
Exploring the Gut Microbiome and Metabolome in Individuals with Alopecia Areata Disease
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients’ Recruitment
2.2. Nutrient Intake
2.3. Samples Collection
2.4. 16S rRNA Gene Sequencing
2.5. Faecal and Urine Volatile Organic Compounds (VOCs)
2.6. Statistical Analyses
3. Results
3.1. Power Analysis, Demographic Data, and Nutrient Intake
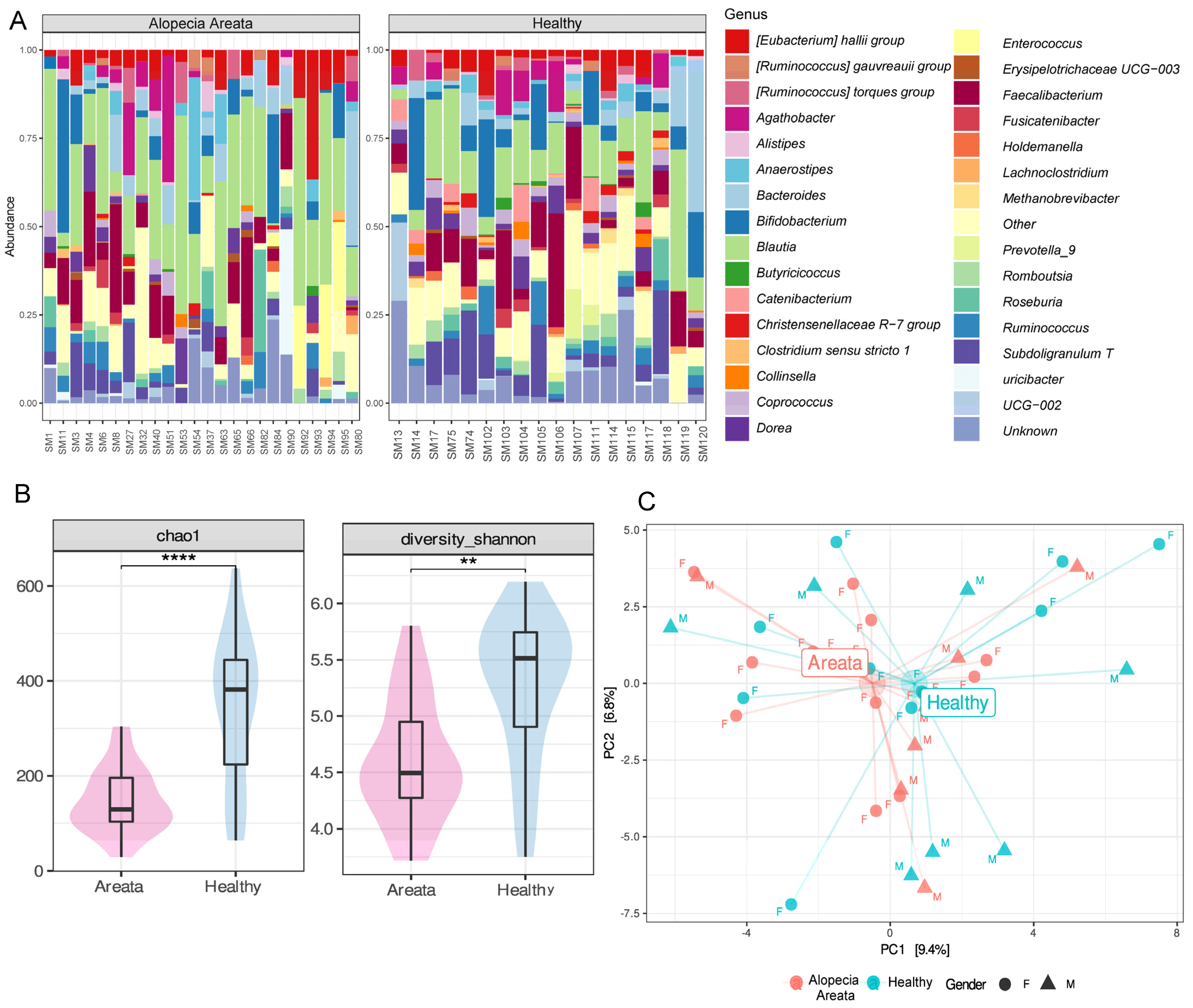
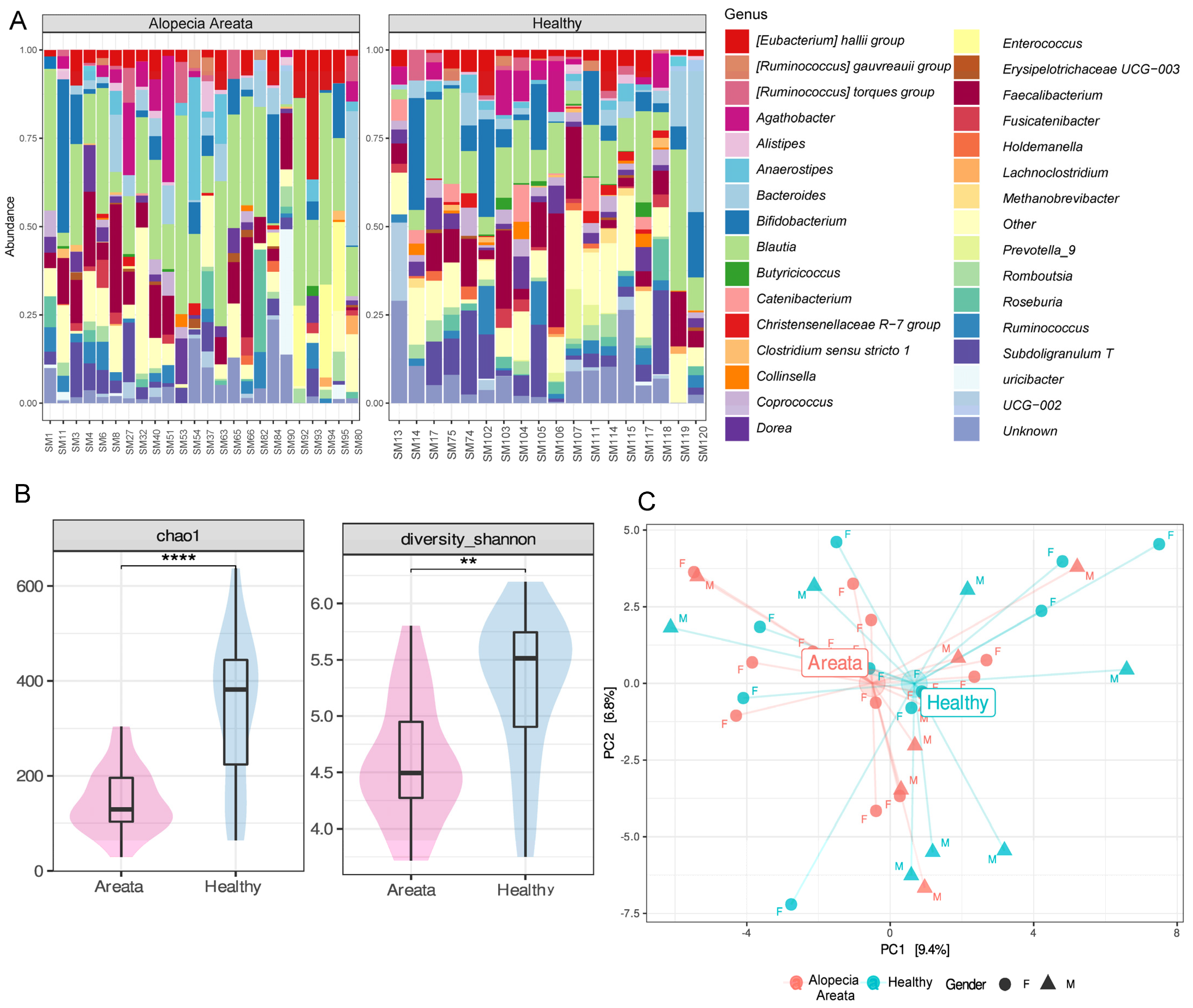
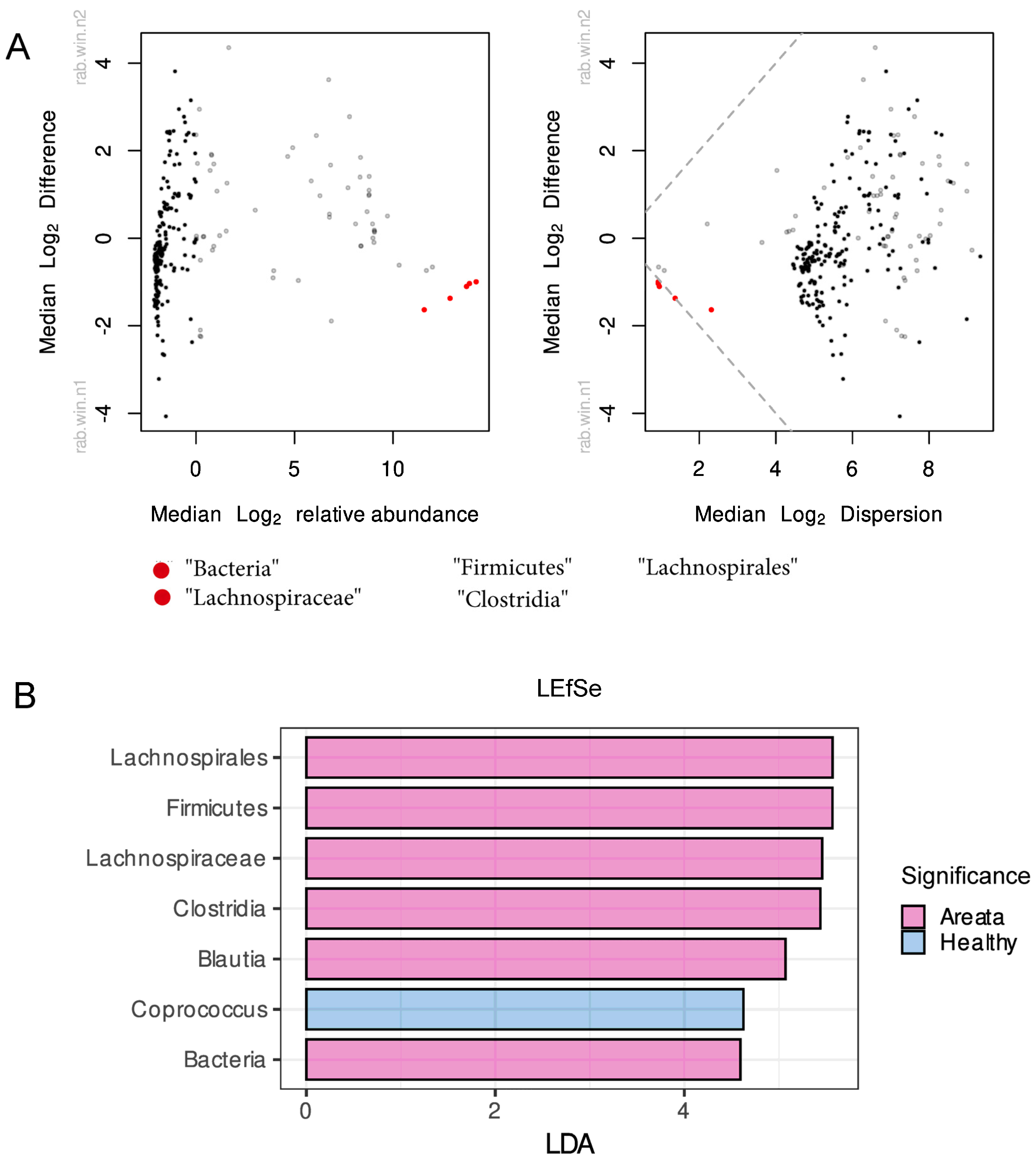
3.2. Gut Microbiome of Healthy and AA Patients
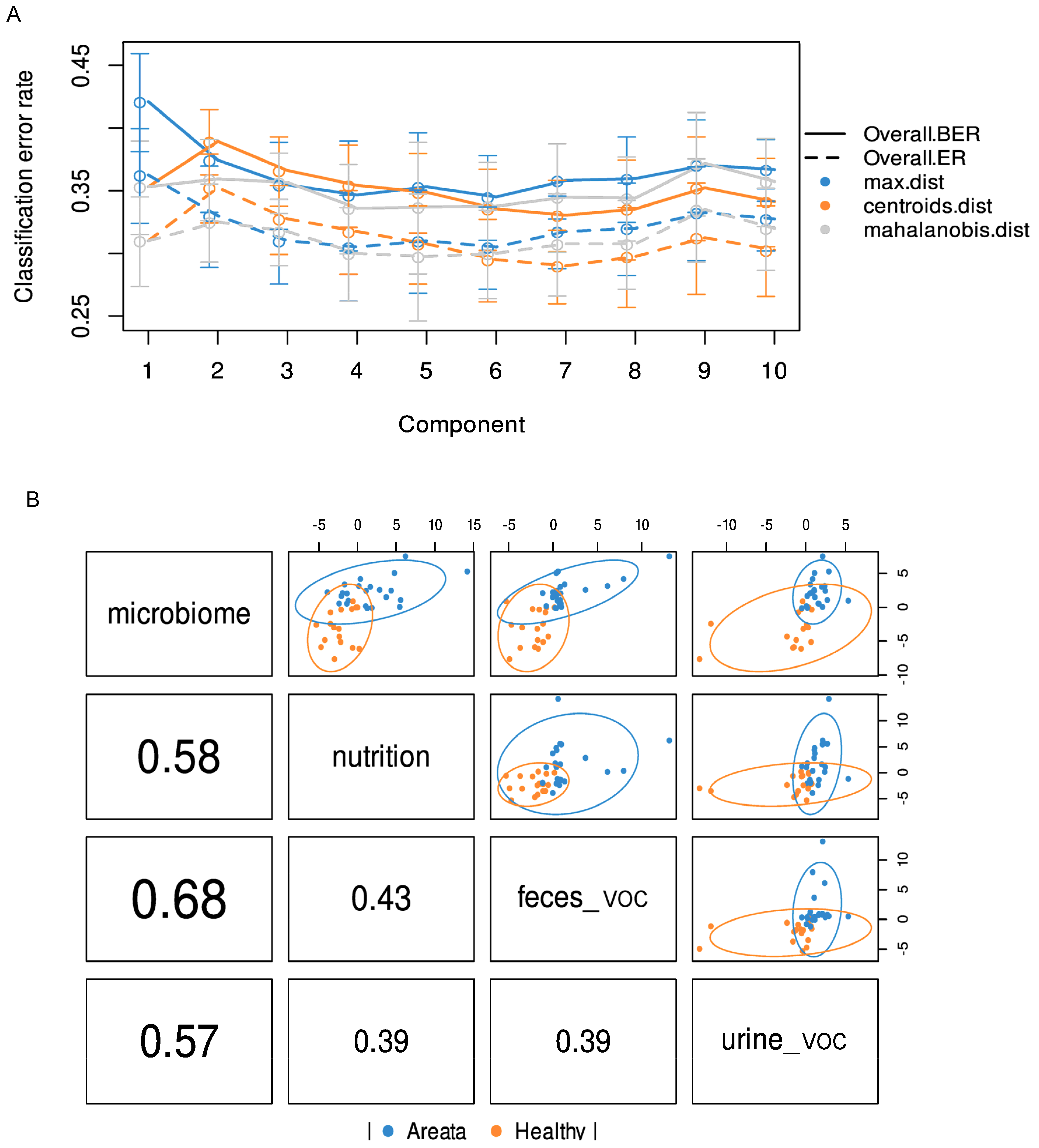
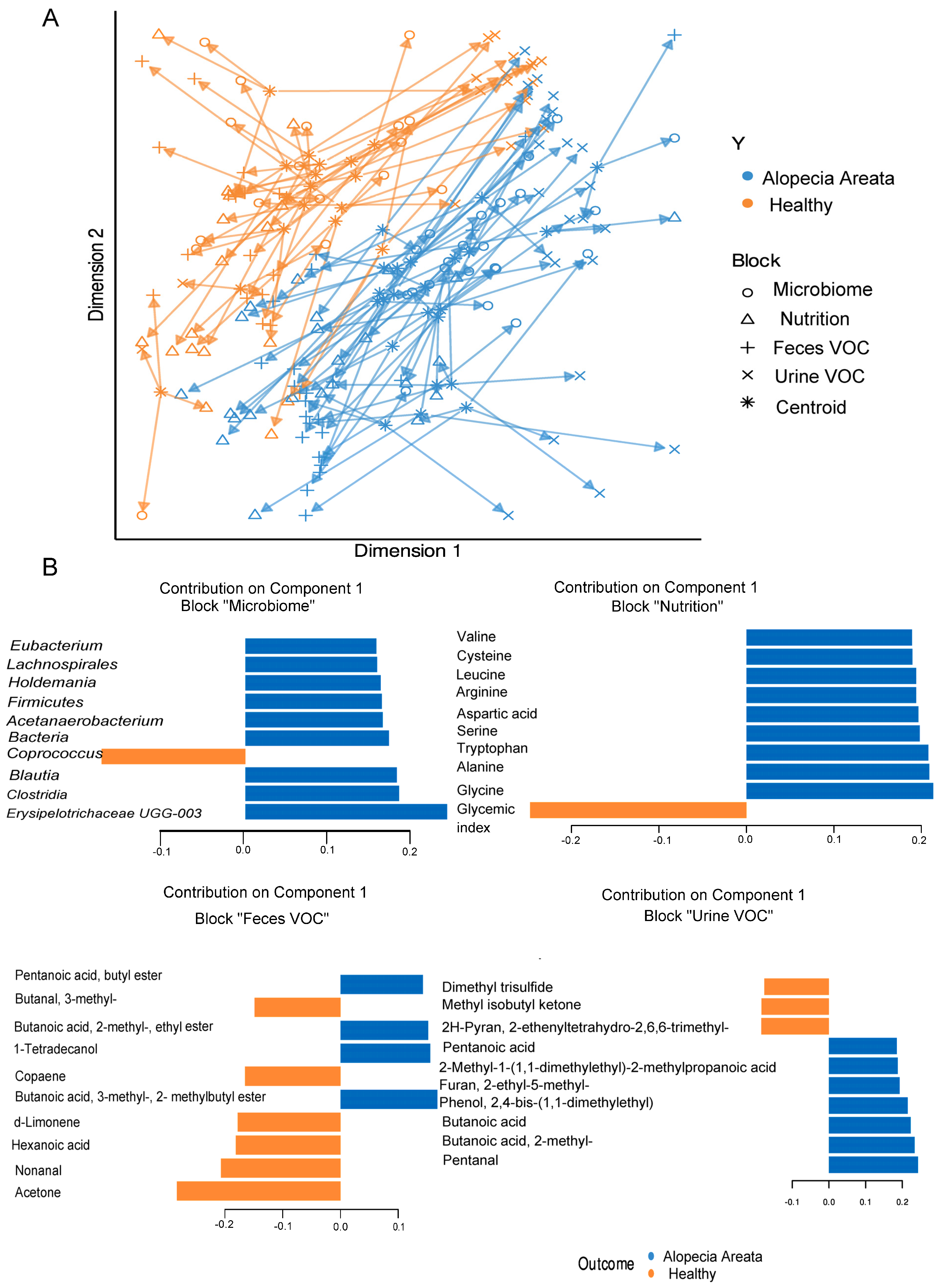
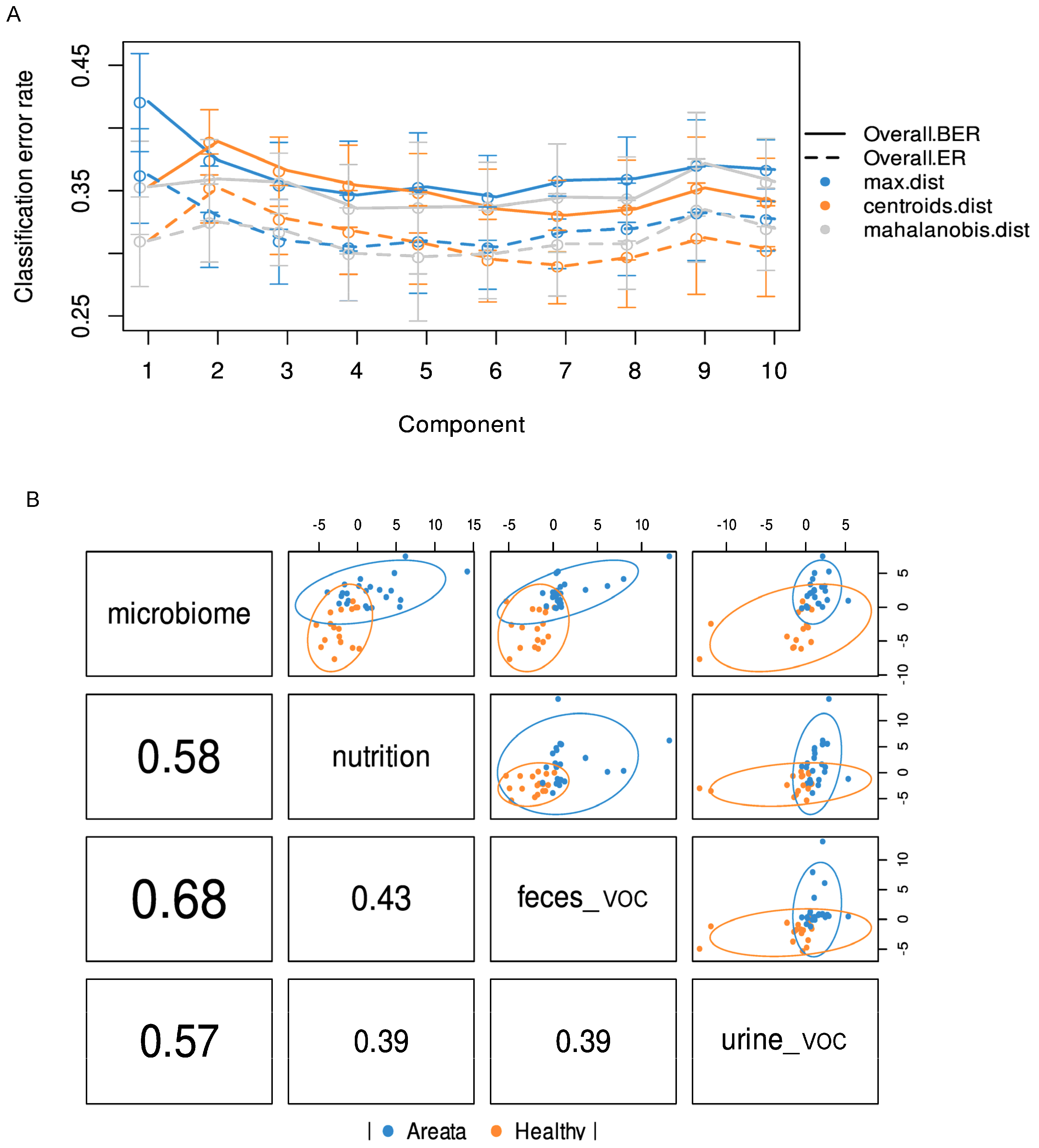
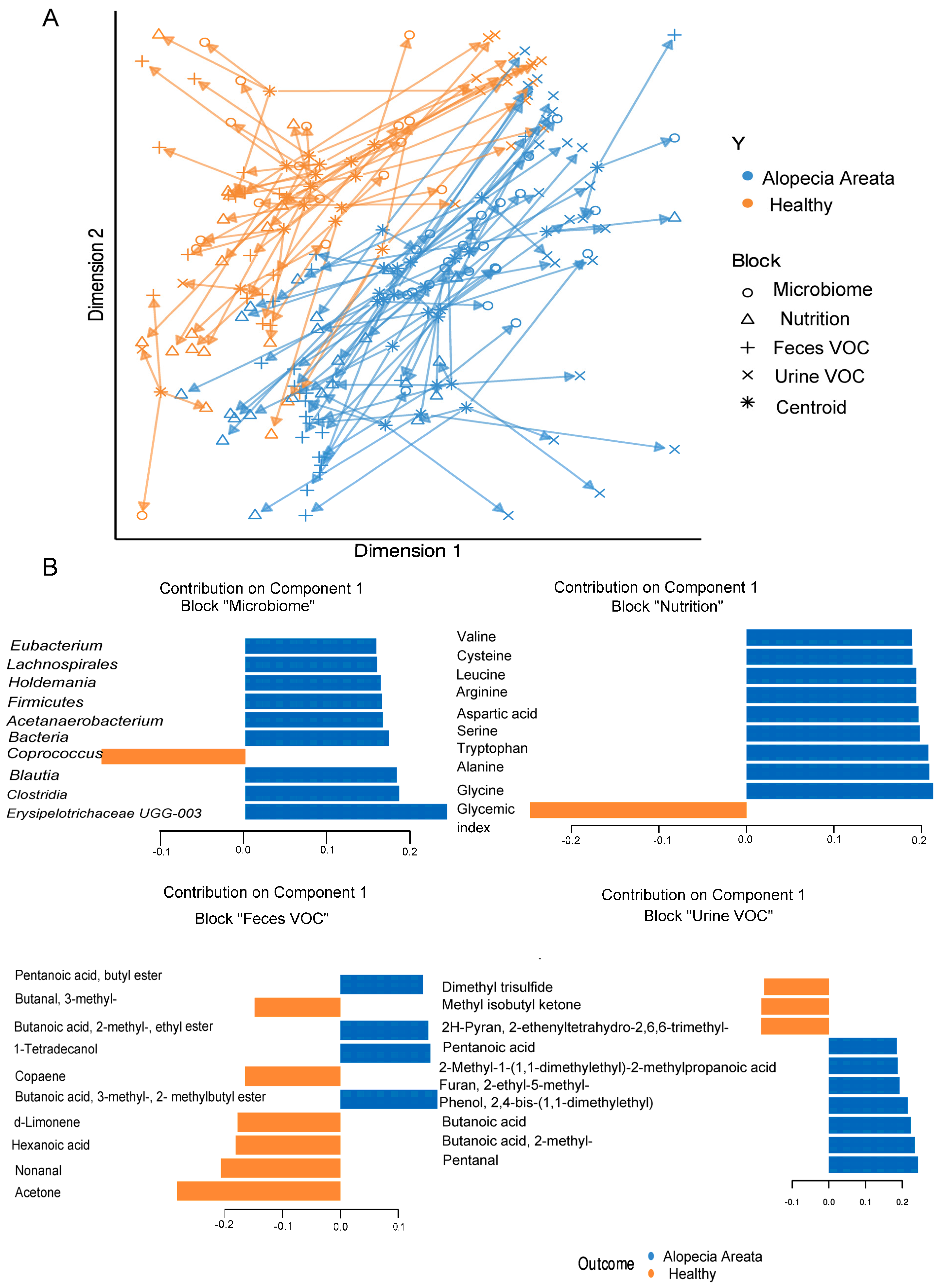
3.3. Integrated Multi-OMICs Analysis on Pathology Regimen (DIABLO)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut Microbiota in the Pathogenesis of Inflammatory Bowel Disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Afzaal, M.; Saeed, F.; Shah, Y.A.; Hussain, M.; Rabail, R.; Socol, C.T.; Hassoun, A.; Pateiro, M.; Lorenzo, J.M.; Rusu, A.V.; et al. Human Gut Microbiota in Health and Disease: Unveiling the Relationship. Front. Microbiol. 2022, 13, 999001. [Google Scholar] [CrossRef]
- Gomaa, E.Z. Human Gut Microbiota/Microbiome in Health and Diseases: A Review. Antonie Van Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef] [PubMed]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal Microbiota, Microbial Translocation, and Systemic Inflammation in Chronic HIV Infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.; Al-Suhaimi, E.A.; Wahid, F.; Shehzad, O.; Shehzad, A. Therapeutic Potential of Curcumin for Multiple Sclerosis. Neurol. Sci. 2018, 39, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the Gut Microbiota in Disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, W.A.; Quraishi, M.N.; Iqbal, T.H. Gut Microbiome and Autoimmune Disorders. Clin. Exp. Immunol. 2022, 209, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, M.; Cao, J.; Li, X.; Fan, D.; Xia, Y.; Lu, X.; Li, J.; Ju, D.; Zhao, H. The Dynamic Interplay between the Gut Microbiota and Autoimmune Diseases. J. Immunol. Res. 2019, 2019, 7546047. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.H.; King, L.E.; Messenger, A.G.; Christiano, A.M.; Sundberg, J.P. Alopecia Areata. Nat. Rev. Dis. Prim. 2017, 3, 17011. [Google Scholar] [CrossRef] [PubMed]
- Darwin, E.; Hirt, P.A.; Fertig, R.; Doliner, B.; Delcanto, G.; Jimenez, J.J. Alopecia Areata: Review of Epidemiology, Clinical Features, Pathogenesis, and New Treatment Options. Int. J. Trichology 2018, 10, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sibbald, C. Alopecia Areata: An Updated Review for 2023. J. Cutan. Med. Surg. 2023, 27, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Paus, R.; Bulfone-Paus, S.; Bertolini, M. Hair Follicle Immune Privilege Revisited: The Key to Alopecia Areata Management. J. Investig. Dermatol. Symp. Proc. 2018, 19, S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, M.; McElwee, K.; Gilhar, A.; Bulfone-Paus, S.; Paus, R. Hair Follicle Immune Privilege and Its Collapse in Alopecia Areata. Exp. Dermatol. 2020, 29, 703–725. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, M.; Zilio, F.; Rossi, A.; Kleditzsch, P.; Emelianov, V.E.; Gilhar, A.; Keren, A.; Meyer, K.C.; Wang, E.; Funk, W.; et al. Abnormal Interactions between Perifollicular Mast Cells and CD8+ T-Cells May Contribute to the Pathogenesis of Alopecia Areata. PLoS ONE 2014, 9, e94260. [Google Scholar] [CrossRef]
- Alli, R.; Nguyen, P.; Boyd, K.; Sundberg, J.P.; Geiger, T.L. A Mouse Model of Clonal CD8+ T Lymphocyte-Mediated Alopecia Areata Progressing to Alopecia Universalis. J. Immunol. 2012, 188, 477–486. [Google Scholar] [CrossRef]
- Ito, T.; Ito, N.; Bettermann, A.; Tokura, Y.; Takigawa, M.; Paus, R. Collapse and Restoration of MHC Class-I-Dependent Immune Privilege: Exploiting the Human Hair Follicle as a Model. Am. J. Pathol. 2004, 164, 623–634. [Google Scholar] [CrossRef]
- Peters, E.M.J.; Liotiri, S.; Bodó, E.; Hagen, E.; Bíró, T.; Arck, P.C.; Paus, R. Probing the Effects of Stress Mediators on the Human Hair Follicle: Substance P Holds Central Position. Am. J. Pathol. 2007, 171, 1872–1886. [Google Scholar] [CrossRef] [PubMed]
- Gilhar, A.; Keren, A.; Shemer, A.; d’Ovidio, R.; Ullmann, Y.; Paus, R. Autoimmune Disease Induction in a Healthy Human Organ: A Humanized Mouse Model of Alopecia Areata. J. Investig. Dermatol. 2013, 133, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, P.; Hu, R.; Qi, S.; Zhao, Y.; Miao, Y.; Han, Y.; Zhou, L.; Yang, Q. Gut Microbiota Characterization in Chinese Patients with Alopecia Areata. J. Dermatol. Sci. 2021, 102, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Loh, S.; Moon, H.; Lew, B.; Sim, W. Role of T Helper 17 Cells and T Regulatory Cells in Alopecia Areata: Comparison of Lesion and Serum Cytokine between Controls and Patients. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- De Pessemier, B.; Grine, L.; Debaere, M.; Maes, A.; Paetzold, B.; Callewaert, C. Gut–Skin Axis: Current Knowledge of the Interrelationship between Microbial Dysbiosis and Skin Conditions. Microorganisms 2021, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Thye, A.Y.; Bah, Y.-R.; Law, J.W.; Tan, L.T.; He, Y.-W.; Wong, S.-H.; Thurairajasingam, S.; Chan, K.-G.; Lee, L.-H.; Letchumanan, V. Gut–Skin Axis: Unravelling the Connection between the Gut Microbiome and Psoriasis. Biomedicines 2022, 10, 1037. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Calabrese, F.M.; De Angelis, M.; Celano, G.; Giuliani, G.; Rinaldi, F. Lichen Planopilaris: The First Biopsy Layer Microbiota Inspection. PLoS ONE 2022, 17, e0269933. [Google Scholar] [CrossRef]
- Colucci, R.; Moretti, S. Implication of Human Bacterial Gut Microbiota on Immune-Mediated and Autoimmune Dermatological Diseases and Their Comorbidities: A Narrative Review. Dermatol. Ther. 2021, 11, 363–384. [Google Scholar] [CrossRef]
- Moreno-Arrones, O.M.; Serrano-Villar, S.; Perez-Brocal, V.; Saceda-Corralo, D.; Morales-Raya, C.; Rodrigues-Barata, R.; Moya, A.; Jaen-Olasolo, P.; Vano-Galvan, S. Analysis of the Gut Microbiota in Alopecia Areata: Identification of Bacterial Biomarkers. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 400–405. [Google Scholar] [CrossRef]
- Rangu, S.; Lee, J.-J.; Hu, W.; Bittinger, K.; Castelo-Soccio, L. Understanding the Gut Microbiota in Pediatric Patients with Alopecia Areata and Their Siblings: A Pilot Study. JID Innov. 2021, 1, 100051. [Google Scholar] [CrossRef]
- Olsen, E.A.; Hordinsky, M.K.; Price, V.H.; Roberts, J.L.; Shapiro, J.; Canfield, D.; Duvic, M.; King, L.E., Jr.; McMichael, A.J.; Randall, V.A.; et al. Alopecia Areata Investigational Assessment Guidelines–Part II. J. Am. Acad. Dermatol. 2004, 51, 440–447. [Google Scholar] [CrossRef]
- SINU (Italian Society of Human Nutrition). Nutrients and Energy Reference Intake for Italian Population, 4th ed.; SINU (Italian Society of Human Nutrition): Milan, Italy, 2014. [Google Scholar]
- Vitellio, P.; Celano, G.; Bonfrate, L.; Gobbetti, M.; Portincasa, P.; De Angelis, M. Effects of Bifidobacterium Longum and Lactobacillus Rhamnosus on Gut Microbiota in Patients with Lactose Intolerance and Persisting Functional Gastrointestinal Symptoms: A Randomised, Double-Blind, Cross-Over Study. Nutrients 2019, 11, 886. [Google Scholar] [CrossRef]
- Gilmore, R.A.; Hutchins, S.; Zhang, X.; Vallender, E. MicrobiomeR: An R Package for Simplified and Standardized Microbiome Analysis Workflows. J. Open Source Softw. 2019, 4, 1299. [Google Scholar] [CrossRef]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Lê Cao, K.-A. DIABLO: An Integrative Approach for Identifying Key Molecular Drivers from Multi-Omics Assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Migacz-Gruszka, K.; Branicki, W.; Obtulowicz, A.; Pirowska, M.; Gruszka, K.; Wojas-Pelc, A. What’s New in the Pathophysiology of Alopecia Areata? The Possible Contribution of Skin and Gut Microbiome in the Pathogenesis of Alopecia–Big Opportunities, Big Challenges, and Novel Perspectives. Int. J. Trichology 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Brzychcy, K.; Dróżdż, I.; Skoczylas, S.; Płoszaj, T.; Sobolewska-Sztychny, D.; Skibińska, M.; Narbutt, J.; Lesiak, A. Gut Microbiota in Alopecia Areata. Adv. Dermatol. Allergol. Dermatol. Alergol. 2022, 39, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pellicer, P.; Navarro-Moratalla, L.; Núñez-Delegido, E.; Agüera-Santos, J.; Navarro-López, V. How Our Microbiome Influences the Pathogenesis of Alopecia Areata. Genes 2022, 13, 1860. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; Heilig, H.G.H.J.; Tims, S.; Zoetendal, E.G.; de Vos, W.M. Long-Term Monitoring of the Human Intestinal Microbiota Composition. Environ. Microbiol. 2013, 15, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Dixit, K.; Chaudhari, D.; Dhotre, D.; Shouche, Y.; Saroj, S. Restoration of Dysbiotic Human Gut Microbiome for Homeostasis. Life Sci. 2021, 278, 119622. [Google Scholar] [CrossRef]
- King, C.H.; Desai, H.; Sylvetsky, A.C.; LoTempio, J.; Ayanyan, S.; Carrie, J.; Crandall, K.A.; Fochtman, B.C.; Gasparyan, L.; Gulzar, N.; et al. Baseline Human Gut Microbiota Profile in Healthy People and Standard Reporting Template. PLoS ONE 2019, 14, e0206484. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Jung, D.-R.; Yoo, H.-Y.; Kim, M.-J.; Singh, V.; Park, S.-H.; Jeong, M.; Park, B.-J.; Shin, J.-H. Comparative Analysis of Scalp and Gut Microbiome in Androgenetic Alopecia: A Korean Cross-Sectional Study. Front. Microbiol. 2022, 13, 1076242. [Google Scholar] [CrossRef] [PubMed]
- de la Cuesta-Zuluaga, J.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, J.; Liang, H.; Ye, L.; Lan, L.; Lu, F.; Wang, Q.; Lei, T.; Yang, X.; Cui, P.; et al. Differences in Alpha Diversity of Gut Microbiota in Neurological Diseases. Front. Neurosci. 2022, 16, 879318. [Google Scholar] [CrossRef] [PubMed]
- Won, E.J.; Jang, H.H.; Park, H.; Kim, S.J. A Potential Predictive Role of the Scalp Microbiome Profiling in Patients with Alopecia Areata: Staphylococcus Caprae, Corynebacterium, and Cutibacterium Species. Microorganisms 2022, 10, 864. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Wu, C.; Zeng, X.; Wang, Q. The Role of Gut Microbiota in the Pathogenesis of Rheumatic Diseases. Clin. Rheumatol. 2018, 37, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bain, K.A.; Nichols, B.; Moffat, F.; Kerbiriou, C.; Ijaz, U.Z.; Gerasimidis, K.; McInnes, I.B.; Åstrand, A.; Holmes, S.; Milling, S.W.F. Stratification of Alopecia Areata Reveals Involvement of CD4 T Cell Populations and Altered Faecal Microbiota. Clin. Exp. Immunol. 2022, 210, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, M.; Chen, S.; Khosrovi-Eghbal, A.; Ekelem, C.; Landaverde, Y.; Baldi, P.; Mesinkovska, N. Characterizing the Skin and Gut Microbiome of Alopecia Areata Patients. Ski. J. Cutan. Med. 2020, 4, 23. [Google Scholar] [CrossRef]
- Gong, X.; Liu, X.; Chen, C.; Lin, J.; Li, A.; Guo, K.; An, D.; Zhou, D.; Hong, Z. Alteration of Gut Microbiota in Patients with Epilepsy and the Potential Index as a Biomarker. Front. Microbiol. 2020, 11, 517797. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, J.; Li, Z.; Huang, Y.; Yuan, Y.; Wang, J.; Zhang, M.; Hu, S.; Liang, Y. Analysis of Gut Microbiota Diversity and Auxiliary Diagnosis as a Biomarker in Patients with Schizophrenia: A Cross-Sectional Study. Schizophr. Res. 2018, 197, 470–477. [Google Scholar] [CrossRef]
- Zhao, F.; Feng, J.; Li, J.; Zhao, L.; Liu, Y.; Chen, H.; Jin, Y.; Zhu, B.; Wei, Y. Alterations of the Gut Microbiota in Hashimoto’s Thyroiditis Patients. Thyroid 2018, 28, 175–186. [Google Scholar] [CrossRef]
- Brunkwall, L.; Ericson, U.; Nilsson, P.M.; Orho-Melander, M.; Ohlsson, B. Self-Reported Bowel Symptoms Are Associated with Differences in Overall Gut Microbiota Composition and Enrichment of Blautia in a Population-Based Cohort. J. Gastroenterol. Hepatol. 2021, 36, 174–180. [Google Scholar] [CrossRef]
- Borde, A.; Åstrand, A. Alopecia Areata and the Gut—The Link Opens up for Novel Therapeutic Interventions. Expert Opin. Ther. Targets 2018, 22, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Notting, F.; Pirovano, W.; Sybesma, W.; Kort, R. The Butyrate-Producing and Spore-Forming Bacterial Genus Coprococcus as a Potential Biomarker for Neurological Disorders. Gut Microbiome 2023, 4, e16. [Google Scholar] [CrossRef]
- Mukherjee, A.; Lordan, C.; Ross, R.P.; Cotter, P.D. Gut Microbes from the Phylogenetically Diverse Genus Eubacterium and Their Various Contributions to Gut Health. Gut Microbes 2020, 12, 1802866. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Liu, R.; Jia, H.; Sun, X.; Gong, Y.; Ma, L.; Qiu, W.; Wang, X. Alterations of the Gut Microbiota in Type 2 Diabetics with or without Subclinical Hypothyroidism. PeerJ 2023, 11, e15193. [Google Scholar] [CrossRef] [PubMed]
- Salachan, P.V.; Sørensen, K.D. Dysbiotic Microbes and How to Find Them: A Review of Microbiome Profiling in Prostate Cancer. J. Exp. Clin. Cancer Res. 2022, 41, 31. [Google Scholar] [CrossRef] [PubMed]
- Indrayan, A.; Mishra, A. The Importance of Small Samples in Medical Research. J. Postgrad. Med. 2021, 67, 219. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Committee on Strategies for Small-Number-Participant Clinical Research Trials. Small Clinical Trials: Issues and Challenges; Charles, H., Evans, J., Ildstad, S.T., Eds.; The National Academies Press: Washington, DC, USA, 2001; ISBN 978-0-309-07333-2.
- Dreier, M.; Meola, M.; Berthoud, H.; Shani, N.; Wechsler, D.; Junier, P. High-Throughput QPCR and 16S RRNA Gene Amplicon Sequencing as Complementary Methods for the Investigation of the Cheese Microbiota. BMC Microbiol. 2022, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 294209. [Google Scholar] [CrossRef]
- Wu, J.; Wang, K.; Wang, X.; Pang, Y.; Jiang, C. The Role of the Gut Microbiome and Its Metabolites in Metabolic Diseases. Protein Cell 2021, 12, 360–373. [Google Scholar] [CrossRef]
- Rios-Covian, D.; González, S.; Nogacka, A.M.; Arboleya, S.; Salazar, N.; Gueimonde, M.; de los Reyes-Gavilán, C.G. An Overview on Fecal Branched Short-Chain Fatty Acids along Human Life and as Related with Body Mass Index: Associated Dietary and Anthropometric Factors. Front. Microbiol. 2020, 11, 513909. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, M.; Eck, A.; Koenen, M.E.; Savelkoul, P.H.M.; Budding, A.E.; Venema, K. Diet Drives Quick Changes in the Metabolic Activity and Composition of Human Gut Microbiota in a Validated In Vitro Gut Model. Res. Microbiol. 2016, 167, 114–125. [Google Scholar] [CrossRef]
- Oyerinde, A.S.; Selvaraju, V.; Babu, J.R.; Geetha, T. Potential Role of Oxidative Stress in the Production of Volatile Organic Compounds in Obesity. Antioxidants 2023, 12, 129. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Sun, Z.; Li, Y.-M.; Xu, H. Oxidative Stress and Alopecia Areata. Front. Med. 2023, 10, 1181572. [Google Scholar] [CrossRef] [PubMed]
- Tsoukalas, D.; Fragoulakis, V.; Papakonstantinou, E.; Antonaki, M.; Vozikis, A.; Tsatsakis, A.; Buga, A.M.; Mitroi, M.; Calina, D. Prediction of Autoimmune Diseases by Targeted Metabolomic Assay of Urinary Organic Acids. Metabolites 2020, 10, 502. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Garcia, V.; Ten-Doménech, I.; Moreno-Giménez, A.; Campos-Berga, L.; Parra-Llorca, A.; Solaz-García, Á.; Lara-Cantón, I.; Pinilla-Gonzalez, A.; Gormaz, M.; Vento, M.; et al. GC-MS Analysis of Short Chain Fatty Acids and Branched Chain Amino Acids in Urine and Faeces Samples from Newborns and Lactating Mothers. Clin. Chim. Acta 2022, 532, 172–180. [Google Scholar] [CrossRef]
- Masuda, H.; Inouye, M. Toxins of Prokaryotic Toxin-Antitoxin Systems with Sequence-Specific Endoribonuclease Activity. Toxins 2017, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Kavisri, M.; Malathy, B.R.; Lavanya, G.; Seema, S.; Christy, H.J.; Anand, D.A.; Jenifer, D.R. Molecular Structure and Bioactivities of 2, 4-Ditert Butyl Phenol Extracted from Plumbago Zeylanica, Investigated Using HPLC and NMR. Biomass Convers. Biorefin. 2023. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, J.; Liang, X.; Wang, Y.; Liu, R.; Zhang, R.; Zha, J.; Martyniuk, C.J. Environmental Concentrations of 2, 4-DTBP Cause Immunotoxicity in Zebrafish (Danio Rerio) and May Elicit Ecological Risk to Wildlife. Chemosphere 2022, 308, 136465. [Google Scholar] [CrossRef]
- Harris, J.E. Chemical-Induced Vitiligo. Dermatol. Clin. 2017, 35, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.; Lee, K.-G.; Lee, Y.-K.; Woo, G.-J.; Park, Y.S.; Lee, S.-J. Correlation of Urinary Furan with Plasma γ-Glutamyltranspeptidase Levels in Healthy Men and Women. Food Chem. Toxicol. 2008, 46, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Uchikawa, M.; Kato, M.; Nagata, A.; Sanada, S.; Yoshikawa, Y.; Tsunematsu, Y.; Sato, M.; Suzuki, T.; Hashidume, T.; Watanabe, K.; et al. Elevated Levels of Proinflammatory Volatile Metabolites in Feces of High Fat Diet Fed KK-Ay Mice. Sci. Rep. 2020, 10, 5681. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic Data | AA | HI |
|---|---|---|
| n = 24 | n = 18 | |
| Men n, (%) | 8 (33.33%) | 7 (38.88%) |
| Women n, (%) | 16 (66.67%) | 11 (61.12%) |
| Age (y, mean ± DS) | 40.00 ± 11.76 | 45.00 ± 9.86 |
| Ethnicity (n, %) | ||
| White | 24 (100%) | 18 (100%) |
| Salt Score (mean ± DS) | 87.46 ± 1.87 | n.a. * |
| AA subtype (mean ± DS) | n.a. | |
| Alopecia areata | 24 (100%) | |
| Comorbidities * | n.a. | |
| Celiac disease | 2 (8.33%) | |
| Allergic rhinitis | 1 (4.16%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikoloudaki, O.; Pinto, D.; Acin Albiac, M.; Celano, G.; Da Ros, A.; De Angelis, M.; Rinaldi, F.; Gobbetti, M.; Di Cagno, R. Exploring the Gut Microbiome and Metabolome in Individuals with Alopecia Areata Disease. Nutrients 2024, 16, 858. https://doi.org/10.3390/nu16060858
Nikoloudaki O, Pinto D, Acin Albiac M, Celano G, Da Ros A, De Angelis M, Rinaldi F, Gobbetti M, Di Cagno R. Exploring the Gut Microbiome and Metabolome in Individuals with Alopecia Areata Disease. Nutrients. 2024; 16(6):858. https://doi.org/10.3390/nu16060858
Chicago/Turabian StyleNikoloudaki, Olga, Daniela Pinto, Marta Acin Albiac, Giuseppe Celano, Alessio Da Ros, Maria De Angelis, Fabio Rinaldi, Marco Gobbetti, and Raffaella Di Cagno. 2024. "Exploring the Gut Microbiome and Metabolome in Individuals with Alopecia Areata Disease" Nutrients 16, no. 6: 858. https://doi.org/10.3390/nu16060858
APA StyleNikoloudaki, O., Pinto, D., Acin Albiac, M., Celano, G., Da Ros, A., De Angelis, M., Rinaldi, F., Gobbetti, M., & Di Cagno, R. (2024). Exploring the Gut Microbiome and Metabolome in Individuals with Alopecia Areata Disease. Nutrients, 16(6), 858. https://doi.org/10.3390/nu16060858