
Rescue of Methionine Dependence by Cobalamin in a Human Colorectal Cancer Cell Line


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Proliferation
2.3. Apoptosis
2.4. Gene Expression
2.5. Protein Expression
2.6. Cell Cycle
2.7. Data Analysis
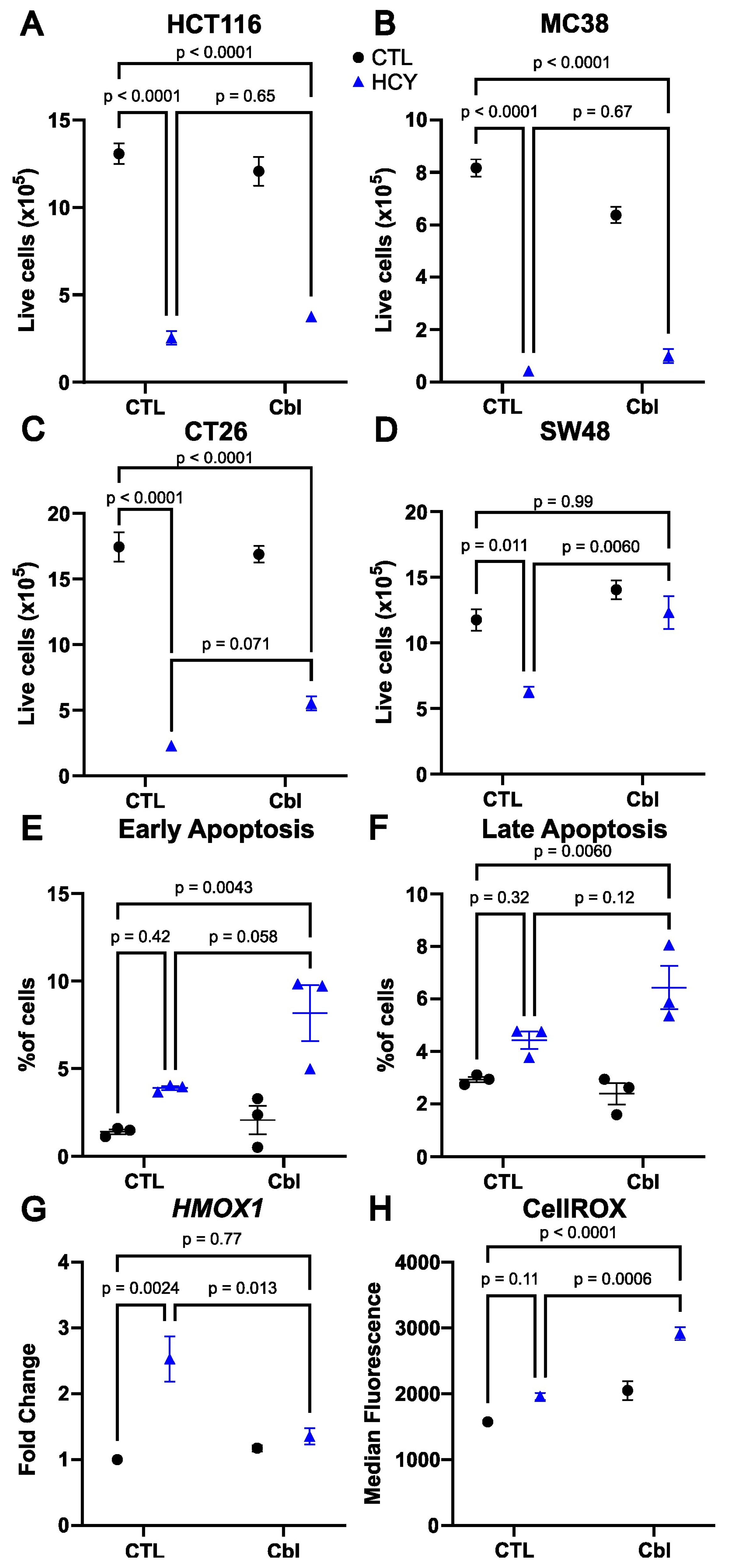
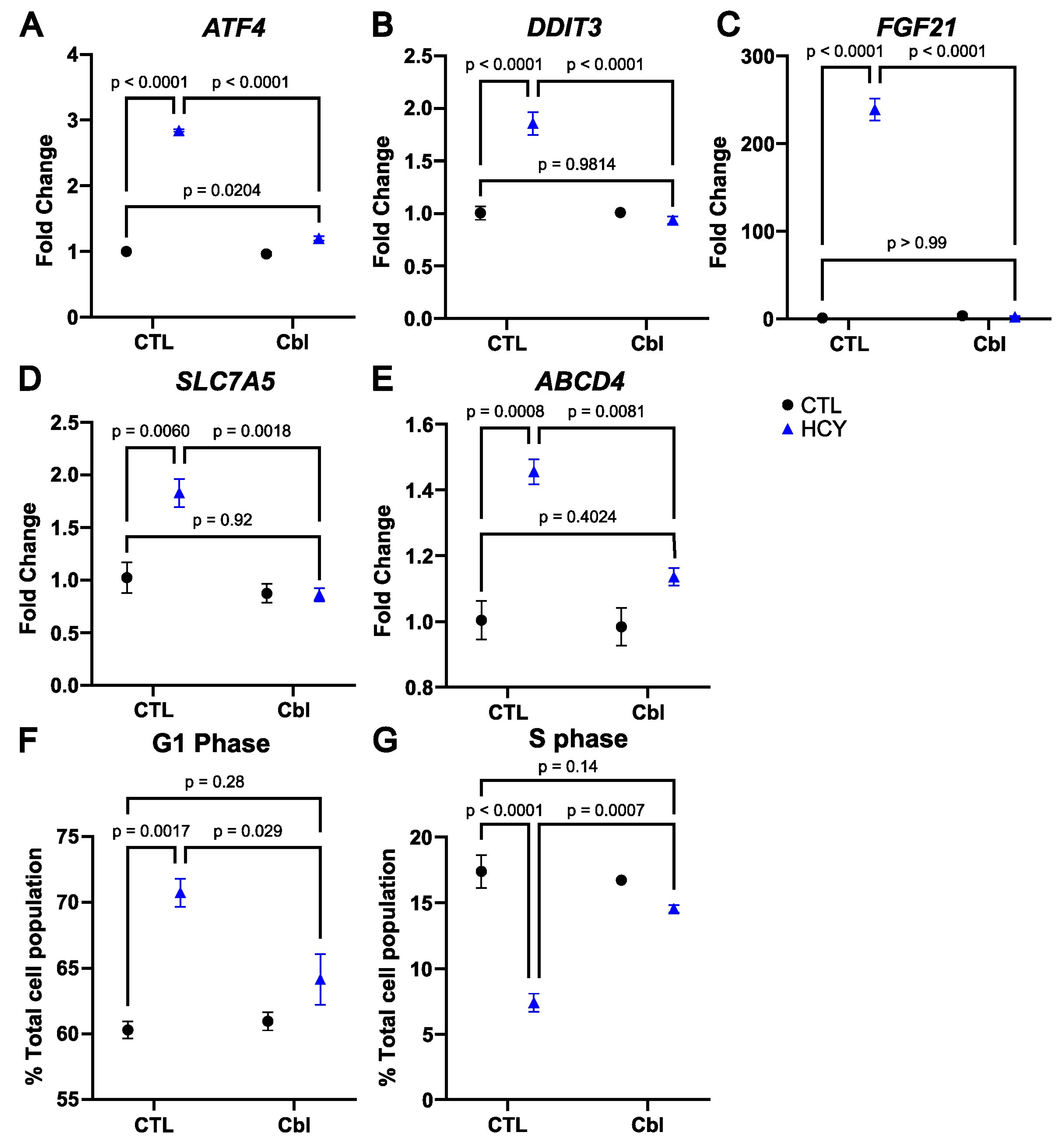
3. Results
3.1. Cobalamin Supplementation Rescues Cell Number in Methionine-Starved SW48 Human Colorectal Cancer Cells
3.2. Apoptosis Is Not Induced by Homocystine Treatment in SW48 Cells
3.3. Cobalamin Does Not Reduce Oxidative Stress
3.4. Cobalamin Prevents the Activation of the Integrated Stress Response in Homocystine-Treated SW48 Cells
3.5. Cobalamin Rescues Cell Cycle Arrest in Methionine-Starved SW48 Cells
3.6. Effects on Protein Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor Suppressor Complex with GAP Activity for the Rag GTPases That Signal Amino Acid Sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef]
- Morehead, L.C.; Garg, S.; Wallis, K.F.; Siegel, E.R.; Tackett, A.J.; Miousse, I.R. Increased Response to Immune Checkpoint Inhibitors with Dietary Methionine Restriction. Cancers 2023, 15, 4467. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary Methionine Influences Therapy in Mouse Cancer Models and Alters Human Metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Kim, J.H.; Lee, E.; Jang, Y.J.; Son, J.E.; Kwon, J.Y.; Lim, T.-G.; Kim, S.; Park, J.H.Y.; Kim, J.-E.; et al. Methionine Deprivation Suppresses Triple-Negative Breast Cancer Metastasis in Vitro and in Vivo. Oncotarget 2016, 7, 67223–67234. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Han, Q.; Murakami, T.; Kiyuna, T.; Kawaguchi, K.; Igarashi, K.; Lwin, T.M.; Miyake, M.; Yamamoto, J.; Bouvet, M.; et al. Colon-Cancer Liver Metastasis Is Effectively Targeted by Recombinant Methioninase (rMETase) in an Orthotopic Mouse Model. Tissue Cell 2023, 83, 102125. [Google Scholar] [CrossRef] [PubMed]
- Masaki, N.; Han, Q.; Wu, N.F.; Samonte, C.; Wu, J.; Hozumi, C.; Obara, K.; Kubota, Y.; Aoki, Y.; Miyazaki, J.; et al. Oral-Recombinant Methioninase Lowers the Effective Dose and Eliminates Toxicity of Cisplatinum for Primary Osteosarcoma of the Mammary Gland in a Patient-Derived Orthotopic Xenograft Mouse Model. In Vivo 2022, 36, 2598–2603. [Google Scholar] [CrossRef]
- Lim, H.I.; Yamamoto, J.; Han, Q.; Sun, Y.U.; Nishino, H.; Tashiro, Y.; Sugisawa, N.; Tan, Y.; Choi, H.J.; Nam, S.J.; et al. Response of Triple-Negative Breast Cancer Liver Metastasis to Oral Recombinant Methioninase in a Patient-Derived Orthotopic Xenograft (PDOX) Model. In Vivo 2020, 34, 3163–3169. [Google Scholar] [CrossRef]
- Garg, S.; Morehead, L.C.; Bird, J.T.; Graw, S.; Gies, A.; Storey, A.J.; Tackett, A.J.; Edmondson, R.D.; Mackintosh, S.G.; Byrum, S.; et al. Characterization of Methionine Dependence in Melanoma Cells. Mol. Omics 2023, 20, 37–47. [Google Scholar] [CrossRef]
- Pettit, A.P.; Jonsson, W.O.; Bargoud, A.R.; Mirek, E.T.; Peelor, F.F.; Wang, Y.; Gettys, T.W.; Kimball, S.R.; Miller, B.F.; Hamilton, K.L.; et al. Dietary Methionine Restriction Regulates Liver Protein Synthesis and Gene Expression Independently of Eukaryotic Initiation Factor 2 Phosphorylation in Mice. J. Nutr. 2017, 147, 1031–1040. [Google Scholar] [CrossRef]
- Wanders, D.; Stone, K.P.; Forney, L.A.; Cortez, C.C.; Dille, K.N.; Simon, J.; Xu, M.; Hotard, E.C.; Nikonorova, I.A.; Pettit, A.P.; et al. Role of GCN2-Independent Signaling through a Non-Canonical PERK/NRF2 Pathway in the Physiological Responses to Dietary Methionine Restriction. Diabetes 2016, 65, 1499–1510. [Google Scholar] [CrossRef]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The Roles of DNA, RNA and Histone Methylation in Ageing and Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef]
- Erichsen, L.; Thimm, C.; Santourlidis, S. Methyl Group Metabolism in Differentiation, Aging, and Cancer. Int. J. Mol. Sci. 2022, 23, 8378. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Erbe, R.W. High in Vivo Rates of Methionine Biosynthesis in Transformed Human and Malignant Rat Cells Auxotrophic for Methionine. Proc. Natl. Acad. Sci. USA 1976, 73, 1523–1527. [Google Scholar] [CrossRef]
- Borrego, S.L.; Fahrmann, J.; Datta, R.; Stringari, C.; Grapov, D.; Zeller, M.; Chen, Y.; Wang, P.; Baldi, P.; Gratton, E.; et al. Metabolic Changes Associated with Methionine Stress Sensitivity in MDA-MB-468 Breast Cancer Cells. Cancer Metab. 2016, 4, 9. [Google Scholar] [CrossRef]
- Fiskerstrand, T.; Riedel, B.; Ueland, P.M.; Seetharam, B.; Pezacka, E.H.; Gulati, S.; Bose, S.; Banerjee, R.; Berge, R.K.; Refsum, H. Disruption of a Regulatory System Involving Cobalamin Distribution and Function in a Methionine-Dependent Human Glioma Cell Line*. J. Biol. Chem. 1998, 273, 20180–20184. [Google Scholar] [CrossRef]
- Loewy, A.D.; Niles, K.M.; Anastasio, N.; Watkins, D.; Lavoie, J.; Lerner-Ellis, J.P.; Pastinen, T.; Trasler, J.M.; Rosenblatt, D.S. Epigenetic Modification of the Gene for the Vitamin B(12) Chaperone MMACHC Can Result in Increased Tumorigenicity and Methionine Dependence. Mol. Genet. Metab. 2009, 96, 261–267. [Google Scholar] [CrossRef]
- Wallis, K.F.; Morehead, L.C.; Bird, J.T.; Byrum, S.; Miousse, I.R. Differences in Cell Death in Methionine versus Cysteine Depletion. Environ. Mol. Mutagen 2021, 62, 216–226. [Google Scholar] [CrossRef]
- Lu, S.; Hoestje, S.M.; Choo, E.M.; Epner, D.E. Methionine Restriction Induces Apoptosis of Prostate Cancer Cells via the C-Jun N-Terminal Kinase-Mediated Signaling Pathway. Cancer Lett. 2002, 179, 51–58. [Google Scholar] [CrossRef]
- Lu, S.; Hoestje, S.M.; Choo, E.; Epner, D.E. Induction of Caspase-Dependent and -Independent Apoptosis in Response to Methionine Restriction. Int. J. Oncol. 2003, 22, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Mazor, K.M.; Stipanuk, M.H. GCN2- and eIF2α-Phosphorylation-Independent, but ATF4-Dependent, Induction of CARE-Containing Genes in Methionine-Deficient Cells. Amino Acids 2016, 48, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Rajanala, S.H.; Ringquist, R.; Cryns, V.L. Methionine Restriction Activates the Integrated Stress Response in Triple-Negative Breast Cancer Cells by a GCN2- and PERK-Independent Mechanism. Am. J. Cancer Res. 2019, 9, 1766–1775. [Google Scholar]
- Tang, X.; Keenan, M.M.; Wu, J.; Lin, C.-A.; Dubois, L.; Thompson, J.W.; Freedland, S.J.; Murphy, S.K.; Chi, J.-T. Comprehensive Profiling of Amino Acid Response Uncovers Unique Methionine-Deprived Response Dependent on Intact Creatine Biosynthesis. PLoS Genet. 2015, 11, e1005158. [Google Scholar] [CrossRef]
- Deme, J.C.; Hancock, M.A.; Xia, X.; Shintre, C.A.; Plesa, M.; Kim, J.C.; Carpenter, E.P.; Rosenblatt, D.S.; Coulton, J.W. Purification and Interaction Analyses of Two Human Lysosomal Vitamin B12 Transporters: LMBD1 and ABCD4. Mol. Membr. Biol. 2014, 31, 250–261. [Google Scholar] [CrossRef]
- Inoue, Y.; Kawachi, S.; Ohkubo, T.; Nagasaka, M.; Ito, S.; Fukuura, K.; Itoh, Y.; Ohoka, N.; Morishita, D.; Hayashi, H. The CDK Inhibitor P21 Is a Novel Target Gene of ATF4 and Contributes to Cell Survival under ER Stress. FEBS Lett. 2017, 591, 3682–3691. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Darnell, A.M.; Sapp, K.M.; Vander Heiden, M.G.; Spencer, S.L. Cells Use Multiple Mechanisms for Cell-Cycle Arrest upon Withdrawal of Individual Amino Acids. Cell Rep. 2023, 42, 113539. [Google Scholar] [CrossRef] [PubMed]
- Worgan, L.C.; Niles, K.; Tirone, J.C.; Hofmann, A.; Verner, A.; Sammak, A.; Kucic, T.; Lepage, P.; Rosenblatt, D.S. Spectrum of Mutations in Mut Methylmalonic Acidemia and Identification of a Common Hispanic Mutation and Haplotype. Hum. Mutat. 2006, 27, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, P.; Mi, W.; Li, L.; Anderson, N.M.; Lesner, N.P.; Burrows, M.; Plesset, J.; Majer, A.; Wang, G.; et al. Blocking Methionine Catabolism Induces Senescence and Confers Vulnerability to GSK3 Inhibition in Liver Cancer. Nat. Cancer 2024, 5, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lishko, V.K.; Herrera, H.; Groce, A.; Kubota, T.; Hoffman, R.M. Therapeutic Tumor-Specific Cell Cycle Block Induced by Methionine Starvation in Vivo. Cancer Res. 1993, 53, 5676–5679. [Google Scholar] [PubMed]
- Booher, K.; Lin, D.-W.; Borrego, S.L.; Kaiser, P. Downregulation of Cdc6 and Pre-Replication Complexes in Response to Methionine Stress in Breast Cancer Cells. Cell Cycle 2012, 11, 4414–4423. [Google Scholar] [CrossRef]
- Yamada, K.; Kawata, T.; Wada, M.; Isshiki, T.; Onoda, J.; Kawanishi, T.; Kunou, A.; Tadokoro, T.; Tobimatsu, T.; Maekawa, A.; et al. Extremely Low Activity of Methionine Synthase in Vitamin B-12-Deficient Rats May Be Related to Effects on Coenzyme Stabilization Rather than to Changes in Coenzyme Induction. J. Nutr. 2000, 130, 1894–1900. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; Latasa, M.U.; Varela-Rey, M.; Lu, S.C.; García-Trevijano, E.R.; Mato, J.M.; Avila, M.A. L-Methionine Availability Regulates Expression of the Methionine Adenosyltransferase 2A Gene in Human Hepatocarcinoma Cells: Role of S-Adenosylmethionine. J. Biol. Chem. 2003, 278, 19885–19890. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, S.; Miousse, I.R. Rescue of Methionine Dependence by Cobalamin in a Human Colorectal Cancer Cell Line. Nutrients 2024, 16, 997. https://doi.org/10.3390/nu16070997
Garg S, Miousse IR. Rescue of Methionine Dependence by Cobalamin in a Human Colorectal Cancer Cell Line. Nutrients. 2024; 16(7):997. https://doi.org/10.3390/nu16070997
Chicago/Turabian StyleGarg, Sarita, and Isabelle R. Miousse. 2024. "Rescue of Methionine Dependence by Cobalamin in a Human Colorectal Cancer Cell Line" Nutrients 16, no. 7: 997. https://doi.org/10.3390/nu16070997
APA StyleGarg, S., & Miousse, I. R. (2024). Rescue of Methionine Dependence by Cobalamin in a Human Colorectal Cancer Cell Line. Nutrients, 16(7), 997. https://doi.org/10.3390/nu16070997