. Circulation 2020, 141, e750–e772. [Google Scholar] [CrossRef]
- Sabzwari, S.R.A.; Garg, L.; Lakkireddy, D.; Day, J. Ten Lifestyle Modification Approaches to Treat Atrial Fibrillation. Cureus 2018, 10, e2682. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, A.V.; Pennella, S.; Farinetti, A.; Manenti, A. Energy Drinks and atrial fibrillation in young adults. Clin. Nutr. 2018, 37, 1073–1074. [Google Scholar] [CrossRef] [PubMed]
- Bochukova, E.; Schoenmakers, N.; Agostini, M.; Schoenmakers, E.; Rajanayagam, O.; Keogh, J.M.; Henning, E.; Reinemund, J.; Gevers, E.; Sarri, M.; et al. A mutation in the thyroid hormone receptor alpha gene. N. Engl. J. Med. 2012, 366, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.; Agostini, M.; Visser, W.E.; Schoenmakers, E.; Schoenmakers, N.; Offiah, A.C.; Poole, K.; Rajanayagam, O.; Lyons, G.; Halsall, D.; et al. Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)α1 and TRα2: Clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol. 2014, 2, 619–626. [Google Scholar] [CrossRef]
- Moran, C.; Agostini, M.; McGowan, A.; Schoenmakers, E.; Fairall, L.; Lyons, G.; Rajanayagam, O.; Watson, L.; Offiah, A.; Barton, J.; et al. Contrasting Phenotypes in Resistance to Thyroid Hormone Alpha Correlate with Divergent Properties of Thyroid Hormone Receptor α1 Mutant Proteins. Thyroid 2017, 27, 973–982. [Google Scholar] [CrossRef]
- van Mullem, A.; van Heerebeek, R.; Chrysis, D.; Visser, E.; Medici, M.; Andrikoula, M.; Tsatsoulis, A.; Peeters, R.; Visser, T.J. Clinical phenotype and mutant TRα1. N. Engl. J. Med. 2012, 366, 1451–1453. [Google Scholar] [CrossRef]
- van Gucht, A.L.M.; Meima, M.E.; Moran, C.; Agostini, M.; Tylki-Szymanska, A.; Krajewska, M.W.; Chrzanowska, K.; Efthymiadou, A.; Chrysis, D.; Demir, K.; et al. Anemia in Patients with Resistance to Thyroid Hormone α: A Role for Thyroid Hormone Receptor α in Human Erythropoiesis. J. Clin. Endocrinol. Metab. 2017, 102, 3517–3525. [Google Scholar] [CrossRef]
- van Gucht, A.L.; Meima, M.E.; Zwaveling-Soonawala, N.; Visser, W.E.; Fliers, E.; Wennink, J.M.; Henny, C.; Visser, T.J.; Peeters, R.P.; van Trotsenburg, A.S. Resistance to Thyroid Hormone Alpha in an 18-Month-Old Girl: Clinical, Therapeutic, and Molecular Characteristics. Thyroid 2016, 26, 338–346. [Google Scholar] [CrossRef]
- Han, C.R.; Wang, H.; Hoffmann, V.; Zerfas, P.; Kruhlak, M.; Cheng, S.Y. Thyroid Hormone Receptor α Mutations Cause Heart Defects in Zebrafish. Thyroid 2021, 31, 315–326. [Google Scholar] [CrossRef]
- Demir, K.; van Gucht, A.L.; Büyükinan, M.; Çatlı, G.; Ayhan, Y.; Baş, V.N.; Dündar, B.; Özkan, B.; Meima, M.E.; Visser, W.E.; et al. Diverse Genotypes and Phenotypes of Three Novel Thyroid Hormone Receptor-α Mutations. J. Clin. Endocrinol. Metab. 2016, 101, 2945–2954. [Google Scholar] [CrossRef]
- Dahll, L.K.; Westbye, A.B.; Vinorum, K.; Sejersted, Y.; Barøy, T.; Thorsby, P.M.; Hammerstad, S.S. Clinical and Biochemical Characteristics of Untreated Adult Patients with Resistance to Thyroid Hormone Alpha. J. Endocr. Soc. 2023, 7, bvad089. [Google Scholar] [CrossRef]
- Espiard, S.; Savagner, F.; Flamant, F.; Vlaeminck-Guillem, V.; Guyot, R.; Munier, M.; d’Herbomez, M.; Bourguet, W.; Pinto, G.; Rose, C.; et al. A Novel Mutation in THRA Gene Associated with an Atypical Phenotype of Resistance to Thyroid Hormone. J. Clin. Endocrinol. Metab. 2015, 100, 2841–2848. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.; Schoenmakers, N.; Agostini, M.; Schoenmakers, E.; Offiah, A.; Kydd, A.; Kahaly, G.; Mohr-Kahaly, S.; Rajanayagam, O.; Lyons, G.; et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor α. J. Clin. Endocrinol. Metab. 2013, 98, 4254–4261. [Google Scholar] [CrossRef] [PubMed]
- van Mullem, A.A.; Chrysis, D.; Eythimiadou, A.; Chroni, E.; Tsatsoulis, A.; de Rijke, Y.B.; Visser, W.E.; Visser, T.J.; Peeters, R.P. Clinical phenotype of a new type of thyroid hormone resistance caused by a mutation of the TRα1 receptor: Consequences of LT4 treatment. J. Clin. Endocrinol. Metab. 2013, 98, 3029–3038. [Google Scholar] [CrossRef] [PubMed]
- Dore, R.; Watson, L.; Hollidge, S.; Krause, C.; Sentis, S.C.; Oelkrug, R.; Geißler, C.; Johann, K.; Pedaran, M.; Lyons, G.; et al. Resistance to thyroid hormone induced tachycardia in RTHα syndrome. Nat. Commun. 2023, 14, 3312. [Google Scholar] [CrossRef]
- Korkmaz, O.; Ozen, S.; Ozdemir, T.R.; Goksen, D.; Darcan, S. A novel thyroid hormone receptor alpha gene mutation, clinic characteristics, and follow-up findings in a patient with thyroid hormone resistance. Hormones 2019, 18, 223–227. [Google Scholar] [CrossRef]
- Erbaş, I.M.; Çakır, M.D.; Yener, A.S.; Demir, K. Long-term follow-up results and treatment outcomes of children and adults with resistance to thyroid hormone alpha. J. Endocrinol. Investig. 2023, 46, 1855–1863. [Google Scholar] [CrossRef]
- Roberts, L.M.; Woodford, K.; Zhou, M.; Black, D.S.; Haggerty, J.E.; Tate, E.H.; Grindstaff, K.K.; Mengesha, W.; Raman, C.; Zerangue, N. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 2008, 149, 6251–6261. [Google Scholar] [CrossRef]
- Schreiner, F.; Vollbach, H.; Sonntag, N.; Schempp, V.; Gohlke, B.; Friese, J.; Woelfle, J.; Braun, D.; Schweizer, U. Phenylbutyrate Treatment in a Boy with MCT8 Deficiency: Improvement of Thyroid Function Tests and Possible Hepatotoxicity. J. Clin. Endocrinol. Metab. 2025, 110, e992–e999. [Google Scholar] [CrossRef]
- van Geest, F.S.; Groeneweg, S.; van den Akker, E.L.T.; Bacos, I.; Barca, D.; van den Berg, S.A.A.; Bertini, E.; Brunner, D.; Brunetti-Pierri, N.; Cappa, M.; et al. Long-Term Efficacy of T3 Analogue Triac in Children and Adults with MCT8 Deficiency: A Real-Life Retrospective Cohort Study. J. Clin. Endocrinol. Metab. 2022, 107, e1136–e1147. [Google Scholar] [CrossRef]
- Groeneweg, S.; Peeters, R.P.; Moran, C.; Stoupa, A.; Auriol, F.; Tonduti, D.; Dica, A.; Paone, L.; Rozenkova, K.; Malikova, J.; et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: An international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Wilpert, N.M.; Tonduti, D.; Vaia, Y.; Krude, H.; Sarret, C.; Schuelke, M. Establishing Patient-Centered Outcomes for MCT8 Deficiency: Stakeholder Engagement and Systematic Literature Review. Neuropsychiatr. Dis. Treat. 2023, 19, 2195–2216. [Google Scholar] [CrossRef] [PubMed]
- van Geest, F.S.; Groeneweg, S.; Popa, V.M.; Stals, M.A.M.; Visser, W.E. Parent Perspectives on Complex Needs in Patients with MCT8 Deficiency: An International, Prospective, Registry Study. J. Clin. Endocrinol. Metab. 2023, 109, e330–e335. [Google Scholar] [CrossRef] [PubMed]
- Scholtes, N.; Jelesch, E.; Diesener, P.; Stoffels, J.C.; Völkl, T.M.K. Swallowing Assessment in a Pediatric Case of Allan-Herndon-Dudley Syndrome (MCT8 Deficiency): Advanced Insights into Dysphagia via Flexible Endoscopic Evaluation of Swallowing. Neuropediatrics 2025, 56, 204–207. [Google Scholar] [CrossRef]
- Tonduti, D.; Vanderver, A.; Berardinelli, A.; Schmidt, J.L.; Collins, C.D.; Novara, F.; Genni, A.D.; Mita, A.; Triulzi, F.; Brunstrom-Hernandez, J.E.; et al. MCT8 deficiency: Extrapyramidal symptoms and delayed myelination as prominent features. J. Child. Neurol. 2013, 28, 795–800. [Google Scholar] [CrossRef]
- Remerand, G.; Boespflug-Tanguy, O.; Tonduti, D.; Touraine, R.; Rodriguez, D.; Curie, A.; Perreton, N.; Des Portes, V.; Sarret, C. Expanding the phenotypic spectrum of Allan-Herndon-Dudley syndrome in patients with SLC16A2 mutations. Dev. Med. Child Neurol. 2019, 61, 1439–1447. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Carlson, B.; Agostini, M.; Moran, C.; Rajanayagam, O.; Bochukova, E.; Tobe, R.; Peat, R.; Gevers, E.; Muntoni, F.; et al. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J. Clin. Investig. 2016, 126, 992–996. [Google Scholar] [CrossRef]
- Hamajima, T.; Mushimoto, Y.; Kobayashi, H.; Saito, Y.; Onigata, K. Novel compound heterozygous mutations in the SBP2 gene: Characteristic clinical manifestations and the implications of GH and triiodothyronine in longitudinal bone growth and maturation. Eur. J. Endocrinol. 2012, 166, 757–764. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Marelli, F.; Jørgensen, H.F.; Visser, W.E.; Moran, C.; Groeneweg, S.; Avalos, C.; Jurgens, S.J.; Figg, N.; Finigan, A.; et al. Selenoprotein deficiency disorder predisposes to aortic aneurysm formation. Nat. Commun. 2023, 14, 7994. [Google Scholar] [CrossRef]
- Di Cosmo, C.; McLellan, N.; Liao, X.H.; Khanna, K.K.; Weiss, R.E.; Papp, L.; Refetoff, S. Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J. Clin. Endocrinol. Metab. 2009, 94, 4003–4009. [Google Scholar] [CrossRef]
- Azevedo, M.F.; Barra, G.B.; Naves, L.A.; Ribeiro Velasco, L.F.; Godoy Garcia Castro, P.; de Castro, L.C.; Amato, A.A.; Miniard, A.; Driscoll, D.; Schomburg, L.; et al. Selenoprotein-related disease in a young girl caused by nonsense mutations in the SBP2 gene. J. Clin. Endocrinol. Metab. 2010, 95, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Shichiri, M.; Hamajima, T.; Ishida, N.; Mita, Y.; Nakao, S.; Hagihara, Y.; Yoshida, Y.; Takahashi, K.; Niki, E.; et al. Enhancement of lipid peroxidation and its amelioration by vitamin E in a subject with mutations in the SBP2 gene. J. Lipid Res. 2015, 56, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Reffin, J.; Holmes, S.; Chatfield, S.; Narayan, S. Exercising with a Muscle Wasting Condition. Available online: https://www.musculardystrophyuk.org/support/information/your-condition/exercise) (accessed on 12 May 2025).
- Schomburg, L.; Dumitrescu, A.M.; Liao, X.H.; Bin-Abbas, B.; Hoeflich, J.; Köhrle, J.; Refetoff, S. Selenium supplementation fails to correct the selenoprotein synthesis defect in subjects with SBP2 gene mutations. Thyroid 2009, 19, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Rebouche, C.J. Ascorbic acid and carnitine biosynthesis. Am. J. Clin. Nutr. 1991, 54, 1147S–1152S. [Google Scholar] [CrossRef]
- Chung, E.; Mo, H.; Wang, S.; Zu, Y.; Elfakhani, M.; Rios, S.R.; Chyu, M.C.; Yang, R.S.; Shen, C.L. Potential roles of vitamin E in age-related changes in skeletal muscle health. Nutr. Res. 2018, 49, 23–36. [Google Scholar] [CrossRef]
- Mason, S.A.; Morrison, D.; McConell, G.K.; Wadley, G.D. Muscle redox signalling pathways in exercise. Role of antioxidants. Free Radic. Biol. Med. 2016, 98, 29–45. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Disorder | Resistance to Thyroid Hormone Beta (RTHβ) | Resistance to Thyroid Hormone Alpha (RTHα) | Monocarboxylate Transporter 8 (MCT8) Deficiency | Selenoprotein Deficiency |
|---|---|---|---|---|
| Gene | THRB | THRA | SLC16A2 | SECISBP2 or TRU-TCA 1-1 |
| Free T4 | High | Low–normal or low | Low–normal or low | High |
| Free T3 | High | High–normal or high | Usually high or high–normal | Low or normal |
| Reverse T3 | High | Normal or low | Low | High |
| TSH | Normal or high | Normal (or mildly raised) | Normal (or mildly raised) | Normal |
| Main cardiovascular manifestations | Tachycardia Atrial fibrillation Cardiac insufficiency | Bradycardia Low BP | Tachycardia Systolic hypertension | Thoracic aortic aneurysm |
| Main metabolic manifestations | Failure to thrive High REE Dyslipidemia MASLD Insulin resistance Osteopenia | Low REE | High REE Osteopenia/osteoporosis | Increased fat mass Increased systemic insulin sensitivity |
| Other relevant or targetable manifestations | Anxiety, behavioral disorders, neurocognitive impairment | Anemia, neurocognitive impairment, constipation | Neurocognitive impairment, hypotonia/dystonia, gastroesophageal reflux, feeding problems, constipation | Photosensitivity Axial and limb muscular dystrophy Male infertility |
| Useful lifestyle/dietary interventions | Mediterranean diet, optimize vitamin D and calcium intake Mind–body practices and psychological support Antioxidants Silymarin, cynarine, curcumin for MASLD | Regular aerobic exercise Hypocaloric diet Optimize vitamin D and calcium intake Optimize iron, vitamin B12 and folate intake Probiotics and liquid-rich diet | Optimize vitamin D and calcium intake Psychological and physiotherapeutic support Dietologist/dietician support to avoid malnutrition, feeding tubes, gastrostomy Probiotics | Optimize vitamin D and calcium intake Antioxidants Physiotherapeutic support/regular exercise Hypo-sodium diet |
| Useful treatments [1] | TRIAC, cardioselective beta-blockers, anti-arrhythmic drugs | L-T4 | TRIAC, cardioselective beta-blockers, anti-emesis drugs, spasmolytic and/or anti-cholinergic drugs, anti-constipation remedies | Antioxidants UV skin protection |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodolfi, S.; Rurale, G.; Marelli, F.; Persani, L.; Campi, I. Lifestyle Interventions to Tackle Cardiovascular Risk in Thyroid Hormone Signaling Disorders. Nutrients 2025, 17, 2053. https://doi.org/10.3390/nu17132053
Rodolfi S, Rurale G, Marelli F, Persani L, Campi I. Lifestyle Interventions to Tackle Cardiovascular Risk in Thyroid Hormone Signaling Disorders. Nutrients. 2025; 17(13):2053. https://doi.org/10.3390/nu17132053
Chicago/Turabian StyleRodolfi, Simone, Giuditta Rurale, Federica Marelli, Luca Persani, and Irene Campi. 2025. "Lifestyle Interventions to Tackle Cardiovascular Risk in Thyroid Hormone Signaling Disorders" Nutrients 17, no. 13: 2053. https://doi.org/10.3390/nu17132053
APA StyleRodolfi, S., Rurale, G., Marelli, F., Persani, L., & Campi, I. (2025). Lifestyle Interventions to Tackle Cardiovascular Risk in Thyroid Hormone Signaling Disorders. Nutrients, 17(13), 2053. https://doi.org/10.3390/nu17132053