

Rickets Types and Treatment with Vitamin D and Analogues




Abstract
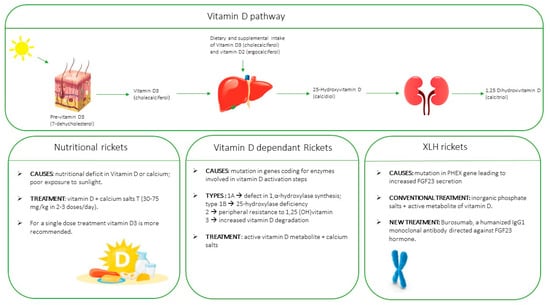
1. Introduction
2. Materials and Methods
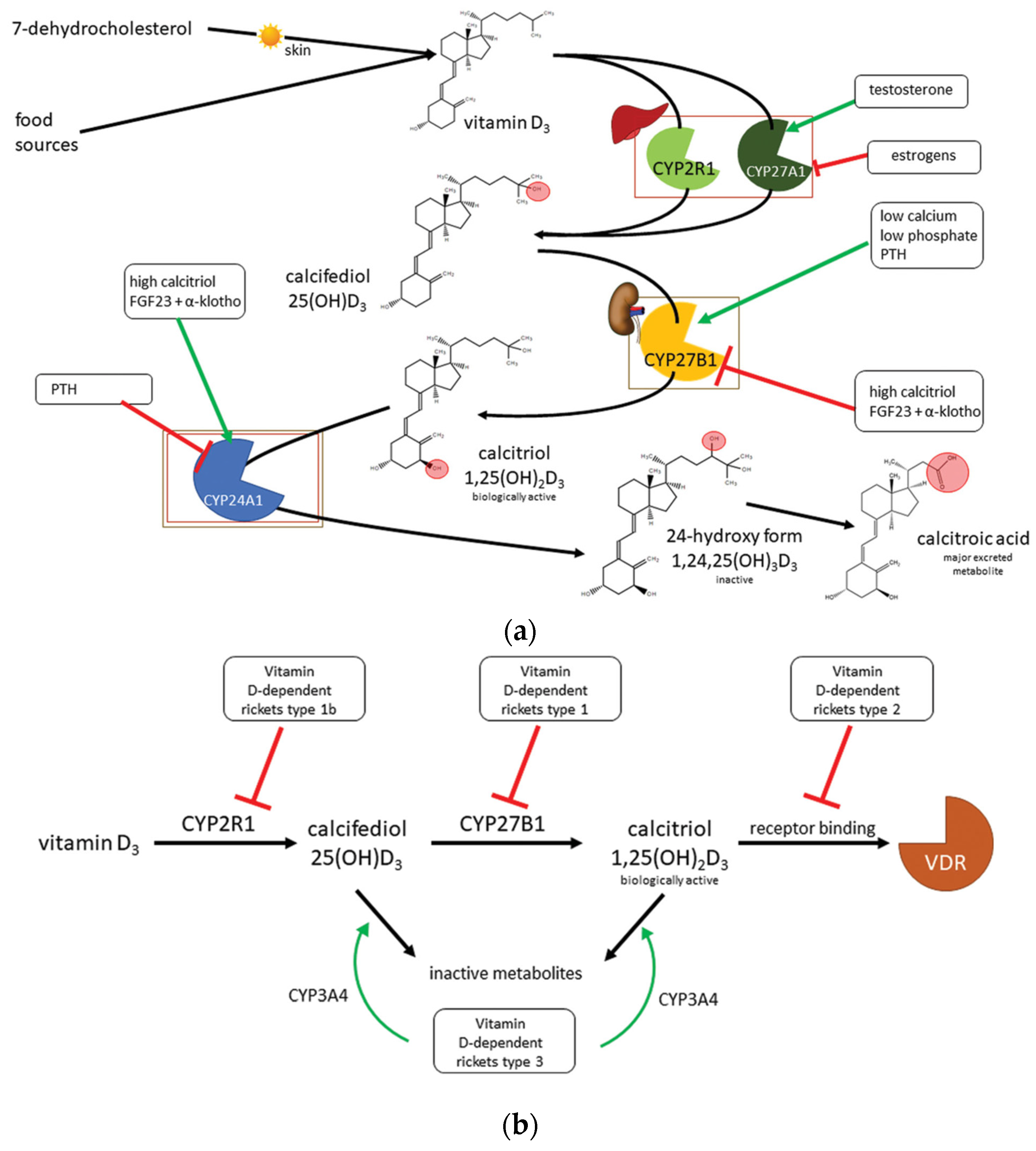
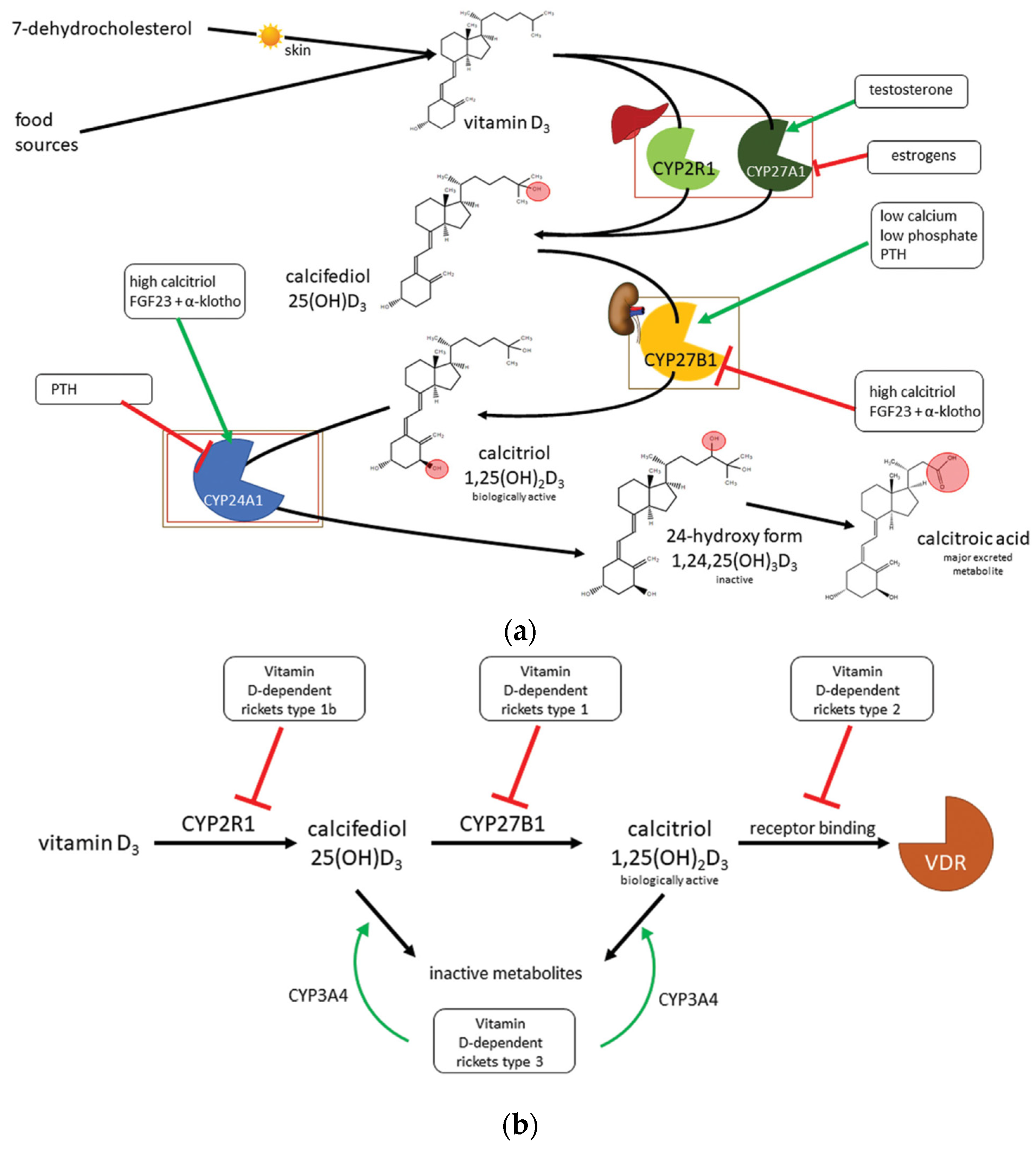
3. Vitamin D
4. Vitamin D Deficiency
5. Types of Rickets
6. Clinical Manifestation and Radiologic Features
7. Diagnostic Approach to Suspected Rickets
8. Treatment
8.1. Nutritional Rickets due to Vitamin D Deficiency
8.2. Genetic Vitamin-D Dependent Rickets
8.3. X-Linked Hypophosphatemic Rickets (XLH)
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chanchlani, R.; Nemer, P.; Sinha, R.; Nemer, L.; Krishnappa, V.; Sochett, E.; Safadi, F.; Raina, R. An Overview of Rickets in Children. Kidney Int. Rep. 2020, 5, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Simm, P.J.; Munns, C.F.; Jefferies, C.A.; Wheeler, B.J. Editorial: Childhood Rickets—New Developments in Epidemiology, Prevention, and Treatment. Front. Endocrinol. 2020, 11, 621734. [Google Scholar] [CrossRef]
- Munns, C.F.; Shaw, N.; Kiely, M.; Specker, B.L.; Thacher, T.D.; Ozono, K.; Michigami, T.; Tiosano, D.; Mughal, M.Z.; Mäkitie, O.; et al. Global Consensus Recommendations on Prevention and Management of Nutritional Rickets. J. Clin. Endocrinol. Metab. 2016, 101, 394–415. [Google Scholar] [CrossRef] [PubMed]
- Janoušek, J.; Pilařová, V.; Macáková, K.; Nomura, A.; Veiga-Matos, J.; Silva, D.D.D.; Remião, F.; Saso, L.; Malá-Ládová, K.; Malý, J.; et al. Vitamin D: Sources, physiological role, biokinetics, deficiency, therapeutic use, toxicity, and overview of analytical methods for detection of vitamin D and its metabolites. Crit. Rev. Clin. Lab. Sci. 2022, 59, 517–554. [Google Scholar] [CrossRef]
- Khazai, N.; Judd, S.E.; Tangpricha, V. Calcium and vitamin D: Skeletal and extraskeletal health. Curr. Rheumatol. Rep. 2008, 10, 110–117. [Google Scholar] [CrossRef]
- Waldron, J.L.; Ashby, H.L.; Cornes, M.P.; Bechervaise, J.; Razavi, C.; Thomas, O.L.; Chugh, S.; Deshpande, S.; Ford, C.; Gama, R. Vitamin D: A negative acute phase reactant. J. Clin. Pathol. 2013, 66, 620–622. [Google Scholar] [CrossRef]
- Holick, M.F. The vitamin D deficiency pandemic: Approaches for diagnosis, treatment and prevention. Rev. Endocr. Metab. Disord. 2017, 18, 153–165. [Google Scholar] [CrossRef]
- Płudowski, P.; Kos-Kudła, B.; Walczak, M.; Fal, A.; Zozulińska-Ziółkiewicz, D.; Sieroszewski, P.; Peregud-Pogorzelski, J.; Lauterbach, R.; Targowski, T.; Lewiński, A.; et al. Guidelines for Preventing and Treating Vitamin D Deficiency: A 2023 Update in Poland. Nutrients 2023, 15, 695. [Google Scholar] [CrossRef]
- Thacher, T.D.; Pludowski, P.; Shaw, N.J.; Mughal, M.Z.; Munns, C.F.; Högler, W. Nutritional rickets in immigrant and refugee children. Public. Health Rev. 2016, 37, 3. [Google Scholar] [CrossRef]
- Byrne, S.N. How much sunlight is enough? Photochem. Photobiol. Sci. 2014, 13, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Saggese, G.; Vierucci, F.; Prodam, F.; Cardinale, F.; Cetin, I.; Chiappini, E.; De’ Angelis, G.L.; Massari, M.; Miraglia Del Giudice, E.; Miraglia Del Giudice, M.; et al. Vitamin D in pediatric age: Consensus of the Italian Pediatric Society and the Italian Society of Preventive and Social Pediatrics, jointly with the Italian Federation of Pediatricians. Ital. J. Pediatr. 2018, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Acar, S.; Demir, K.; Shi, Y. Genetic Causes of Rickets. J. Clin. Res. Pediatr. Endocrinol. 2018, 9, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Shaw, N.J.; Portale, A.A.; Ward, L.M.; Abrams, S.A.; Pettifor, J.M. Rickets. Nat. Rev. Dis. Primers 2017, 3, 17101. [Google Scholar] [CrossRef]
- Roizen, J.D.; Li, D.; O’Lear, L.; Javaid, M.K.; Shaw, N.J.; Ebeling, P.R.; Nguyen, H.H.; Rodda, C.P.; Thummel, K.E.; Thacher, T.D.; et al. CYP3A4 mutation causes vitamin D–dependent rickets type 3. J. Clin. Investig. 2018, 128, 1913–1918. [Google Scholar] [CrossRef]
- Mughal, M.Z. Rickets. Curr. Osteoporos. Rep. 2011, 9, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, B.J.; Dickson, N.P.; Houghton, L.A.; Ward, L.M.; Taylor, B.J. Incidence and characteristics of vitamin D deficiency rickets in New Zealand children: A New Zealand Paediatric Surveillance Unit study. Aust. N. Z. J. Public. Health 2015, 39, 380–383. [Google Scholar] [CrossRef]
- Basatemur, E.; Sutcliffe, A. Incidence of Hypocalcemic Seizures Due to Vitamin D Deficiency in Children in the United Kingdom and Ireland. J. Clin. Endocrinol. Metab. 2015, 100, E91–E95. [Google Scholar] [CrossRef]
- Maiya, S.; Sullivan, I.; Allgrove, J.; Yates, R.; Malone, M.; Brain, C.; Archer, N.; Mok, Q.; Daubeney, P.; Tulloh, R.; et al. Hypocalcaemia and vitamin D deficiency: An important, but preventable, cause of life-threatening infant heart failure. Heart 2008, 94, 581–584. [Google Scholar] [CrossRef]
- Baroncelli, G.I.; Zampollo, E.; Manca, M.; Toschi, B.; Bertelloni, S.; Michelucci, A.; Isola, A.; Bulleri, A.; Peroni, D.; Giuca, M.R. Pulp chamber features, prevalence of abscesses, disease severity, and PHEX mutation in X-linked hypophosphatemic rickets. J. Bone Miner Metab 2021, 39, 212–223. [Google Scholar] [CrossRef]
- Chaussain-Miller, C.; Sinding, C.; Wolikow, M.; Lasfargues, J.-J.; Godeau, G.; Garabédian, M. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: Prevention by early treatment with 1-hydroxyvitamin D. J. Pediatr. 2003, 142, 324–331. [Google Scholar] [CrossRef]
- Thacher, T.D.; Fischer, P.R.; Pettifor, J.M.; Lawson, J.O.; Manaster, B.J.; Reading, J.C. Radiographic scoring method for the assessment of the severity of nutritional rickets. J. Trop. Pediatr. 2000, 46, 132–139. [Google Scholar] [CrossRef]
- Cannalire, G.; Pilloni, S.; Esposito, S.; Biasucci, G.; Di Franco, A.; Street, M.E. Alkaline phosphatase in clinical practice in childhood: Focus on rickets. Front. Endocrinol. 2023, 14, 1111445. [Google Scholar] [CrossRef]
- Gentile, C.; Chiarelli, F. Rickets in Children: An Update. Biomedicines 2021, 9, 738. [Google Scholar] [CrossRef] [PubMed]
- Chesher, D.; Oddy, M.; Darbar, U.; Sayal, P.; Casey, A.; Ryan, A. Outcome of adult patients with X-linked hypophosphatemia caused by PHEX gene mutations. J. Inher. Metab. Dis. 2018, 41, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.R.; Tripkovic, L.; Hart, K.H.; Lanham-New, S.A. Vitamin D deficiency as a public health issue: Using vitamin D 2 or vitamin D 3 in future fortification strategies. Proc. Nutr. Soc. 2017, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Thacher, T.D.; Fischer, P.R.; Pettifor, J.M. Vitamin D treatment in calcium-deficiency rickets: A randomised controlled trial. Arch. Dis. Child. 2014, 99, 807–811. [Google Scholar] [CrossRef]
- Shaw, N.J. Prevention and treatment of nutritional rickets. J. Steroid Biochem. Mol. Biol. 2016, 164, 145–147. [Google Scholar] [CrossRef]
- Misra, M.; Pacaud, D.; Petryk, A.; Collett-Solberg, P.F.; Kappy, M.; on behalf of the Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Vitamin D Deficiency in Children and Its Management: Review of Current Knowledge and Recommendations. Pediatrics 2008, 122, 398–417. [Google Scholar] [CrossRef]
- Cesur, Y.; Caksen, H.; Gündem, A.; Kirimi, E.; Odabas, D. Comparison of low and high dose of vitamin D treatment in nutritional vitamin D deficiency rickets. J. Pediatr. Endocrinol. Metab. 2003, 16, 1105–1109. [Google Scholar] [CrossRef]
- Mittal, H.; Rai, S.; Shah, D.; Madhu, S.V.; Mehrotra, G.; Malhotra, R.K.; Gupta, P. 300,000 IU or 600,000 IU of oral vitamin D3fortreatmentofnutritional rickets: A randomized con trolled trial. Indian Pediatr. 2014, 51, 265–272. [Google Scholar] [CrossRef]
- Zabihiyeganeh, M.; Jahed, A.; Nojomi, M. Treatment of hypovitaminosis D with pharmacologic doses of cholecalciferol, oral vs intramuscular; an open labeled RCT. Clin. Endocrinol. 2013, 78, 210–216. [Google Scholar] [CrossRef]
- Armas, L.A.G.; Hollis, B.W.; Heaney, R.P. Vitamin D 2 Is Much Less Effective than Vitamin D 3 in Humans. J. Clin. Endocrinol. Metab. 2004, 89, 5387–5391. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.L.; Greer, F.R.; the Section on Breastfeeding and Committee on Nutrition. Prevention of Rickets and Vitamin D Deficiency in Infants, Children, and Adolescents. Pediatrics 2008, 122, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.A. Diagnosis and Management of Vitamin D Dependent Rickets. Front. Pediatr. 2020, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Molin, A.; Wiedemann, A.; Demers, N.; Kaufmann, M.; Do Cao, J.; Mainard, L.; Dousset, B.; Journeau, P.; Abeguile, G.; Coudray, N.; et al. Vitamin D–Dependent Rickets Type 1B (25-Hydroxylase Deficiency): A Rare Condition or a Misdiagnosed Condition? J. Bone Miner. Res. 2017, 32, 1893–1899. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Gomez, J.M.; Bouillon, R. Is calcifediol better than cholecalciferol for vitamin D supplementation? Osteoporos. Int. 2018, 29, 1697–1711. [Google Scholar] [CrossRef]
- Guidelines of ISPED Study Group on Bone Metabolism and Paediatric Osteoporosis. Available online: https://www.siedp.it/files/PDTARachitismiFINALE.pdf (accessed on 15 March 2022).
- Haffner, D.; Leifheit-Nestler, M.; Grund, A.; Schnabel, D. Rickets guidance: Part II—Management. Pediatr. Nephrol. 2022, 37, 2289–2302. [Google Scholar] [CrossRef]
- Trombetti, A.; Al-Daghri, N.; Brandi, M.L.; Cannata-Andía, J.B.; Cavalier, E.; Chandran, M.; Chaussain, C.; Cipullo, L.; Cooper, C.; Haffner, D.; et al. Interdisciplinary management of FGF23-related phosphate wasting syndromes: A Consensus Statement on the evaluation, diagnosis and care of patients with X-linked hypophosphataemia. Nat. Rev. Endocrinol. 2022, 18, 366–384. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Portale, A.A.; Högler, W.; Simmons, J.H.; Gottesman, G.S.; Padidela, R.; Namba, N.; et al. Effect of Burosumab Compared with Conventional Therapy on Younger vs Older Children with X-linked Hypophosphatemia. J. Clin. Endocrinol. Metab. 2022, 107, e3241–e3253. [Google Scholar] [CrossRef] [PubMed]
- Imel, E.A.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Ward, L.M.; Nilsson, O.; Simmons, J.H.; Padidela, R.; Namba, N.; Cheong, H.I.; et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: A randomised, active-controlled, open-label, phase 3 trial. Lancet 2019, 393, 2416–2427. [Google Scholar] [CrossRef]
- Whyte, M.P.; Carpenter, T.O.; Gottesman, G.S.; Mao, M.; Skrinar, A.; San Martin, J.; Imel, E.A. Efficacy and safety of burosumab in children aged 1–4 years with X-linked hypophosphataemia: A multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Linglart, A.; Biosse-Duplan, M.; Briot, K.; Chaussain, C.; Esterle, L.; Guillaume-Czitrom, S.; Kamenicky, P.; Nevoux, J.; Prié, D.; Rothenbuhler, A.; et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr. Connect. 2014, 3, R13–R30. [Google Scholar] [CrossRef] [PubMed]
- Brandi, M.L.; Jan de Beur, S.; Briot, K.; Carpenter, T.; Cheong, H.I.; Cohen-Solal, M.; Crowley, R.K.; Eastell, R.; Imanishi, Y.; Imel, E.A.; et al. Efficacy of Burosumab in Adults with X-linked Hypophosphatemia (XLH): A Post Hoc Subgroup Analysis of a Randomized Double-Blind Placebo-Controlled Phase 3 Study. Calcif. Tissue Int. 2022, 111, 409–418. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Age | Dose IU |
|---|---|
| 0–3 months | 400/die |
| 1–3 years | 600/die |
| 4–10 years * | 600–1000/die |
| 11–18 years * | 1000–2000/die |
| 19–65 years * | 1000–2000/die |
| 65–75 years | 1000–2000/die |
| >75 years | 2000–4000/die |
| Calcipenic Rickets | Causes |
|---|---|
| Nutritional rickets | Calcium deficiency |
| Vitamin D deficiency | |
| Congenital defects of action of Vitamin D | Vitamin D dependent rickets type 1A Vitamin D dependent rickets type 1B Vitamin D dependent rickets type 2A Vitamin D dependent rickets type 2B Vitamin D dependent rickets type 3 |
| Acquired vitamin D deficiency | Hepatic insufficiency |
| Renal insufficiency | |
| Malabsorption | |
| Drugs | |
| Hyperparathyroidism |
| Hypophosphatemic Rickets | Causes |
|---|---|
| Nutritional/reduced intestinal intake/absorption of phosphates | Malnutrition |
| Prematurity | |
| Phosphate chelators Administration of elemental formulas Total parental nutrition Gastrointestinal nutrition (e.g., short bowel syndrome) Cellular re-distribution Insulin therapy in diabetic ketoacidosis Acute respiratory alkalosis Re-feeding syndrome | |
| Increased renal phosphate loss | FGF23-related hypophosphatemia |
| Hypophosphatemic rickets with hypercalciuria | |
| Renal proximal tubule dysfunction/Fanconi’s syndrome | Oculocerebrorenal syndrome |
| X-linked recessive nephrolithiasis | |
| Infantile cystinosis Tyrosinemia Hepatolenticular degeneration |
| Grade | Description | |
|---|---|---|
| Wrist 1 | 0 | normal growth cartilage without signs of rickets |
| 0.5 | metaphyseal margin radiolucency without enlargement or irregularity of the metaphyseal margin | |
| 1 | enlargement of growth cartilage, irregularity of metaphyseal margin | |
| 1.5 | partial metaphyseal concavity or incomplete irregularity of metaphyseal margin | |
| 2 | concave appearance of the metaphyses with fraying of the margins | |
| Knee 2 | 0 | normal growth cartilage without signs of rickets |
| 1 | partial radiolucency, regular margins of metaphyses | |
| 2 | partial radiolucency, irregular margins of the metaphyses | |
| 3 | complete radiolucency, the epiphyses are clearly separated from metaphyses |
| Ca | P | ALP | uCa | FGF23 | PTH | 25OHD | 1,25(OH)2D | |
|---|---|---|---|---|---|---|---|---|
| Nutritional Rickets with vitamin D deficiency | N, < | N, < | > | < | N, < | N | < | >, N, < |
| Nutritional Rickets with Ca deficiency | N, < | N, < | > | < | > | N, > | N, < | > |
| XLH rickets | N | < | > | N | > | N, > | N | N, < |
| Hereditary hypophosphatemic Rickets with hypercalciuria | N | < | > | > | < | N, < | N | > |
| Hypophosphatemic Rickets with Hyperparathyroidism | N | < | > | N | > | > | N | N |
| Vitamin D-dependent R. type 1A | < | < | > | < | N, < | > | N, > | < |
| Vitamin D-dependent R. type 1B | N, < | < | > | < | ? | > | < | < |
| Vitamin D-dependent R. type 2A | < | < | > | < | N, < | > | N, > | > |
| Vitamin D-dependent R. type 2B | < | < | > | < | ? | > | N, > | > |
| vitamin D-dependent R. type 3 | < | < | > | < | ? | > | < | < |
| Society/Organization | Year | Severe Deficiency | Deficiency | Insufficiency | Sufficiency/ Adequacy |
|---|---|---|---|---|---|
| Canadian Pediatric Society | 2007 | - | <10 ng/mL | 10–29 ng/mL | ≥30 ng/mL |
| Lawson Wilkins Pediatric Endocrine Society | 2008 | <5 ng/mL | 5–14 ng/mL | 15–19 ng/mL | ≥20 ng/mL |
| Institute of Medicine | 2011 | - | <12 ng/mL | 12–20 ng/mL (a) | ≥20 ng/mL |
| The Endocrine Society | 2011 | - | <20 ng/mL | 21–29 ng/mL | ≥30 ng/mL |
| British Paediatric and Adolescent Bone Group | 2012 | - | < 10 ng/mL | 10–19 ng/mL | ≥20 ng/mL |
| French Society of Paediatrics | 2012 | - | < 20 ng/mL | - | ≥20 ng/mL |
| Asociación Espanola de Pediatría (Spain) | 2012 | - | <20 ng/mL | - | ≥20 ng/mL |
| Federal Commission for Nutrition (Switzerland) | 2012 | <10 ng/mL | <20 ng/mL | - | ≥20 ng/mL |
| Nordic Nutrition Recommendations | 2012 | - | <12 ng/mL | 12–20 ng/mL | ≥20 ng/mL |
| German Nutrition Society | 2012 | - | - | - | ≥20 ng/mL |
| Health council of the Netherlands | 2012 | - | - | - | ≥12 ng/mL |
| European Society for Paediatric Gastroenterology Hepatology and Nutrition | 2013 | <10 ng/mL | <20 ng/mL | - | ≥20 ng/mL |
| Central Europe | 2013 | - | <20 ng/mL | 20–29 ng/mL | ≥30 ng/mL |
| Society for Adolescent Health and Medicine | 2013 | - | <20 ng/mL | 20–29 ng/mL | ≥30 ng/mL |
| Australia/New Zealand | 2013 | <5 ng/mL | 5–11 ng/mL | 12–19 ng/mL | ≥20 ng/mL |
| American Academy of Pediatrics | 2014 | - | <20 ng/mL | - | ≥20 ng/mL |
| Japanese Society for Bone and Mineral Research, Japan Endocrine Society (b) | 2015 | - | <20 ng/mL | - | - |
| Scientific Advisory Committee on Nutrition | 2016 | - | - | - | ≥10 ng/mL |
| European Food Safety Authority | 2016 | - | - | - | ≥20 ng/mL |
| United Arab Emirates | 2016 | - | <20 ng/mL | 20–29 ng/mL | ≥30 ng/mL |
| Global Consensus for rickets | 2016 | - | <12 ng/mL | 12–19 ng/mL | ≥20 ng/mL |
| Japanese Society for Bone and Mineral Research, Japan Endocrine Society (c) | 2017 | - | <20 ng/mL | 20–29 ng/mL | ≥30 ng/mL |
| European Academy of Pediatrics | 2017 | Definition of vitamin D status is unclear due to a lack of consensus |
| 2009 Polish Recommendations | 2013 Central European Recommendations | 2018 Polish Recommendations | |
|---|---|---|---|
| Diagnostics Thresholds Defining Vitamin D Status on the Basis of Serum 25(OH)D Concentration [ng/mL] * | |||
| Sufficiency | Children: 20–60 Adults: 30–80 | 30–50 | 30–50 |
| Insufficiency | Not defined | 20–30 | 20–30 |
| Deficiency | <10 | <20 | 10–20 deficiency <10 severe deficiency |
| Toxicity | Not defined | >100 | >100 |
| Age | Dose IU | Duration |
|---|---|---|
| <3 months | 2000 | 3 months |
| 3–12 months | 2000 | 3 months |
| >1–12 years | 3000–6000 | 3 months |
| >12 years | 6000 | 3 months |
| Age | Dose IU | Duration |
|---|---|---|
| 3–12 months | 50,000 | Single dose |
| >1–12 years | 150,000 | Single dose |
| >12 years | 300,000 | Single dose |
| Vitamin D or Metabolites | Calcium Salt | |
|---|---|---|
| Vitamin D dependent rickets type 1A | Alfacalcidiol or calcitriol 10–100 ng/kg/day | 0.5–3 g/day in 2–3 doses |
| Vitamin D dependent rickets type 1 B | Homozygous patient: vitamin D 600,000 IU/every 3 months | 0.5–2 g/day in 2–3 doses |
| Heterozygous patients: vitamin D 5000–10,000 IU/day or | ||
| Calcifediol 15–50 µg/day | ||
| Vitamin D dependet rickets type 2 | Calcitriol or alfacalcidol 10–400 ng/kg/day | 3–6 g/day in 2–3 doses |
| Vitamin D dependent rickets type 3 | Vitamin D 20,000–50,000/day |
| Treatment Before the Development of Clinical or Radiological Signs and Symptoms | Treatment in Presence of Clinical Signs or Symptoms | |
|---|---|---|
| Start dose | Alfacalcidiol: 25–40 ng/kg/day | Alfacalcidiol: 40–80 ng/kg/day |
| Calcitriol: 20–30 ng/kg/day | Calcitriol: 20–40 ng/kg/day | |
| Inorganic phosphate salts: 40–60 mg/kg/day (divided into 4–6 doses/day) | Inorganic phosphate salts: 40–60 mg/kg/day (divided into 4–6 doses/day) | |
| Retention dose | Alfacalcidiol: 25–40 ng/kg/day (1–2 µg/day) Calcitriol: 20–40 ng/kg/day Inorganic phosphate salts: 30–60 mg/kg/day (divided into 4–6 doses/day) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biasucci, G.; Donini, V.; Cannalire, G. Rickets Types and Treatment with Vitamin D and Analogues. Nutrients 2024, 16, 416. https://doi.org/10.3390/nu16030416
Biasucci G, Donini V, Cannalire G. Rickets Types and Treatment with Vitamin D and Analogues. Nutrients. 2024; 16(3):416. https://doi.org/10.3390/nu16030416
Chicago/Turabian StyleBiasucci, Giacomo, Valentina Donini, and Giuseppe Cannalire. 2024. "Rickets Types and Treatment with Vitamin D and Analogues" Nutrients 16, no. 3: 416. https://doi.org/10.3390/nu16030416
APA StyleBiasucci, G., Donini, V., & Cannalire, G. (2024). Rickets Types and Treatment with Vitamin D and Analogues. Nutrients, 16(3), 416. https://doi.org/10.3390/nu16030416