
Dihydromyricetin May Attenuate Skin Aging as a RAGE Inhibitor


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Evaluation of Skin Wrinkles, Elasticity and Hydration
2.3. Histological Analysis
2.4. Immunohistochemical Staining
2.5. Cell Culture and Treatments
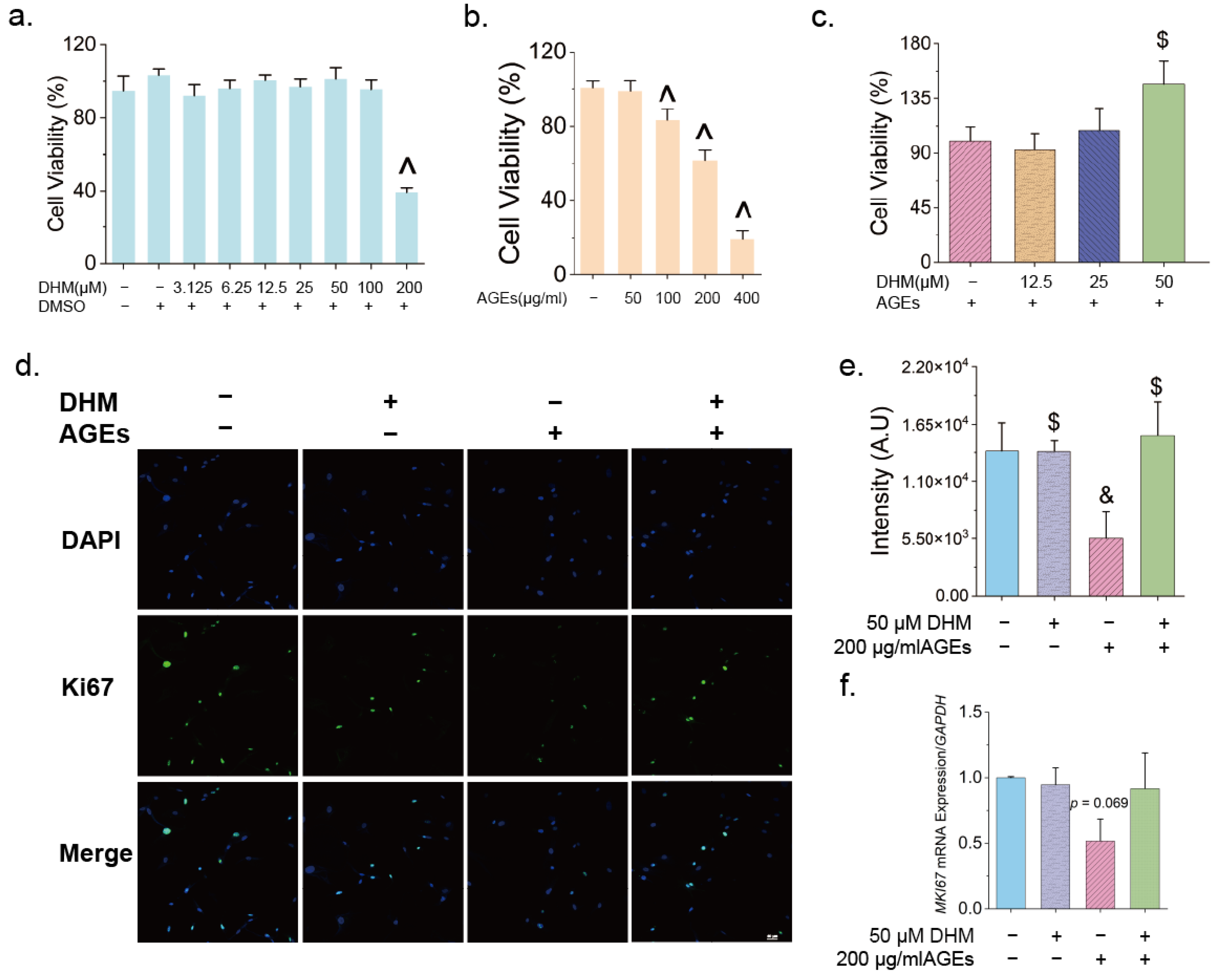
2.6. Cell Viability Assay
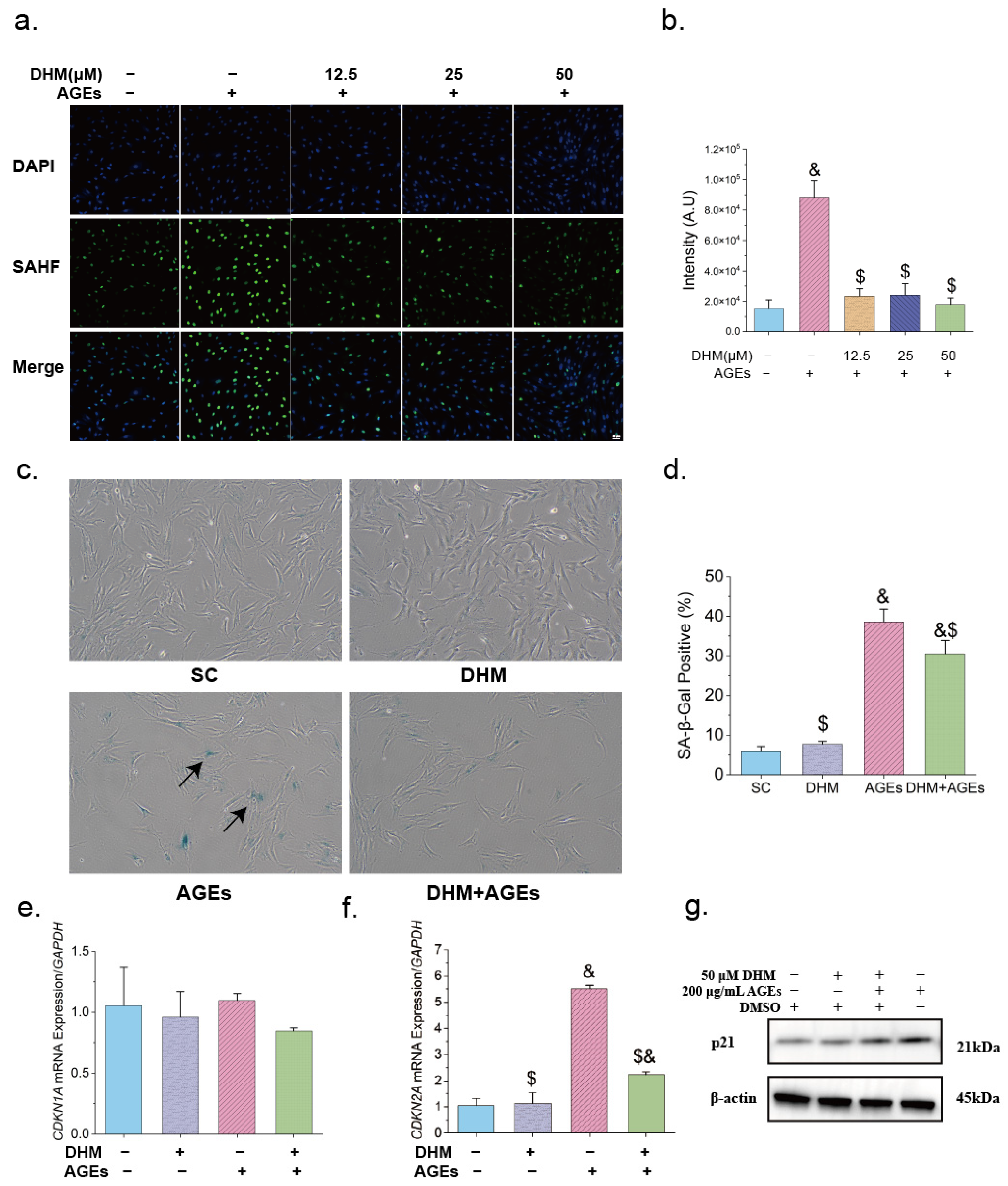
2.7. Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
2.8. Immunofluorescence Staining
2.9. Western Blot
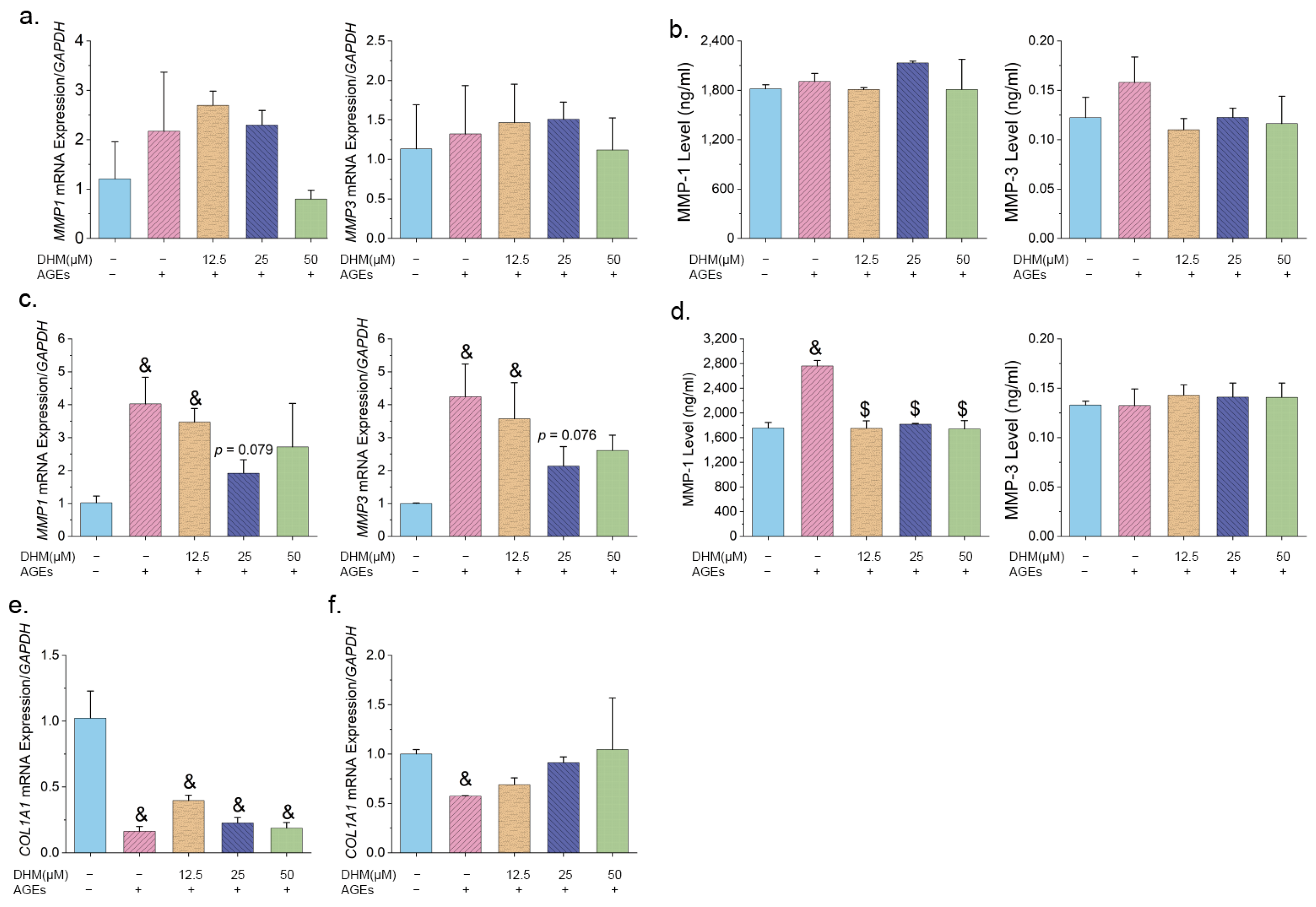
2.10. Measurement of MMPs in Cultured Cells
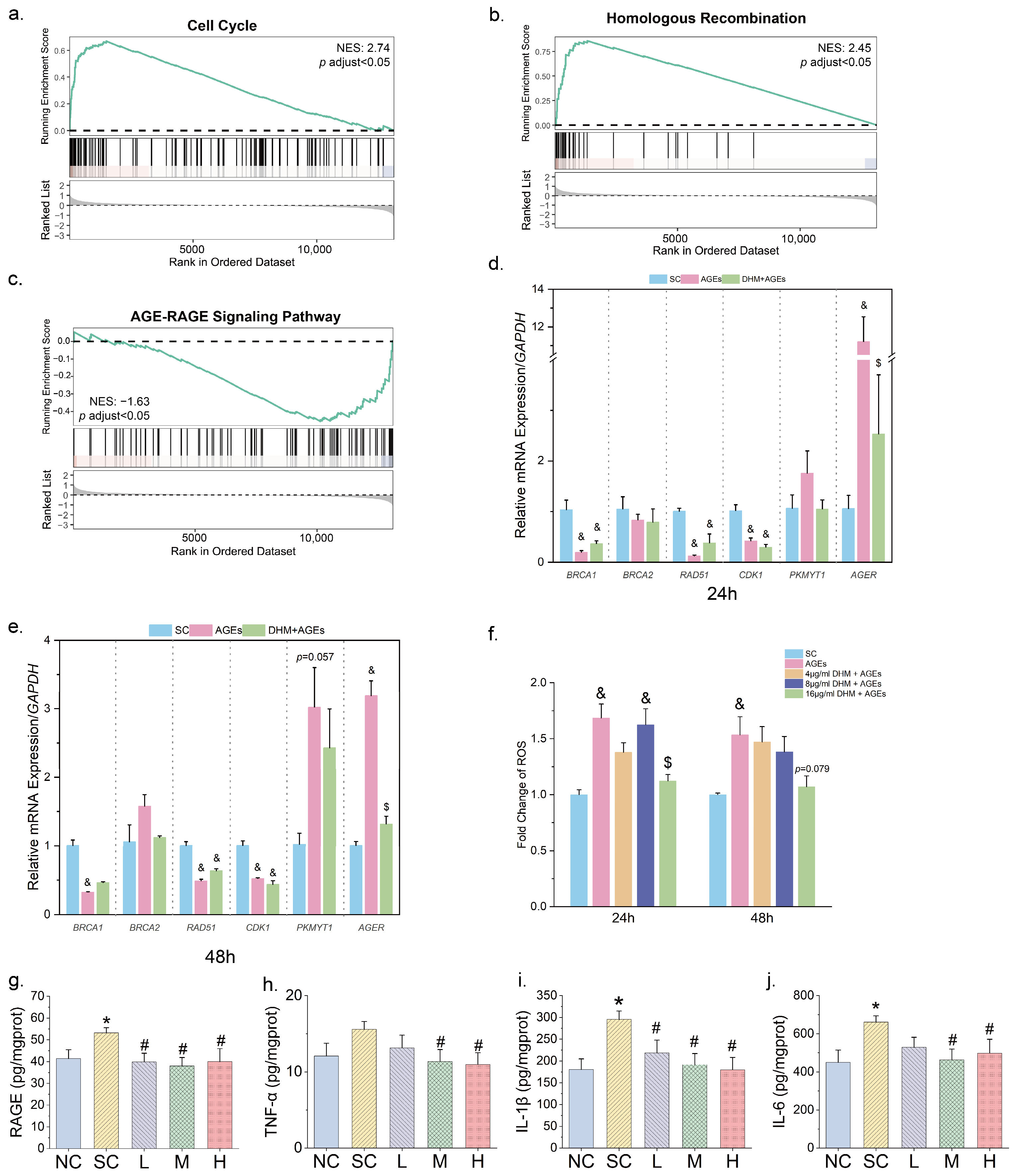
2.11. Transcriptome Sequencing and Analysis
2.12. Quantitative Real-Time PCR (qRT-PCR)
2.13. Measurement of Reactive Oxygen Species
2.14. RAGE and Inflammatory Chemokines Analysis
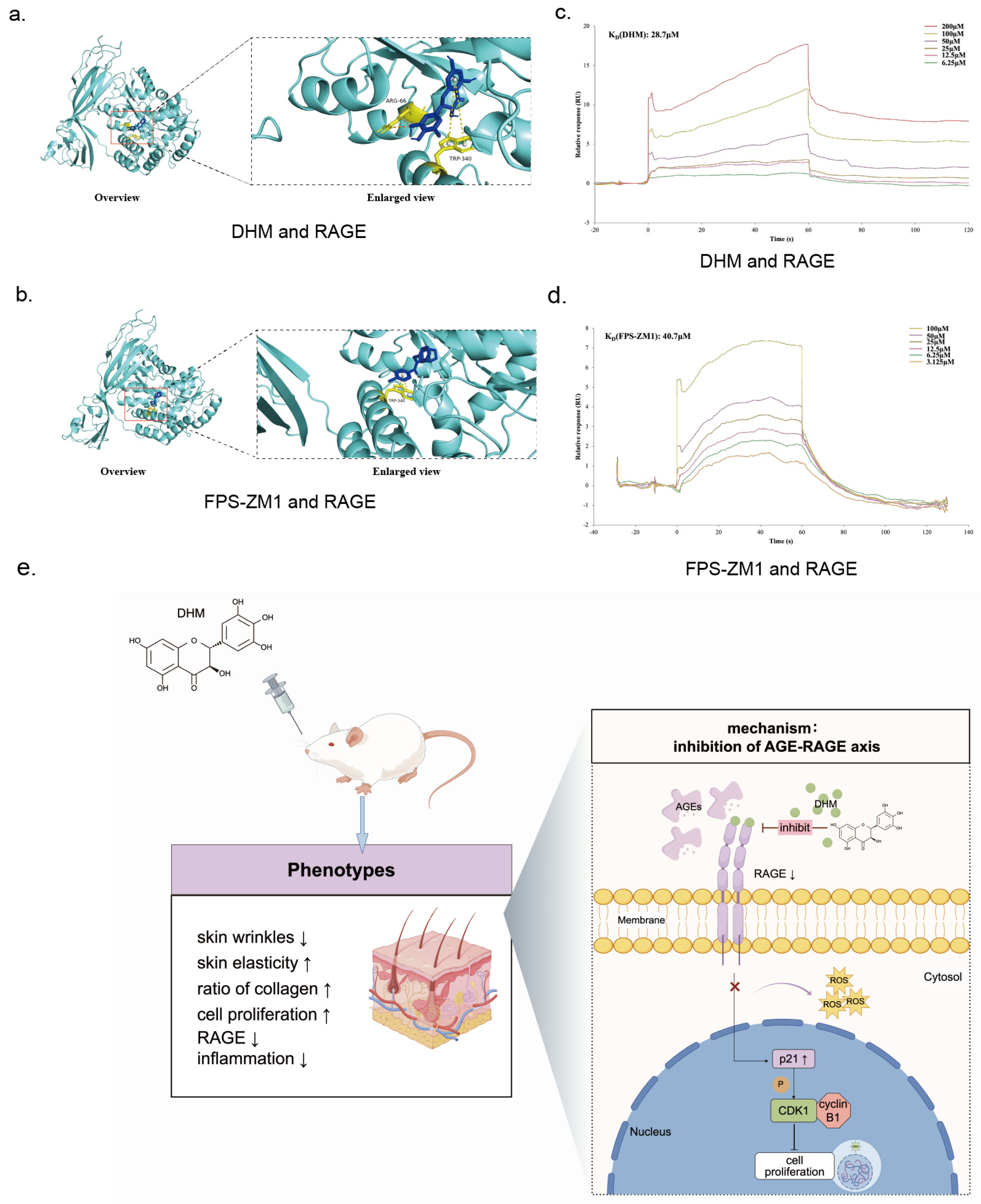
2.15. Molecular Docking
2.16. Surface Plasmon Resonance (SPR)
2.17. Statistical Analysis
3. Results
3.1. DHM Attenuated Skin Aging in Glycated Rats
3.2. DHM Delayed AGE-Induced Cellular Senescence
3.3. Effects of DHM on MMPs and COL1A1 Expression Induced by AGEs in HFF-1 Cells
3.4. DHM Might Mtigate Cellular Senescence by Targeting AGE-RAGE Signaling
3.5. DHM May Function as an Inhibitor of RAGE Against Aging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | advanced glycation end products |
| BCA | Bicinchoninic Acid Assay |
| BRCA1 | breast cancer type 1 susceptibility protein |
| BRCA2 | breast cancer type 2 susceptibility protein |
| CDKI | Cyclin-dependent kinase inhibitor |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| COL1A1 | collagen type I alpha 1 chain |
| DAPI | 4′,6-Diamidino-2′-phenylindole |
| DEG | differentially expressed gene |
| DHM | dihydromyricetin |
| D-loop | displacement loop |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DSB | DNA double-strand break |
| ECM | extracellular matrix |
| FBS | fetal bovine serum |
| FcɛRI | the high-affinity IgE Fc receptor |
| FC | fold change |
| FDR | false discovery rate |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GSEA | Gene Set Enrichment Analysis |
| H&E | hematoxylin and eosin |
| HR | homologous recombination |
| IHC | immunohistochemistry |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| JNK | c-Jun N-terminal kinase |
| KD | dissociation constant |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MKI67 | marker of proliferation Ki-67 |
| MMP | matrix metalloproteinase |
| MMP-1 | matrix metallopeptidase-1 |
| MMP-3 | matrix metallopeptidase-3 |
| NF-κB | nuclear factor-κB |
| PFA | paraformaldehyde |
| PKMYT1 | protein kinase membrane associated tyrosine/threonine 1 |
| pre-MPF | pre-Mitosis Promoting Factor |
| PVDF | polyvinylidene fluoride |
| RAD51 | RAD51 recombinase |
| ROS | reactive oxygen species |
| SAHF | senescence-associated heterochromatin foci |
| SA-β-Gal | senescence-associated β-galactosidase |
| SPF | specific pathogen-free |
| SPR | surface plasmon resonance |
| TBST | Tris-buffered saline with 0.1% Tween-20 |
| TNF-α | tumor necrosis factor-alpha |
| UV | ultraviolet |
References
- Zouboulis, C.C.; Ganceviciene, R.; Liakou, A.I.; Theodoridis, A.; Elewa, R.; Makrantonaki, E. Aesthetic aspects of skin aging, prevention, and local treatment. Clin. Dermatol. 2019, 37, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Chen, S.X.; Guerrero-Juarez, C.F.; Li, F.; Tong, Y.; Liang, Y.; Liggins, M.; Chen, X.; Chen, H.; Li, M.; et al. Age-Related Loss of Innate Immune Antimicrobial Function of Dermal Fat Is Mediated by Transforming Growth Factor Beta. Immunity 2019, 50, 121–136.e125. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H., 3rd; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef]
- Gunn, D.A.; de Craen, A.J.M.; Dick, J.L.; Tomlin, C.C.; van Heemst, D.; Catt, S.D.; Griffiths, T.; Ogden, S.; Maier, A.B.; Murray, P.G.; et al. Facial Appearance Reflects Human Familial Longevity and Cardiovascular Disease Risk in Healthy Individuals. J. Gerontol. Ser. A 2013, 68, 145–152. [Google Scholar] [CrossRef]
- Waaijer, M.E.; Parish, W.E.; Strongitharm, B.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.; Sedivy, J.M.; Westendorp, R.G.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef]
- Skobowiat, C.; Slominski, A.T. UVB Activates Hypothalamic-Pituitary-Adrenal Axis in C57BL/6 Mice. J. Investig. Dermatol. 2015, 135, 1638–1648. [Google Scholar] [CrossRef]
- Gu, Y.; Han, J.; Jiang, C.; Zhang, Y. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 2020, 59, 101036. [Google Scholar] [CrossRef]
- Hussein, R.S.; Bin Dayel, S.; Abahussein, O.; El-Sherbiny, A.A. Influences on Skin and Intrinsic Aging: Biological, Environmental, and Therapeutic Insights. J. Cosmet. Dermatol. 2025, 24, e16688. [Google Scholar] [CrossRef]
- Wong, Q.Y.A.; Chew, F.T. Defining skin aging and its risk factors: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 22075. [Google Scholar] [CrossRef]
- D’Cunha, N.M.; Sergi, D.; Lane, M.M.; Naumovski, N.; Gamage, E.; Rajendran, A.; Kouvari, M.; Gauci, S.; Dissanayka, T.; Marx, W.; et al. The Effects of Dietary Advanced Glycation End-Products on Neurocognitive and Mental Disorders. Nutrients 2022, 14, 2421. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Sell, D.R.; Abdul-Karim, F.W.; Emancipator, S.N. Collagen browning and cross-linking are increased in chronic experimental hyperglycemia. Relevance to diabetes and aging. Diabetes 1988, 37, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M. Toward a Maillard reaction theory of aging. Prog. Clin. Biol. Res. 1989, 304, 1–22. [Google Scholar]
- Zhu, J.; Wang, Z.; Lv, C.; Li, M.; Wang, K.; Chen, Z. Advanced Glycation End Products and Health: A Systematic Review. Ann. Biomed. Eng. 2024, 52, 3145–3156. [Google Scholar] [CrossRef]
- Dozio, E.; Caldiroli, L.; Molinari, P.; Castellano, G.; Delfrate, N.W.; Romanelli, M.M.C.; Vettoretti, S. Accelerated AGEing: The Impact of Advanced Glycation End Products on the Prognosis of Chronic Kidney Disease. Antioxidants 2023, 12, 584. [Google Scholar] [CrossRef] [PubMed]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Singh, S.; Siva, B.V.; Ravichandiran, V. Advanced Glycation End Products: Key player of the pathogenesis of atherosclerosis. Glycoconj. J. 2022, 39, 547–563. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, Y.; Zhao, C. The effects of advanced glycation end-products on skin and potential anti-glycation strategies. Exp. Dermatol. 2024, 33, e15065. [Google Scholar] [CrossRef]
- Chen, C.-y.; Zhang, J.-Q.; Li, L.; Guo, M.-m.; He, Y.-f.; Dong, Y.-m.; Meng, H.; Yi, F. Advanced Glycation End Products in the Skin: Molecular Mechanisms, Methods of Measurement, and Inhibitory Pathways. Front. Med. 2022, 9, 837222. [Google Scholar] [CrossRef]
- Liu, D.; Mao, Y.; Ding, L.; Zeng, X.A. Dihydromyricetin: A review on identification and quantification methods, biological activities, chemical stability, metabolism and approaches to enhance its bioavailability. Trends Food Sci. Technol. 2019, 91, 586–597. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Zhang, Y.; Xiong, T.; Niu, Y.; Huang, Y. Extracting myricetin and dihydromyricetin simultaneously from Hovenia acerba seed by Ultrasound-Assisted extraction on a lab and small Pilot-Scale. Ultrason. Sonochem. 2023, 93, 106304. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, F.; Fei, T.; Lin, X.; Wang, L.; Liu, Z. Natural alpha-glucosidase inhibitors from Aquilaria sinensis leaf-tea: Targeted bio-affinity screening, identification, and inhibition mechanism. Food Chem. 2025, 463 Pt 2, 141329. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Xiang, H.; Ding, P.; Wu, T.; Ji, G. Recent update on application of dihydromyricetin in metabolic related diseases. Biomed. Pharmacother. 2022, 148, 112771. [Google Scholar] [CrossRef]
- Fan, X.; Zeng, Y.; Fan, Z.; Cui, L.; Song, W.; Wu, Q.; Gao, Y.; Yang, D.; Mao, X.; Zeng, B.; et al. Dihydromyricetin promotes longevity and activates the transcription factors FOXO and AOP in Drosophila. Aging 2020, 13, 460–476. [Google Scholar] [CrossRef]
- Sun, C.C.; Yin, Z.P.; Chen, J.G.; Wang, W.J.; Zheng, G.D.; Li, J.E.; Chen, L.L.; Zhang, Q.F. Dihydromyricetin Improves Cognitive Impairments in d-Galactose-Induced Aging Mice through Regulating Oxidative Stress and Inhibition of Acetylcholinesterase. Mol. Nutr. Food Res. 2022, 66, e2101002. [Google Scholar] [CrossRef]
- Qian, J.; Wang, X.; Cao, J.; Zhang, W.; Lu, C.; Chen, X. Dihydromyricetin attenuates D-galactose-induced brain aging of mice via inhibiting oxidative stress and neuroinflammation. Neurosci. Lett. 2021, 756, 135963. [Google Scholar] [CrossRef]
- Falckenhayn, C.; Bienkowska, A.; Sohle, J.; Wegner, K.; Raddatz, G.; Kristof, B.; Kuck, D.; Siegner, R.; Kaufmann, R.; Korn, J.; et al. Identification of dihydromyricetin as a natural DNA methylation inhibitor with rejuvenating activity in human skin. Front. Aging 2023, 4, 1258184. [Google Scholar] [CrossRef]
- He, Z.; Zhang, L.; Zhuo, C.; Jin, F.; Wang, Y. Apoptosis inhibition effect of Dihydromyricetin against UVA-exposed human keratinocyte cell line. J. Photochem. Photobiol. B 2016, 161, 40–49. [Google Scholar] [CrossRef]
- Moon, N.R.; Kang, S.; Park, S. Consumption of ellagic acid and dihydromyricetin synergistically protects against UV-B induced photoaging, possibly by activating both TGF-β1 and wnt signaling pathways. J. Photochem. Photobiol. B 2018, 178, 92–100. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, G.D.; Luo, X.B.; Yin, B.; Shu, B.; Guan, J.Z.; Jia, C.Y. Potential of bone marrow mesenchymal stem cells in rejuvenation of the aged skin of rats. Biomed. Rep. 2017, 6, 279–284. [Google Scholar] [CrossRef]
- Yuan, S.; Yang, Y.; Li, J.; Tan, X.; Cao, Y.; Li, S.; Hong, H.D.; Liu, L.; Zhang, Q. Ganoderma lucidum Rhodiola compound preparation prevent D-galactose-induced immune impairment and oxidative stress in aging rat model. Sci. Rep. 2020, 10, 19244. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, N.; Arnold, R.; Equey, A.; Gandhi, A.; Adams, P.D. The role of the dynamic epigenetic landscape in senescence: Orchestrating SASP expression. NPJ Aging 2024, 10, 48. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, F.; Yang, Y.; Yang, Y.; Liu, Z. Preventive effect of human umbilical cord mesenchymal stem cells on skin aging in rats. Heliyon 2024, 10, e24342. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: A key therapeutic target in aging and diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef]
- Franco, A.C.; Aveleira, C.; Cavadas, C. Skin senescence: Mechanisms and impact on whole-body aging. Trends Mol. Med. 2022, 28, 97–109. [Google Scholar] [CrossRef]
- Zheng, D.L.; Wu, Q.R.; Zeng, P.; Li, S.M.; Cai, Y.J.; Chen, S.Z.; Luo, X.S.; Kuang, S.J.; Rao, F.; Lai, Y.Y.; et al. Advanced glycation end products induce senescence of atrial myocytes and increase susceptibility of atrial fibrillation in diabetic mice. Aging Cell 2022, 21, e13734. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chen, S.H.; Lin, T.; Liao, Y.W.; Chang, Y.C.; Chen, C.C.; Yu, C.C.; Chen, C.J. Resveratrol attenuates advanced glycation end product-induced senescence and inflammation in human gingival fibroblasts. J. Dent. Sci. 2024, 19, 580–586. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Fontanella, R.A.; Scisciola, L.; Pesapane, A.; Taktaz, F.; Franzese, M.; Puocci, A.; Ceriello, A.; Prattichizzo, F.; Rizzo, M.R.; et al. Targeting redox imbalance in neurodegeneration: Characterizing the role of GLP-1 receptor agonists. Theranostics 2023, 13, 4872–4884. [Google Scholar] [CrossRef] [PubMed]
- Massacci, G.; Perfetto, L.; Sacco, F. The Cyclin-dependent kinase 1: More than a cell cycle regulator. Br. J. Cancer 2023, 129, 1707–1716. [Google Scholar] [CrossRef]
- Radziszewski, M.; Galus, R.; Luszczynski, K.; Winiarski, S.; Wasowski, D.; Malejczyk, J.; Wlodarski, P.; Sciezynska, A. The RAGE Pathway in Skin Pathology Development: A Comprehensive Review of Its Role and Therapeutic Potential. Int. J. Mol. Sci. 2024, 25, 13570. [Google Scholar] [CrossRef]
- Shu, M.; Cheng, W.; Jia, X.; Bai, X.; Zhao, Y.; Lu, Y.; Zhu, L.; Zhu, Y.; Wang, L.; Shu, Y.; et al. AGEs promote atherosclerosis by increasing LDL transcytosis across endothelial cells via RAGE/NF-kappaB/Caveolin-1 pathway. Mol. Med. 2023, 29, 113. [Google Scholar] [CrossRef]
- Sultana, R.; Parveen, A.; Kang, M.C.; Hong, S.M.; Kim, S.Y. Glyoxal-derived advanced glycation end products (GO-AGEs) with UVB critically induce skin inflammaging: In vitro and in silico approaches. Sci. Rep. 2024, 14, 1843. [Google Scholar] [CrossRef]
- Xie, J.; Mendez, J.D.; Mendez-Valenzuela, V.; Aguilar-Hernandez, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Chen, M.C.; Chen, K.C.; Chang, G.C.; Lin, H.; Wu, C.C.; Kao, W.H.; Teng, C.J.; Hsu, S.L.; Yang, T.Y. RAGE acts as an oncogenic role and promotes the metastasis of human lung cancer. Cell Death Dis. 2020, 11, 265. [Google Scholar] [CrossRef]
- Banerjee, A.; Singh, P.; Sheikh, P.A.; Kumar, A.; Koul, V.; Bhattacharyya, J. Simultaneous regulation of AGE/RAGE signaling and MMP-9 expression by an immunomodulating hydrogel accelerates healing in diabetic wounds. Biomater. Adv. 2024, 163, 213937. [Google Scholar] [CrossRef]
- Pathomthongtaweechai, N.; Chutipongtanate, S. AGE/RAGE signaling-mediated endoplasmic reticulum stress and future prospects in non-coding RNA therapeutics for diabetic nephropathy. Biomed. Pharmacother. 2020, 131, 110655. [Google Scholar] [CrossRef]
- Prasad, K. AGE-RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell. Biochem. 2019, 459, 95–112. [Google Scholar] [CrossRef]
- Sarkar, S. Pathological role of RAGE underlying progression of various diseases: Its potential as biomarker and therapeutic target. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2025, 398, 3467–3487. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; He, X.; Liu, N.; Deng, H. Role of reactive oxygen species in ultraviolet-induced photodamage of the skin. Cell Div. 2024, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, X.; Zhang, W.; Wei, X.; Chen, X.L.; Wang, X. Zn-DHM nanozymes regulate metabolic and immune homeostasis for early diabetic wound therapy. Bioact. Mater. 2025, 49, 63–84. [Google Scholar] [CrossRef]
- Huang, J.; Chen, B.; Wang, H.; Hu, S.; Yu, X.; Reilly, J.; He, Z.; You, Y.; Shu, X. Dihydromyricetin Attenuates Depressive-like Behaviors in Mice by Inhibiting the AGE-RAGE Signaling Pathway. Cells 2022, 11, 3730. [Google Scholar] [CrossRef]
- Wen, X.; Lv, C.; Zhou, R.; Wang, Y.; Zhou, X.; Qin, S. The Molecular Mechanism Underlying the Therapeutic Effect of Dihydromyricetin on Type 2 Diabetes Mellitus Based on Network Pharmacology, Molecular Docking, and Transcriptomics. Foods 2024, 13, 344. [Google Scholar] [CrossRef]
- Amornsupak, K.; Thongchot, S.; Thinyakul, C.; Box, C.; Hedayat, S.; Thuwajit, P.; Eccles, S.A.; Thuwajit, C. HMGB1 mediates invasion and PD-L1 expression through RAGE-PI3K/AKT signaling pathway in MDA-MB-231 breast cancer cells. BMC Cancer 2022, 22, 578. [Google Scholar] [CrossRef]
- Xie, J.; Liu, J.; Chen, T.M.; Lan, Q.; Zhang, Q.Y.; Liu, B.; Dai, D.; Zhang, W.D.; Hu, L.P.; Zhu, R.Z. Dihydromyricetin alleviates carbon tetrachloride-induced acute liver injury via JNK-dependent mechanism in mice. World J. Gastroenterol. 2015, 21, 5473–5481. [Google Scholar] [CrossRef]
- He, C.; Chen, Y.; Xie, J.; Luo, M.; Fisher, D.; Hien, N.T.T.; Musabaev, E.; Dang, Y.; Zhao, L.; Xia, Y. Dihydromyricetin: An emerging compound with comprehensive effects on multiple systems. Front. Pharmacol. 2024, 15, 1488003. [Google Scholar] [CrossRef]
- Fan, L.; Tong, Q.; Dong, W.; Yang, G.; Hou, X.; Xiong, W.; Shi, C.; Fang, J.; Wang, W. Tissue Distribution, Excretion, and Metabolic Profile of Dihydromyricetin, a Flavonoid from Vine Tea (Ampelopsis grossedentata) after Oral Administration in Rats. J. Agric. Food Chem. 2017, 65, 4597–4604. [Google Scholar] [CrossRef] [PubMed]
- Carry, E.; Kshatriya, D.; Silva, J.; Davies, D.L.; Yuan, B.; Wu, Q.; Patel, H.; Park, E.R.; Gilleran, J.; Hao, L.; et al. Identification of Dihydromyricetin and Metabolites in Serum and Brain Associated with Acute Anti-Ethanol Intoxicating Effects in Mice. Int. J. Mol. Sci. 2021, 22, 7460. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′ ) |
|---|---|---|
| CDKN1A | TGTCCGTCAGAACCCATGC | AAAGTCGAAGTTCCATCGCTC |
| CDKN2A | GGGTTTTCGTGGTTCACATCC | CTAGACGCTGGCTCCTCAGTA |
| MKI67 | ACGCCTGGTTACTATCAAAAGG | CAGACCCATTTACTTGTGTTGGA |
| MMP1 | AAAATTACACGCCAGATTTGCC | GGTGTGACATTACTCCAGAGTTG |
| MMP3 | AAGGATACAACAGGGACCA | GTTGGCTGAGTGAAAGAGAC |
| COL1A1 | GTGCGATGACGTGATCTGTGA | CGGTGGTTTCTTGGTCGGT |
| BRCA1 | TTGTTACAAATCACCCCTCAAGG | CCCTGATACTTTTCTGGATGCC |
| BRCA2 | TGCCTGAAAACCAGATGACTATC | AGGCCAGCAAACTTCCGTTTA |
| RAD51 | CAACCCATTTCACGGTTAGAGC | TTCTTTGGCGCATAGGCAACA |
| CDK1 | AAACTACAGGTCAAGTGGTAGCC | TCCTGCATAAGCACATCCTGA |
| PKMYT1 | GCCTGCCAACATCTTCCTG | CCCAGACTGAACACATCCGC |
| AGER | CACCTTCTCCTGTAGCTTCAGC | AGGAGCTACTGCTCCACCTTCT |
| GAPDH | ACAGTCAGCCGCATCTTCTT | GTTAAAAGCAGCCCTGGTGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Jian, Y.; Xia, F.; Kuo, L.; Wang, J. Dihydromyricetin May Attenuate Skin Aging as a RAGE Inhibitor. Nutrients 2025, 17, 1862. https://doi.org/10.3390/nu17111862
Wang F, Jian Y, Xia F, Kuo L, Wang J. Dihydromyricetin May Attenuate Skin Aging as a RAGE Inhibitor. Nutrients. 2025; 17(11):1862. https://doi.org/10.3390/nu17111862
Chicago/Turabian StyleWang, Fei, Yuanzhi Jian, Fangzhi Xia, Liangchun Kuo, and Junbo Wang. 2025. "Dihydromyricetin May Attenuate Skin Aging as a RAGE Inhibitor" Nutrients 17, no. 11: 1862. https://doi.org/10.3390/nu17111862
APA StyleWang, F., Jian, Y., Xia, F., Kuo, L., & Wang, J. (2025). Dihydromyricetin May Attenuate Skin Aging as a RAGE Inhibitor. Nutrients, 17(11), 1862. https://doi.org/10.3390/nu17111862