

Nicotinamide Adenine Dinucleotide Supplementation to Alleviate Heart Failure: A Mitochondrial Dysfunction Perspective


Abstract
1. Introduction
2. Materials and Methods
3. Heart Failure
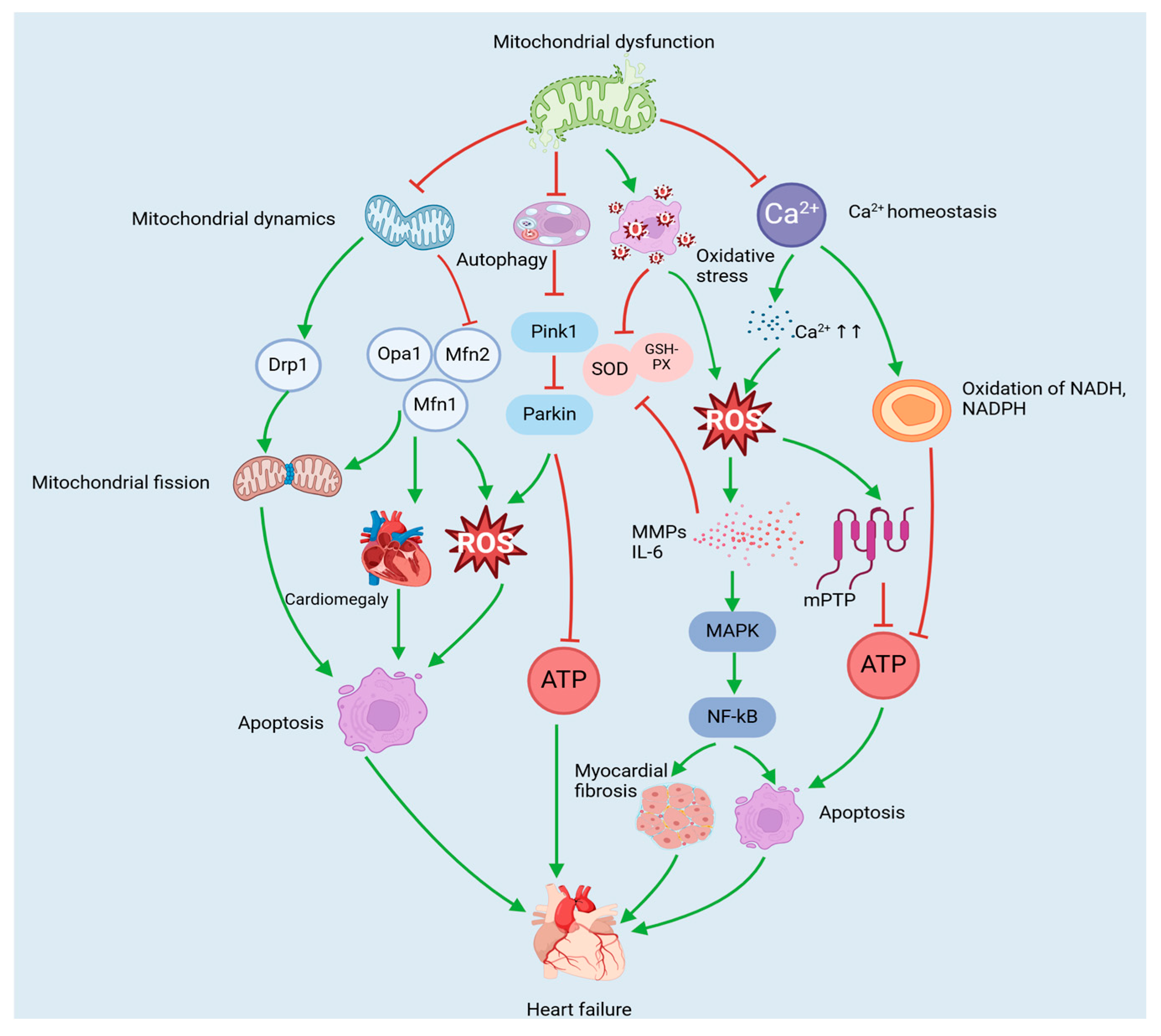
3.1. Mitochondria-Mediated Pathology of Heart Failure
3.1.1. Heart Failure and Mitochondrial Dynamics
3.1.2. Heart Failure and Mitochondrial Autophagy
3.1.3. Heart Failure and Mitochondrial Oxidative Stress
3.1.4. Heart Failure and Mitochondrial Ca2+ Homeostasis
3.2. Effects of Other Metabolic Diseases on Heart Failure
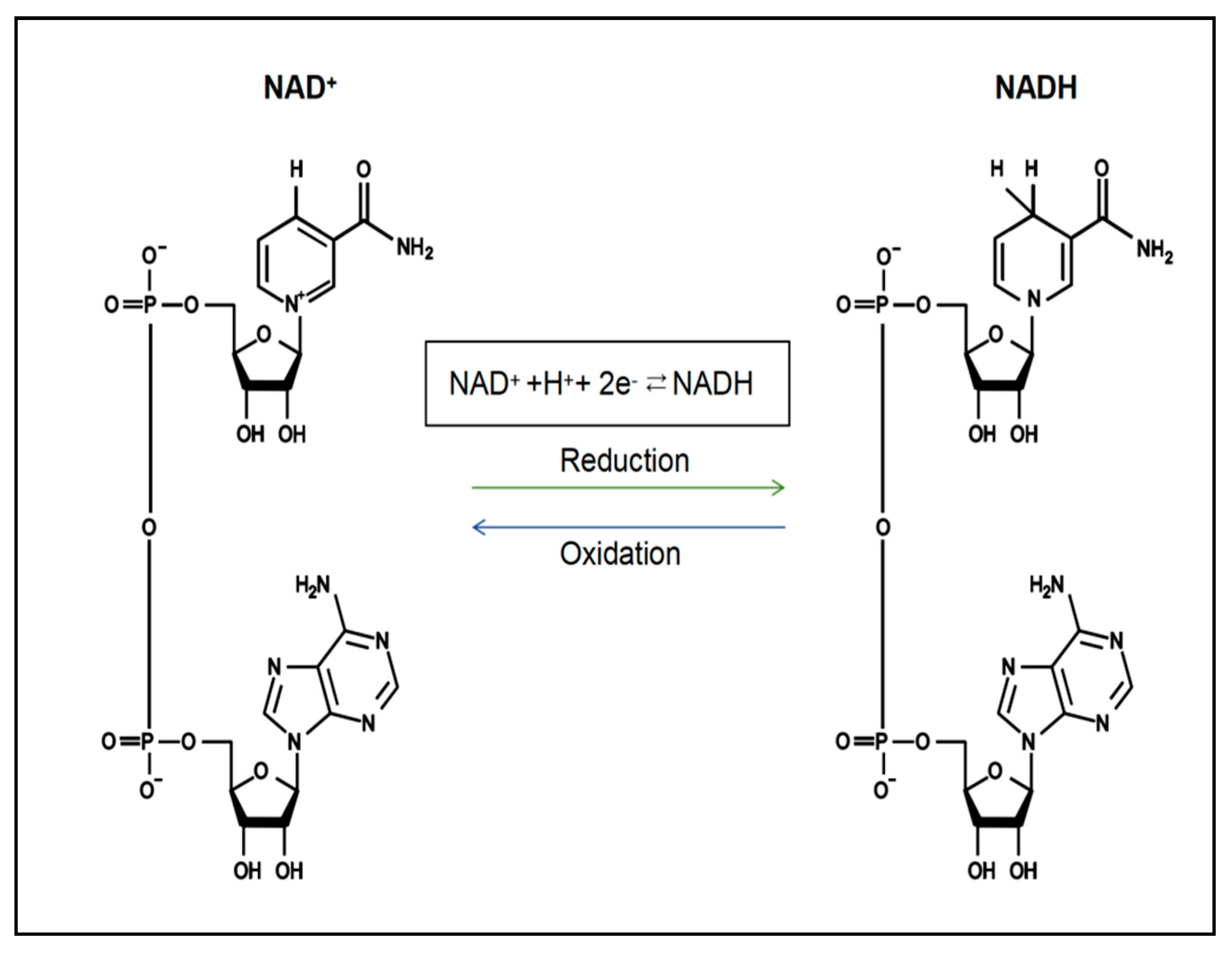
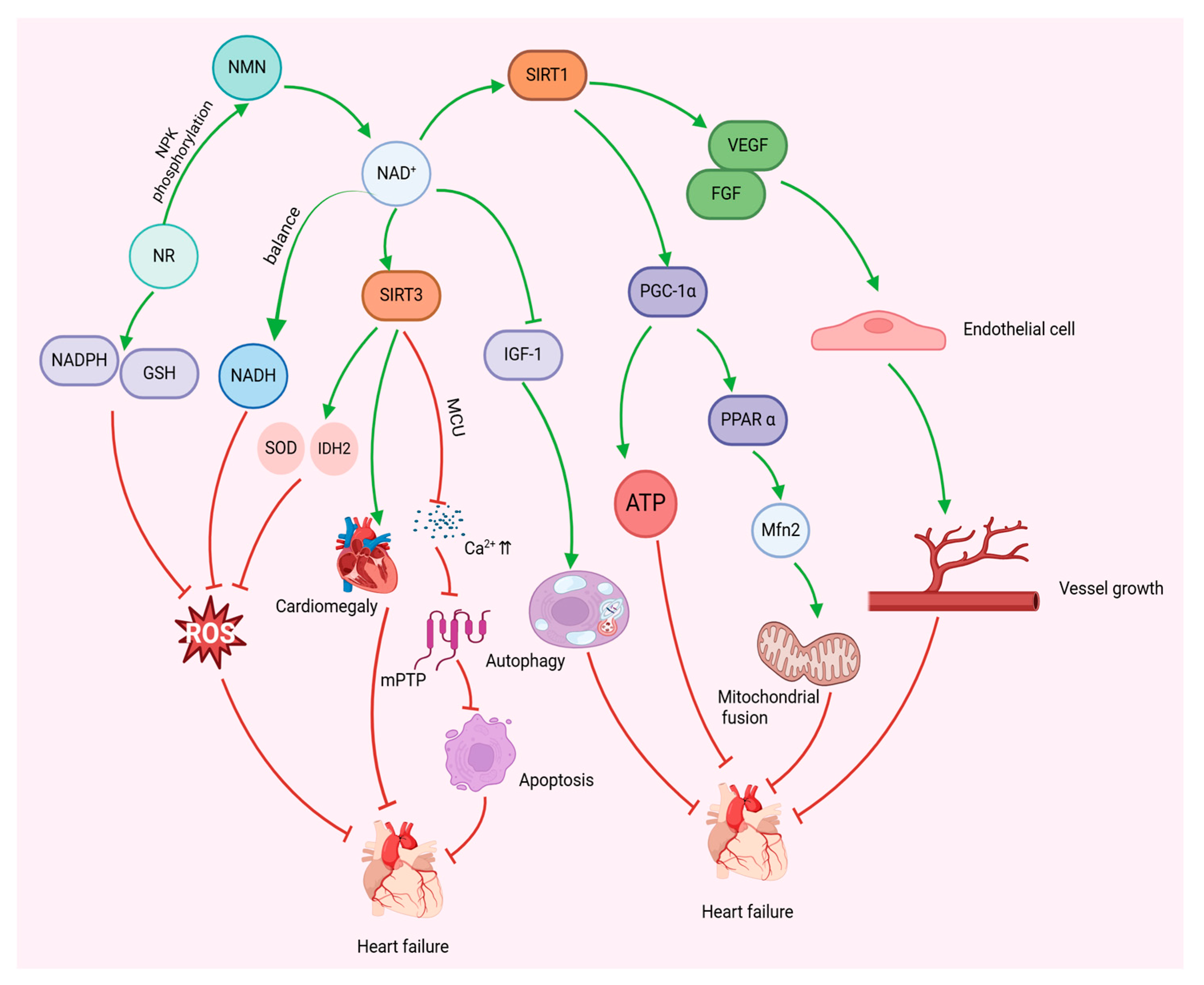
4. NAD+ and Mitochondrial Biogenesis
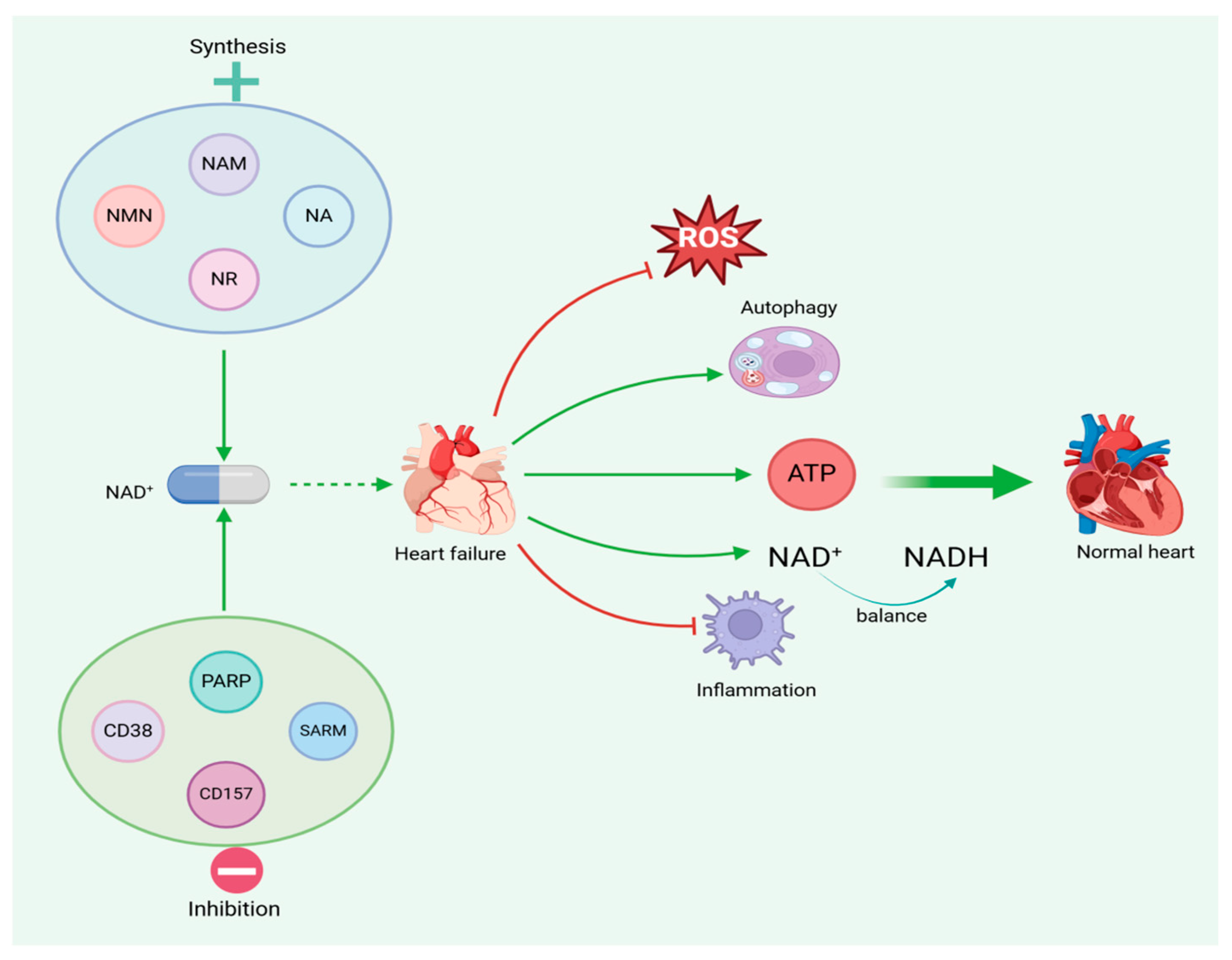
5. NAD+ Modulates Mitochondria to Improve Heart Failure
5.1. Increased Levels of NAD+
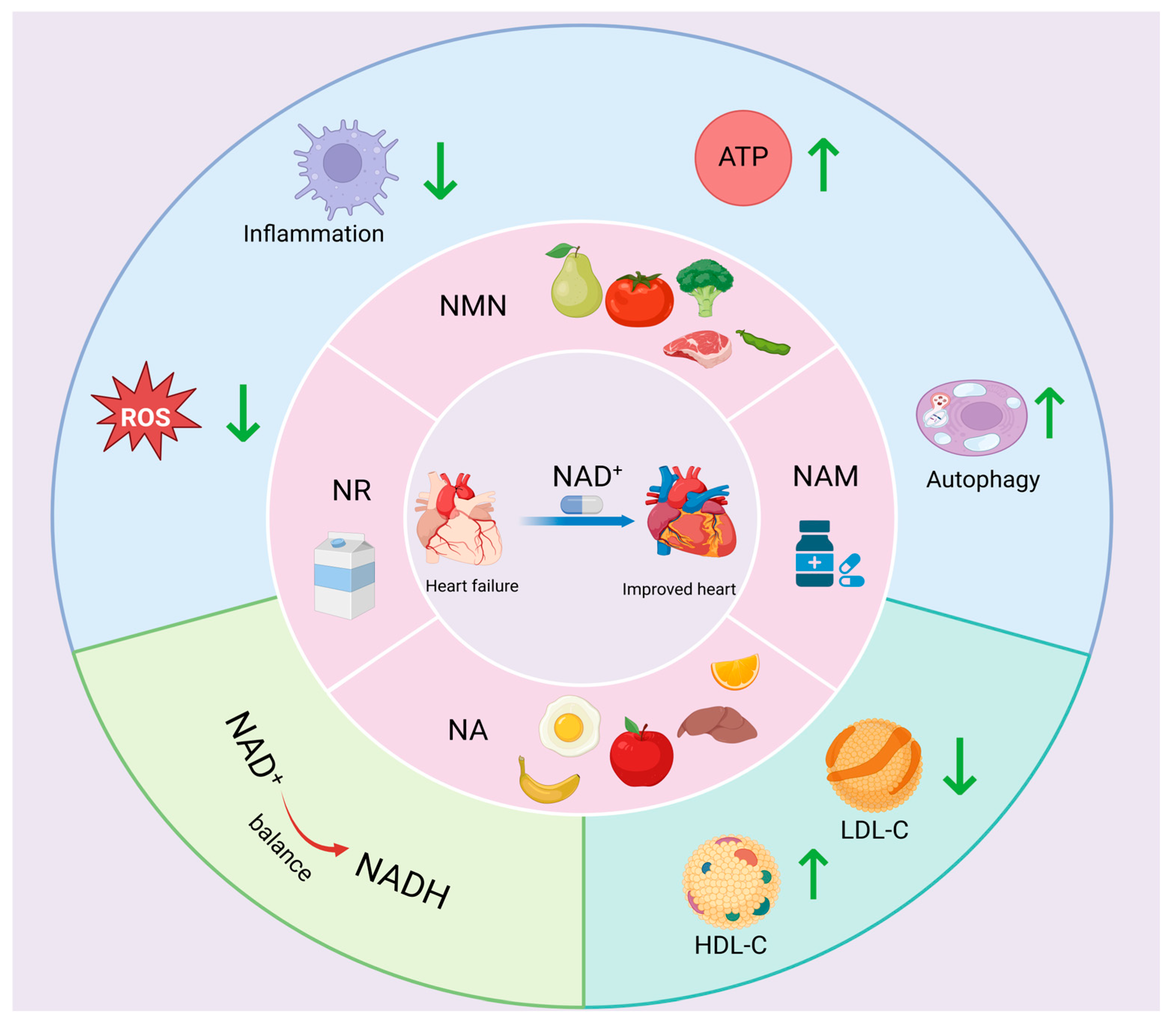
5.2. NAD+ Precursor Supplementation
5.2.1. NR
5.2.2. NMN
5.2.3. NAM
5.2.4. NA
5.2.5. Inhibition of NAD+ Consumption
6. Summary and Outlook
7. Limitations
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Miura, M.; Nochioka, K.; Sakata, Y. Heart failure as a general pandemic in Asia. Eur. J. Heart Fail. 2015, 17, 884–892. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ogden, L.G.; Bazzano, L.A.; Vupputuri, S.; Loria, C.; Whelton, P.K. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch. Intern. Med. 2001, 161, 996–1002. [Google Scholar] [CrossRef]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [CrossRef]
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A.J.S. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2023, 118, 3272–3287. [Google Scholar] [CrossRef]
- Guha, K.; McDonagh, T. Heart failure epidemiology: European perspective. Curr. Cardiol. Rev. 2013, 9, 123–127. [Google Scholar] [CrossRef]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef]
- Lin, L.; Xu, H.; Yao, Z.; Zeng, X.; Kang, L.; Li, Y.; Zhou, G.; Wang, S.; Zhang, Y.; Cheng, D.; et al. Jin-Xin-Kang alleviates heart failure by mitigating mitochondrial dysfunction through the Calcineurin/Dynamin-Related Protein 1 signaling pathway. J. Ethnopharmacol. 2024, 335, 118685. [Google Scholar] [CrossRef]
- Popa, I.P.; Haba MȘ, C.; Mărănducă, M.A.; Tănase, D.M.; Șerban, D.N.; Șerban, L.I.; Iliescu, R.; Tudorancea, I. Modern Approaches for the Treatment of Heart Failure: Recent Advances and Future Perspectives. Pharmaceutics 2022, 14, 1964. [Google Scholar] [CrossRef] [PubMed]
- Zhong, O.; Wang, J.; Tan, Y.; Lei, X.; Tang, Z. Effects of NAD+ precursor supplementation on glucose and lipid metabolism in humans: A meta-analysis. Nutr. Metab. 2022, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Zeidler, J.D.; Kashyap, S.; Warner, G.; Chini, E.N. Evolving concepts in NAD+ metabolism. Cell Metab. 2021, 33, 1076–1087. [Google Scholar] [CrossRef]
- Anderson, R.M.; Bitterman, K.J.; Wood, J.G.; Medvedik, O.; Sinclair, D.A. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 2003, 423, 181–185. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Fang, J.; Chen, W.; Hou, P.; Liu, Z.; Zuo, M.; Liu, S.; Feng, C.; Han, Y.; Li, P.; Shi, Y.; et al. NAD+ metabolism-based immunoregulation and therapeutic potential. Cell Biosci. 2023, 13, 81. [Google Scholar] [CrossRef]
- Hershberger, K.A.; Martin, A.S.; Hirschey, M.D. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 2017, 13, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Luo, C.; Chowdhury, S.; Gao, Z.H.; Liu, J.L. Parp1 deficient mice are protected from streptozotocin-induced diabetes but not caerulein-induced pancreatitis, independent of the induction of Reg family genes. Regul. Pept. 2013, 186, 83–91. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Interacting NAD+ and Cell Senescence Pathways Complicate Antiaging Therapies. Rejuvenation Res. 2019, 22, 261–266. [Google Scholar] [CrossRef]
- Rotllan, N.; Camacho, M.; Tondo, M.; Diarte-Añazco, E.M.G.; Canyelles, M.; Méndez-Lara, K.A.; Benitez, S.; Alonso, N.; Mauricio, D.; Escolà-Gil, J.C.; et al. Therapeutic Potential of Emerging NAD+-Increasing Strategies for Cardiovascular Diseases. Antioxidants 2021, 10, 1939. [Google Scholar] [CrossRef]
- Walker, M.A.; Tian, R. Raising NAD in Heart Failure: Time to Translate? Circulation 2018, 137, 2274–2277. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pei, Z.; Qu, P. NAD+-A Hub of Energy Metabolism in Heart Failure. Int. J. Med. Sci. 2024, 21, 369–375. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of Nicotinamide Mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol. 2017, 112, 64–73. [Google Scholar] [CrossRef]
- Matasic, D.S.; Brenner, C.; London, B. Emerging potential benefits of modulating NAD+ metabolism in cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H839–H852. [Google Scholar] [CrossRef]
- Arrigo, M.; Jessup, M.; Mullens, W.; Reza, N.; Shah, A.M.; Sliwa, K.; Mebazaa, A. Acute heart failure. Nat. Rev. Dis. Primers 2020, 6, 16. [Google Scholar] [CrossRef]
- Cleland, J.G.F.; Pfeffer, M.A.; Clark, A.L.; Januzzi, J.L.; McMurray, J.J.V.; Mueller, C.; Pellicori, P.; Richards, M.; Teerlink, J.R.; Zannad, F.; et al. The struggle towards a Universal Definition of Heart Failure-how to proceed? Eur. Heart J. 2021, 42, 2331–2343. [Google Scholar] [CrossRef]
- Pellicori, P.; Cleland, J.G.; Zhang, J.; Kallvikbacka-Bennett, A.; Urbinati, A.; Shah, P.; Kazmi, S.; Clark, A.L. Cardiac Dysfunction, Congestion and Loop Diuretics: Their Relationship to Prognosis in Heart Failure. Cardiovasc. Drugs Ther. 2016, 30, 599–609. [Google Scholar] [CrossRef]
- Shugg, T.; Hudmon, A.; Overholser, B.R. Neurohormonal Regulation of IKs in Heart Failure: Implications for Ventricular Arrhythmogenesis and Sudden Cardiac Death. J. Am. Heart Assoc. 2020, 9, e016900. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Gona, P.; Vasan, R.S.; Larson, M.G.; Benjamin, E.J.; Wang, T.J.; Tu, J.V.; Levy, D. Relation of disease pathogenesis and risk factors to heart failure with preserved or reduced ejection fraction: Insights from the framingham heart study of the national heart, lung, and blood institute. Circulation 2009, 119, 3070–3077. [Google Scholar] [CrossRef]
- Upadhya, B.; Kitzman, D.W. Heart failure with preserved ejection fraction: New approaches to diagnosis and management. Clin. Cardiol. 2020, 43, 145–155. [Google Scholar] [CrossRef]
- Schwinger, R.H.G. Pathophysiology of heart failure. Cardiovasc. Diagn. Ther. 2021, 11, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Golla, M.S.G.; Hajouli, S.; Ludhwani, D. Heart Failure and Ejection Fraction. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Kapoor, J.R.; Kapoor, R.; Ju, C.; Heidenreich, P.A.; Eapen, Z.J.; Hernandez, A.F.; Butler, J.; Yancy, C.W.; Fonarow, G.C. Precipitating Clinical Factors, Heart Failure Characterization, and Outcomes in Patients Hospitalized With Heart Failure With Reduced, Borderline, and Preserved Ejection Fraction. JACC. Heart Fail. 2016, 4, 464–472. [Google Scholar] [CrossRef]
- Tsuji, K.; Sakata, Y.; Nochioka, K.; Miura, M.; Yamauchi, T.; Onose, T.; Abe, R.; Oikawa, T.; Kasahara, S.; Sato, M.; et al. Characterization of heart failure patients with mid-range left ventricular ejection fraction-a report from the CHART-2 Study. Eur. J. Heart Fail. 2017, 19, 1258–1269. [Google Scholar] [CrossRef]
- Löfman, I.; Szummer, K.; Dahlström, U.; Jernberg, T.; Lund, L.H. Associations with and prognostic impact of chronic kidney disease in heart failure with preserved, mid-range, and reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 1606–1614. [Google Scholar] [CrossRef]
- Wang, N.; Hales, S.; Barin, E.; Tofler, G. Characteristics and outcome for heart failure patients with mid-range ejection fraction. J. Cardiovasc. Med. 2018, 19, 297–303. [Google Scholar] [CrossRef]
- Savarese, G.; Stolfo, D.; Sinagra, G.; Lund, L.H. Heart failure with mid-range or mildly reduced ejection fraction. Nat. Rev. Cardiol. 2022, 19, 100–116. [Google Scholar] [CrossRef]
- D’Amario, D.; Migliaro, S.; Borovac, J.A.; Restivo, A.; Vergallo, R.; Galli, M.; Leone, A.M.; Montone, R.A.; Niccoli, G.; Aspromonte, N.; et al. Microvascular Dysfunction in Heart Failure With Preserved Ejection Fraction. Front. Physiol. 2019, 10, 1347. [Google Scholar] [CrossRef] [PubMed]
- Goidescu, C.M.; Chiorescu, R.M.; Diana, M.L.; Mocan, M.; Stoia, M.A.; Anton, F.P.; Farcaş, A.D. ACE2 and Apelin-13: Biomarkers with a Prognostic Value in Congestive Heart Failure. Dis. Markers 2021, 2021, 5569410. [Google Scholar] [CrossRef]
- Chiorescu, R.M.; Lazar, R.D.; Buksa, S.B.; Mocan, M.; Blendea, D. Biomarkers of Volume Overload and Edema in Heart Failure With Reduced Ejection Fraction. Front. Cardiovasc. Med. 2022, 9, 910100. [Google Scholar] [CrossRef] [PubMed]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef]
- Satomura, H.; Wada, H.; Sakakura, K.; Kubo, N.; Ikeda, N.; Sugawara, Y.; Ako, J.; Momomura, S. Congestive heart failure in the elderly: Comparison between reduced ejection fraction and preserved ejection fraction. J. Cardiol. 2012, 59, 215–219. [Google Scholar] [CrossRef]
- Tsao, C.W.; Lyass, A.; Enserro, D.; Larson, M.G.; Ho, J.E.; Kizer, J.R.; Gottdiener, J.S.; Psaty, B.M.; Vasan, R.S. Temporal Trends in the Incidence of and Mortality Associated With Heart Failure With Preserved and Reduced Ejection Fraction. JACC Heart Fail. 2018, 6, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Dassanayaka, S.; Jones, S.P. Recent Developments in Heart Failure. Circ. Res. 2015, 117, e58–e63. [Google Scholar] [CrossRef]
- He, Y.; Huang, W.; Zhang, C.; Chen, L.; Xu, R.; Li, N.; Wang, F.; Han, L.; Yang, M.; Zhang, D. Energy metabolism disorders and potential therapeutic drugs in heart failure. Acta Pharm. Sin. B 2021, 11, 1098–1116. [Google Scholar] [CrossRef]
- Saddik, M.; Lopaschuk, G.D. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J. Biol. Chem. 1991, 266, 8162–8170. [Google Scholar] [CrossRef]
- Wisneski, J.A.; Stanley, W.C.; Neese, R.A.; Gertz, E.W. Effects of acute hyperglycemia on myocardial glycolytic activity in humans. J. Clin. Investig. 1990, 85, 1648–1656. [Google Scholar] [CrossRef]
- Lee, C.F.; Tian, R. Mitochondrion as a Target for Heart Failure Therapy- Role of Protein Lysine Acetylation. Circ. J. Off. J. Jpn. Circ. Soc. 2015, 79, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Barth, E.; Stämmler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Schaper, J.; Meiser, E.; Stämmler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391. [Google Scholar] [CrossRef]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients With Heart Failure With Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Casademont, J.; Miró, O. Electron transport chain defects in heart failure. Heart Fail. Rev. 2002, 7, 131–139. [Google Scholar] [CrossRef]
- Quigley, A.F.; Kapsa, R.M.; Esmore, D.; Hale, G.; Byrne, E. Mitochondrial respiratory chain activity in idiopathic dilated cardiomyopathy. J. Card. Fail. 2000, 6, 47–55. [Google Scholar] [CrossRef]
- Jansen, J.M. Rong Tian: Finding What Feeds the Heart. Circ. Res. 2017, 120, 1542–1544. [Google Scholar] [CrossRef]
- Fukushima, A.; Milner, K.; Gupta, A.; Lopaschuk, G.D. Myocardial Energy Substrate Metabolism in Heart Failure: From Pathways to Therapeutic Targets. Curr. Pharm. Des. 2015, 21, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Zhou, N.; Wang, D.W. Trimetazidine in Heart Failure. Front. Pharmacol. 2020, 11, 569132. [Google Scholar] [CrossRef]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Lopaschuk, G.D. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim. Biophys. Acta 2013, 1833, 857–865. [Google Scholar] [CrossRef]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef]
- Battogtokh, G.; Choi, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Huh, K.M.; Cho, Y.Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; Böhm, M. Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J. Am. Coll. Cardiol. 2011, 58, 83–86. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Z.; Zhang, W.; Liu, X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol. Res. 2022, 175, 106038. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Abel, E.D. Mitochondrial dynamics and metabolic regulation in cardiac and skeletal muscle. Trans. Am. Clin. Climatol. Assoc. 2018, 129, 266–278. [Google Scholar]
- Wu, D.; Dasgupta, A.; Chen, K.H.; Neuber-Hess, M.; Patel, J.; Hurst, T.E.; Mewburn, J.D.; Lima, P.D.A.; Alizadeh, E.; Martin, A.; et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 1447–1464. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Mileo, A.M.; Matarrese, P. Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer. Cancers 2021, 13, 5812. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. The role of mitochondrial fission in cardiovascular health and disease. Nat. Rev. Cardiol. 2022, 19, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.H.; Li, Y.Y.; Jin, J. The essential functions of mitochondrial dynamics in immune cells. Cell. Mol. Immunol. 2020, 17, 712–721. [Google Scholar] [CrossRef]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W., 2nd. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883.e5. [Google Scholar] [CrossRef]
- Hinton, A., Jr.; Claypool, S.M.; Neikirk, K.; Senoo, N.; Wanjalla, C.N.; Kirabo, A.; Williams, C.R. Mitochondrial Structure and Function in Human Heart Failure. Circ. Res. 2024, 135, 372–396. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Clark, C.F.; Eschenbacher, W.H.; Kang, M.Y.; Engelhard, J.T.; Warner, S.J.; Matkovich, S.J.; Jowdy, C.C. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ. Res. 2011, 108, 12–17. [Google Scholar] [CrossRef]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef]
- Sun, D.; Li, C.; Liu, J.; Wang, Z.; Liu, Y.; Luo, C.; Chen, Y.; Wen, S. Expression Profile of microRNAs in Hypertrophic Cardiomyopathy and Effects of microRNA-20 in Inducing Cardiomyocyte Hypertrophy Through Regulating Gene MFN2. DNA Cell Biol. 2019, 38, 796–807. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging. Oxidative Med. Cell. Longev. 2019, 2019, 9825061. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Aghanejad, A.; Zhang, Y.; Ren, J. Mitophagy Receptors and Mediators: Therapeutic Targets in the Management of Cardiovascular Ageing. Ageing Res. Rev. 2020, 62, 101129. [Google Scholar] [CrossRef]
- Morales, P.E.; Arias-Durán, C.; Ávalos-Guajardo, Y.; Aedo, G.; Verdejo, H.E.; Parra, V.; Lavandero, S. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol. Asp. Med. 2020, 71, 100822. [Google Scholar] [CrossRef]
- Shires, S.E.; Gustafsson, Å.B. Mitophagy and heart failure. J. Mol. Med. 2015, 93, 253–262. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Kura, B.; Szeiffova Bacova, B.; Kalocayova, B.; Sykora, M.; Slezak, J. Oxidative Stress-Responsive MicroRNAs in Heart Injury. Int. J. Mol. Sci. 2020, 21, 358. [Google Scholar] [CrossRef]
- Dolivo, D.; Weathers, P.; Dominko, T. Artemisinin and artemisinin derivatives as anti-fibrotic therapeutics. Acta Pharm. Sinica B 2021, 11, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Chen, M.J.; Wei, J. Involvement of phosphatase and tensin homolog-induced putative kinase 1/Parkin-mediated autophagy in angiotensin II-induced cardiac hypertrophy in C57BL/6 mice. J. Int. Med. Res. 2020, 48, 300060519896143. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lv, J.; Pan, Z.; Wang, D.; Zhao, L.; Guo, X. Mitochondrial dysfunction in heart failure and its therapeutic implications. Front. Cardiovasc. Med. 2022, 9, 945142. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Shah, A.M. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J. Biol. Chem. 2003, 278, 12094–12100. [Google Scholar] [CrossRef]
- Hishikawa, K.; Lüscher, T.F. Pulsatile stretch stimulates superoxide production in human aortic endothelial cells. Circulation 1997, 96, 3610–3616. [Google Scholar] [CrossRef]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Cappola, T.P.; Kass, D.A.; Nelson, G.S.; Berger, R.D.; Rosas, G.O.; Kobeissi, Z.A.; Marbán, E.; Hare, J.M. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 2001, 104, 2407–2411. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Hill, M.F.; Singal, P.K. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am. J. Pathol. 1996, 148, 291–300. [Google Scholar]
- Khaper, N.; Singal, P.K. Effects of afterload-reducing drugs on pathogenesis of antioxidant changes and congestive heart failure in rats. J. Am. Coll. Cardiol. 1997, 29, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Khaper, N.; Kaur, K.; Li, T.; Farahmand, F.; Singal, P.K. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol. Cell. Biochem. 2003, 251, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef]
- Al Ghouleh, I.; Khoo, N.K.; Knaus, U.G.; Griendling, K.K.; Touyz, R.M.; Thannickal, V.J.; Barchowsky, A.; Nauseef, W.M.; Kelley, E.E.; Bauer, P.M.; et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011, 51, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Lehnart, S.E.; Maier, L.S.; Hasenfuss, G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail. Rev. 2009, 14, 213–224. [Google Scholar] [CrossRef]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef]
- Weisleder, N.; Ma, J. Altered Ca2+ sparks in aging skeletal and cardiac muscle. Ageing Res. Rev. 2008, 7, 177–188. [Google Scholar] [CrossRef]
- Lombardi, A.A.; Gibb, A.A.; Arif, E.; Kolmetzky, D.W.; Tomar, D.; Luongo, T.S.; Jadiya, P.; Murray, E.K.; Lorkiewicz, P.K.; Hajnóczky, G.; et al. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat. Commun. 2019, 10, 4509. [Google Scholar] [CrossRef]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef]
- Xu, H.X.; Cui, S.M.; Zhang, Y.M.; Ren, J. Mitochondrial Ca2+ regulation in the etiology of heart failure: Physiological and pathophysiological implications. Acta Pharmacol. Sin. 2020, 41, 1301–1309. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Giorgi, C.; Pinton, P.; Rizzuto, R. The versatility of mitochondrial calcium signals: From stimulation of cell metabolism to induction of cell death. Biochim. Biophys. Acta 2008, 1777, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Balaska, D.; Cheng, W.H. The ups and downs of mitochondrial calcium signalling in the heart. Biochim. Biophys. Acta 2010, 1797, 856–864. [Google Scholar] [CrossRef] [PubMed]
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef]
- East, D.A.; Campanella, M. Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013, 9, 1710–1719. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafà, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Aryee, E.K.; Ozkan, B.; Ndumele, C.E. Heart Failure and Obesity: The Latest Pandemic. Prog. Cardiovasc. Dis. 2023, 78, 43–48. [Google Scholar] [CrossRef]
- Nakamura, K.; Fuster, J.J.; Walsh, K. Adipokines: A link between obesity and cardiovascular disease. J. Cardiol. 2014, 63, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Fu, T.; Seok, S.; Kim, D.H.; Yu, E.; Lee, K.W.; Kang, Y.; Li, X.; Kemper, B.; Kemper, J.K. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 2013, 12, 1062–1072. [Google Scholar] [CrossRef]
- Rosano, G.M.; Vitale, C.; Seferovic, P. Heart Failure in Patients with Diabetes Mellitus. Card. Fail. Rev. 2017, 3, 52–55. [Google Scholar] [CrossRef]
- Fukushima, A.; Kinugawa, S.; Takada, S.; Matsushima, S.; Sobirin, M.A.; Ono, T.; Takahashi, M.; Suga, T.; Homma, T.; Masaki, Y.; et al. (Pro)renin receptor in skeletal muscle is involved in the development of insulin resistance associated with postinfarct heart failure in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E503–E514. [Google Scholar] [CrossRef]
- Elendu, C.; Amaechi, D.C.; Elendu, T.C.; Ashna, M.; Ross-Comptis, J.; Ansong, S.O.; Egbunu, E.O.; Okafor, G.C.; Jingwa, K.A.; Akintunde, A.A.; et al. Heart failure and diabetes: Understanding the bidirectional relationship. Medicine 2023, 102, e34906. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Chakraborty, A.D.; Light, C.M.; Tian, R.; Sadoshima, J.; Shi, X.; Gu, H.; Lee, C.F. NAD+ Redox Imbalance in the Heart Exacerbates Diabetic Cardiomyopathy. Circ. Heart Fail. 2021, 14, e008170. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Mo, F.; Zhang, Z.; Huang, M.; Wei, X. Nicotinamide Mononucleotide: A Promising Molecule for Therapy of Diverse Diseases by Targeting NAD+ Metabolism. Front. Cell Dev. Biol. 2020, 8, 246. [Google Scholar] [CrossRef]
- Li, H.; Cai, Z. SIRT3 regulates mitochondrial biogenesis in aging-related diseases. J. Biomed. Res. 2022, 37, 77–88. [Google Scholar] [CrossRef]
- Li, W.; Zhou, Y.; Pang, N.; Hu, Q.; Li, Q.; Sun, Y.; Ding, Y.; Gu, Y.; Xiao, Y.; Gao, M.; et al. NAD Supplement Alleviates Intestinal Barrier Injury Induced by Ethanol Via Protecting Epithelial Mitochondrial Function. Nutrients 2022, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Bugga, P.; Alam, M.J.; Kumar, R.; Pal, S.; Chattopadyay, N.; Banerjee, S.K. Sirt3 ameliorates mitochondrial dysfunction and oxidative stress through regulating mitochondrial biogenesis and dynamics in cardiomyoblast. Cell. Signal. 2022, 94, 110309. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Karamanlidis, G.; Tian, R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016, 8, 326rv323. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Hamaidi, I.; Kim, S. Sirtuins are crucial regulators of T cell metabolism and functions. Exp. Mol. Med. 2022, 54, 207–215. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an Endothelial NAD+-H2S Signaling Network Is a Reversible Cause of Vascular Aging. Cell 2018, 173, 74–89.e20. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Odanovic, N.; Nakada, Y.; Dohi, S.; Zhai, P.; Ivessa, A.; Yang, Z.; Abdellatif, M.; Sadoshima, J. Dietary carbohydrates restriction inhibits the development of cardiac hypertrophy and heart failure. Cardiovasc. Res. 2021, 117, 2365–2376. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.R.; Kong, M.J.; Han, S.J.; Kim, J.I.; Park, K.M. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol. 2020, 34, 101548. [Google Scholar] [CrossRef]
- Kim, H.; Lee, Y.D.; Kim, H.J.; Lee, Z.H.; Kim, H.H. SOD2 and Sirt3 Control Osteoclastogenesis by Regulating Mitochondrial ROS. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 397–406. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef]
- Rimessi, A.; Bonora, M.; Marchi, S.; Patergnani, S.; Marobbio, C.M.; Lasorsa, F.M.; Pinton, P. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy 2013, 9, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Landick, R.; Raman, S. A Regulatory NADH/NAD+ Redox Biosensor for Bacteria. ACS Synth. Biol. 2019, 8, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.L.; Zhang, D.X.; Xiang, F.; Teng, M.; Jiang, X.P.; Hou, J.M.; Zhang, Q.; Huang, Y.S. Nicotinamide pretreatment protects cardiomyocytes against hypoxia-induced cell death by improving mitochondrial stress. Pharmacology 2012, 90, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Bhatnagar, A.; Sadoshima, J. Overview of pyridine nucleotides review series. Circ. Res. 2012, 111, 604–610. [Google Scholar] [CrossRef]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Klimova, N.; Long, A.; Kristian, T. Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3-dependent mechanism in male mice. J. Neurosci. Res. 2019, 97, 975–990. [Google Scholar] [CrossRef]
- Mehmel, M.; Jovanović, N.; Spitz, U. Nicotinamide Riboside-The Current State of Research and Therapeutic Uses. Nutrients 2020, 12, 1616. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Koay, Y.C.; Liu, R.P.; McIntosh, B.; Vigder, N.; Lauren, S.; Bai, A.Y.; Tomita, S.; Li, D.; Harney, D.; Hunter, B.; et al. The Efficacy of Risk Factor Modification Compared to NAD+ Repletion in Diastolic Heart Failure. JACC Basic Transl. Sci. 2024, 9, 733–750. [Google Scholar] [CrossRef]
- Hu, L.; Guo, Y.; Song, L.; Wen, H.; Sun, N.; Wang, Y.; Qi, B.; Liang, Q.; Geng, J.; Liu, X.; et al. Nicotinamide riboside promotes Mfn2-mediated mitochondrial fusion in diabetic hearts through the SIRT1-PGC1α-PPARα pathway. Free Radic. Biol. Med. 2022, 183, 75–88. [Google Scholar] [CrossRef]
- Abdellatif, M.; Vasques-Nóvoa, F.; Trummer-Herbst, V.; Durand, S.; Koser, F.; Islam, M.; Nah, J.; Sung, E.A.; Feng, R.; Aprahamian, F.; et al. Autophagy is required for the therapeutic effects of the NAD+ precursor nicotinamide in obesity-related heart failure with preserved ejection fraction. Eur. Heart J. 2025, 46, 1863–1866. [Google Scholar] [CrossRef] [PubMed]
- Broeks, M.H.; Meijer, N.W.F.; Westland, D.; Bosma, M.; Gerrits, J.; German, H.M.; Ciapaite, J.; van Karnebeek, C.D.M.; Wanders, R.J.A.; Zwartkruis, F.J.T.; et al. The malate-aspartate shuttle is important for de novo serine biosynthesis. Cell Rep. 2023, 42, 113043. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Bertrand, L.; Beauloye, C.R.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.R.; Kula-Alwar, D.; Prag, H.A.; Eric Botker, H.; Dambrova, M.; et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J. Cell. Mol. Med. 2020, 24, 5937–5954. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.T.; Støttrup, N.B.; Løfgren, B.; Bøtker, H.E. Metabolic fingerprint of ischaemic cardioprotection: Importance of the malate-aspartate shuttle. Cardiovasc. Res. 2011, 91, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, Q.; Leng, J.; Zheng, Y.; Li, J. The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases. Life Sci. 2015, 121, 97–103. [Google Scholar] [CrossRef]
- Ussher, J.R.; Jaswal, J.S.; Lopaschuk, G.D. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ. Res. 2012, 111, 628–641. [Google Scholar] [CrossRef]
- Wang, L.; Quan, N.; Sun, W.; Chen, X.; Cates, C.; Rousselle, T.; Zhou, X.; Zhao, X.; Li, J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc. Res. 2018, 114, 805–821. [Google Scholar] [CrossRef]
- Riess, M.L.; Camara, A.K.; Chen, Q.; Novalija, E.; Rhodes, S.S.; Stowe, D.F. Altered NADH and improved function by anesthetic and ischemic preconditioning in guinea pig intact hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H53–H60. [Google Scholar] [CrossRef]
- Sims, C.A.; Guan, Y.; Mukherjee, S.; Singh, K.; Botolin, P.; Davila, A., Jr.; Baur, J.A. Nicotinamide mononucleotide preserves mitochondrial function and increases survival in hemorrhagic shock. JCI Insight 2018, 3, 120182. [Google Scholar] [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Nagana Gowda, G.A.; Tian, R. Targeting NAD+ Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 3073. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Nagana Gowda, G.A.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef] [PubMed]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Deng, Y.; Yang, L.; Ou, W.; Xie, M.; Ding, L.; Jiang, C.; Yu, H.; Li, Q.; et al. Mitochondrial protein hyperacetylation underpins heart failure with preserved ejection fraction in mice. J. Mol. Cell. Cardiol. 2022, 165, 76–85. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Altamirano, F.; Szweda, P.A.; Elnwasany, A.; Lee, D.I.; Yoo, H.; Kass, D.A.; Szweda, L.I.; et al. NAD+ Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circ. Res. 2021, 128, 1629–1641. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Zang, Q.S.; Sadek, H.; Maass, D.L.; Martinez, B.; Ma, L.; Kilgore, J.A.; Williams, N.S.; Frantz, D.E.; Wigginton, J.G.; Nwariaku, F.E.; et al. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1847–H1859. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Hong, G.; Zheng, D.; Zhang, L.; Ni, R.; Wang, G.; Fan, G.C.; Lu, Z.; Peng, T. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free Radic. Biol. Med. 2018, 123, 125–137. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.D.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Guarente, L. SnapShot: Sirtuins, NAD, and aging. Cell Metab. 2014, 20, 192–192.e1. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef]
- Yagi, M.; Do, Y.; Hirai, H.; Miki, K.; Toshima, T.; Fukahori, Y.; Setoyama, D.; Abe, C.; Nabeshima, Y.I.; Kang, D.; et al. Improving lysosomal ferroptosis with NMN administration protects against heart failure. Life Sci. Alliance 2023, 6, e202302116. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef]
- Martin, A.S.; Abraham, D.M.; Hershberger, K.A.; Bhatt, D.P.; Mao, L.; Cui, H.; Liu, J.; Liu, X.; Muehlbauer, M.J.; Grimsrud, P.A.; et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2017, 2, e93885. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Beck, M.E.; Goetzman, E.S. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol. 2019, 26, 101253. [Google Scholar] [CrossRef]
- Nascimben, L.; Ingwall, J.S.; Lorell, B.H.; Pinz, I.; Schultz, V.; Tornheim, K.; Tian, R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004, 44, 662–667. [Google Scholar] [CrossRef]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Wang, Y.T.; Nehrke, K.; Munger, J.; Brookes, P.S. Cardioprotection by nicotinamide mononucleotide (NMN): Involvement of glycolysis and acidic pH. J. Mol. Cell. Cardiol. 2018, 121, 155–162. [Google Scholar] [CrossRef]
- Jaconello, P. Niacin versus niacinamide. CMAJ Can. Med. Assoc. J. J. L’association Medicale Can. 1992, 147, 990. [Google Scholar]
- Jung, M.; Lee, K.M.; Im, Y.; Seok, S.H.; Chung, H.; Kim, D.Y.; Han, D.; Lee, C.H.; Hwang, E.H.; Park, S.Y.; et al. Nicotinamide (niacin) supplement increases lipid metabolism and ROS-induced energy disruption in triple-negative breast cancer: Potential for drug repositioning as an anti-tumor agent. Mol. Oncol. 2022, 16, 1795–1815. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- van Ommen, A.; Canto, E.D.; Cramer, M.J.; Rutten, F.H.; Onland-Moret, N.C.; Ruijter, H.M.D. Diastolic dysfunction and sex-specific progression to HFpEF: Current gaps in knowledge and future directions. BMC Med. 2022, 20, 496. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Trummer-Herbst, V.; Koser, F.; Durand, S.; Adão, R.; Vasques-Nóvoa, F.; Freundt, J.K.; Voglhuber, J.; Pricolo, M.R.; Kasa, M.; et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 2021, 13, eabd7064. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Anand, S.K.; Singh, N.; Dwivedi, U.N.; Kakkar, P. AMP-activated protein kinase: An energy sensor and survival mechanism in the reinstatement of metabolic homeostasis. Exp. Cell Res. 2023, 428, 113614. [Google Scholar] [CrossRef]
- Lai, Y.F.; Wang, L.; Liu, W.Y. Nicotinamide pretreatment alleviates mitochondrial stress and protects hypoxic myocardial cells via AMPK pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1797–1806. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Munson, M.J.; Ganley, I.G. MTOR, PIK3C3, and autophagy: Signaling the beginning from the end. Autophagy 2015, 11, 2375–2376. [Google Scholar] [CrossRef]
- Li, W.; Zhu, L.; Ruan, Z.B.; Wang, M.X.; Ren, Y.; Lu, W. Nicotinamide protects chronic hypoxic myocardial cells through regulating mTOR pathway and inducing autophagy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5503–5511. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhu, M.; Fan, S.; Zhang, J. Niacin intake and mortality (total and cardiovascular disease) in patients with cardiovascular disease: Insights from NHANES 2003–2018. Nutr. J. 2024, 23, 123. [Google Scholar] [CrossRef]
- Gasperi, V.; Sibilano, M.; Savini, I.; Catani, M.V. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 974. [Google Scholar] [CrossRef]
- Saeedi, R.; Frohlich, J. Lipoprotein (a), an independent cardiovascular risk marker. Clin. Diabetes Endocrinol. 2016, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, P.M.; Karas, R.H. The current state of niacin in cardiovascular disease prevention: A systematic review and meta-regression. J. Am. Coll. Cardiol. 2013, 61, 440–446. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, C.; Wu, G. Dietary niacin Intake and its association with all-cause and cardiovascular mortality rates in individuals with metabolic syndrome. Nutr. J. 2024, 23, 90. [Google Scholar] [CrossRef]
- Nizhenkovska, I.; Narokha, V.; Kuznetsova, O. Effects of nicotinic acid on protein oxidative modifications in experimental chronic heart failure. Farmacia 2018, 66, 959–962. [Google Scholar] [CrossRef]
- Chini, E.N.; Chini, C.C.S.; Espindola Netto, J.M.; de Oliveira, G.C.; van Schooten, W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol. Sci. 2018, 39, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.H.; Hong, X.; Zhao, N.; Liu, X.H.; Xiao, Y.F.; Chen, T.T.; Deng, L.B.; Wang, X.L.; Wang, J.B.; Ji, G.J.; et al. CD38 promotes angiotensin II-induced cardiac hypertrophy. J. Cell. Mol. Med. 2017, 21, 1492–1502. [Google Scholar] [CrossRef]
- Boslett, J.; Hemann, C.; Zhao, Y.J.; Lee, H.C.; Zweier, J.L. Luteolinidin Protects the Postischemic Heart through CD38 Inhibition with Preservation of NAD(P)(H). J. Pharmacol. Exp. Ther. 2017, 361, 99–108. [Google Scholar] [CrossRef]
- Kellenberger, E.; Kuhn, I.; Schuber, F.; Muller-Steffner, H. Flavonoids as inhibitors of human CD38. Bioorganic Med. Chem. Lett. 2011, 21, 3939–3942. [Google Scholar] [CrossRef] [PubMed]
- Espírito-Santo, D.A.; Cordeiro, G.S.; Santos, L.S.; Silva, R.T.; Pereira, M.U.; Matos, R.J.B.; Boaventura, G.T.; Barreto-Medeiros, J.M. Cardioprotective effect of the quercetin on cardiovascular remodeling and atherosclerosis in rodents fed a high-fat diet: A systematic review. Chem.-Biol. Interact. 2023, 384, 110700. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD+/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging 2020, 12, 11325–11336. [Google Scholar] [CrossRef]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095.e10. [Google Scholar] [CrossRef]
- Boslett, J.; Reddy, N.; Alzarie, Y.A.; Zweier, J.L. Inhibition of CD38 with the Thiazoloquin(az)olin(on)e 78c Protects the Heart against Postischemic Injury. J. Pharmacol. Exp. Ther. 2019, 369, 55–64. [Google Scholar] [CrossRef]
- Summers, D.W.; Gibson, D.A.; DiAntonio, A.; Milbrandt, J. SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc. Natl. Acad. Sci. USA 2016, 113, E6271–E6280. [Google Scholar] [CrossRef]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Tentori, L.; Ricci-Vitiani, L.; Muzi, A.; Ciccarone, F.; Pelacchi, F.; Calabrese, R.; Runci, D.; Pallini, R.; Caiafa, P.; Graziani, G. Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC Cancer 2014, 14, 151. [Google Scholar] [CrossRef]
- Wicik, Z.; Nowak, A.; Jarosz-Popek, J.; Wolska, M.; Eyileten, C.; Siller-Matula, J.M.; von Lewinski, D.; Sourij, H.; Filipiak, K.J.; Postuła, M. Characterization of the SGLT2 Interaction Network and Its Regulation by SGLT2 Inhibitors: A Bioinformatic Analysis. Front. Pharmacol. 2022, 13, 901340. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Tsai, W.C.; Chiu, C.C.; Chi, N.Y.; Liu, Y.H.; Huang, T.C.; Wu, W.T.; Lin, T.H.; Lai, W.T.; Sheu, S.H.; et al. The Beneficial Effect of the SGLT2 Inhibitor Dapagliflozin in Alleviating Acute Myocardial Infarction-Induced Cardiomyocyte Injury by Increasing the Sirtuin Family SIRT1/SIRT3 and Cascade Signaling. Int. J. Mol. Sci. 2024, 25, 8541. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zuo, Z.; Tian, J.; Ali, Q.; Lin, Y.; Lei, H.; Sun, Z. Activation of SIRT1 Attenuates Klotho Deficiency-Induced Arterial Stiffness and Hypertension by Enhancing AMP-Activated Protein Kinase Activity. Hypertension 2016, 68, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.S.; Vieira, C.P.; McFarland, D.; Sandler, M.; Levitsky, Y.; Dorweiler, T.F.; Lydic, T.A.; Asare-Bediako, B.; Adu-Agyeiwaah, Y.; Sielski, M.S.; et al. Fasting and fasting-mimicking treatment activate SIRT1/LXRα and alleviate diabetes-induced systemic and microvascular dysfunction. Diabetologia 2021, 64, 1674–1689. [Google Scholar] [CrossRef]
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef]
- Stein, S.; Lohmann, C.; Schäfer, N.; Hofmann, J.; Rohrer, L.; Besler, C.; Rothgiesser, K.M.; Becher, B.; Hottiger, M.O.; Borén, J.; et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur. Heart J. 2010, 31, 2301–2309. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathomechanism | Core Media | Function | References |
|---|---|---|---|
| Mitochondrial dynamics | MFN1, MFN2, Drp1, OPA1 | Regulating mitochondrial fusion (MFN1, MFN2, OPA1) and division (Drp1), the imbalance leads to mitochondrial fragmentation, leading to heart failure | [79] |
| Mitochondrial autophagy | PINK1, Parkin | PINK1/Parkin is an important component of mitochondrial autophagy mediated by cardiomyocytes. Its deficiency leads to mitochondrial autophagy disorder and promotes cardiomyocyte apoptosis | [97,99,100] |
| Mitochondrial oxidative stress | SOD, GSH-Px, Catalase | The activity of antioxidant enzymes (SOD scavenging superoxide anion and GPx1/catalase decomposing H2O2) decreased in heart failure | [112,113,114] |
| Mitochondrial Ca2+ homeostasis | Ca2+ | Ca2+ overload stimulates ROS production, inhibits ATP generation, promotes mitochondrial damage and cardiomyocyte apoptosis, and ultimately leads to heart failure | [125] |
| Categories | Dosage | In Vivo/InVitro | Model | Effects | References |
|---|---|---|---|---|---|
| NR | 400 mg/kg/d | In vivo, In vitro | Mouse HFpEF model | NAD+ repletion improved mitochondrial function and reversed HFpEF phenotype | [187] |
| 500 mg/kg/d | In vivo, In vitro | HFpEF mice | NR repletion fully rescued HFpEF | [170] | |
| 100, 300 or 500 mg/kg | In vivo, In vitro | C57BL/6 mice | NR therapy elevated the NAD+ levels, reduced oxidative stress and apoptosis in cardiac tissue, and prevented cardiac injury | [191] | |
| 250 mg, 500 mg, 1000 mg | In vitro | PBMC of stage D HFrEF patients | NR administration enhanced PBMC respiration and reduced proinflammatory cytokine expression in patients with heart failure. | [192] | |
| NMN | 500 mg/kg, twice in three days | In vivo, In vitro | Cardiac-restricted complex I-deficient mouse | NMN supplementation increased the NAD+/NADH ratio in the cKO hearts, decreased the mitochondrial protein acetylation and improved the sensitivity of the mPTP in the cKO mitochondria | [197] |
| 500 mg/kg, twice a week | In vivo, In vitro | Friedreich’s ataxia cardiomyopathy mouse model (FXN-KO) | NMN significantly increased cardiac ejection times and improved cardiac energy generation and utilisation in a SIRT3-dependent manner | [198] | |
| 500 mg/kg/d | In vivo, In vitro | Mouse model of contracted TAC | NMN administration improved cardiac mitochondrial function and rescued heart failure | [27] | |
| 500 mg/kg | In vitro | Cardiac of mice undergoing TAC | NMN administration reversed mitochondrial protein hyperacetylation in control mice after TAC and delayed the progression of heart failure | [157] | |
| A 100× stock in Krebs Henseleit (KH) for 20 min | In vitro | Cardiomyocytes of wild-type C57BL6/J mice and Sirt3−/− mice | NMN stimulated cardiac glycolysis to protect cardiac | [202] | |
| NAM | 2.5, 5, 10, or 20 mmol/L | In vitro | Primary rat neonatal cardiomyocytes | NAM pretreatment protected cardiomyocytes by improving mitochondrial stress | [164] |
| 40 mM | In vivo, In vitro | ZSF1 obese rats | NAM improved diastolic dysfunction in ZSF1 obese rats and alleviated HFpEF risk factors | [207] | |
| 100 μL/tube | In vitro | Primary cardiomyocytes | NAM-induced energy production in hypoxic myocardial cells to protect myocardial cells | [209] | |
| 5, 10, and 20 mM | In vitro | Model of chronic hypoxic myocardial cells | NAM induced autophagy in chronic hypoxic cardiomyocytes and reduced cardiomyocyte apoptosis by regulating the mTOR pathway | [212] | |
| NA | 10 mg/kg | In vivo, In vitro | Model of chronic heart failure experimental model | NA significantly reduced the oxidation intensity of cardiomyocyte proteins in heart failure | [218] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, F.; Zhao, H.; Luo, L.; Wu, W. Nicotinamide Adenine Dinucleotide Supplementation to Alleviate Heart Failure: A Mitochondrial Dysfunction Perspective. Nutrients 2025, 17, 1855. https://doi.org/10.3390/nu17111855
Yu F, Zhao H, Luo L, Wu W. Nicotinamide Adenine Dinucleotide Supplementation to Alleviate Heart Failure: A Mitochondrial Dysfunction Perspective. Nutrients. 2025; 17(11):1855. https://doi.org/10.3390/nu17111855
Chicago/Turabian StyleYu, Fan, Huiying Zhao, Lu Luo, and Wei Wu. 2025. "Nicotinamide Adenine Dinucleotide Supplementation to Alleviate Heart Failure: A Mitochondrial Dysfunction Perspective" Nutrients 17, no. 11: 1855. https://doi.org/10.3390/nu17111855
APA StyleYu, F., Zhao, H., Luo, L., & Wu, W. (2025). Nicotinamide Adenine Dinucleotide Supplementation to Alleviate Heart Failure: A Mitochondrial Dysfunction Perspective. Nutrients, 17(11), 1855. https://doi.org/10.3390/nu17111855