
Sickle Cell Disease Update: New Treatments and Challenging Nutritional Interventions



Abstract
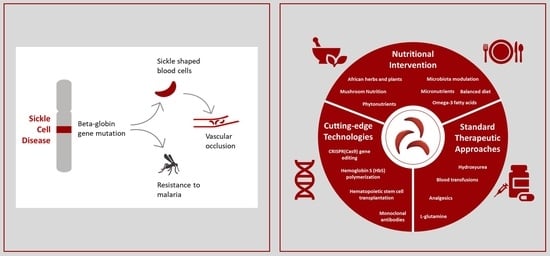
1. Introduction
1.1. The Incidence of Sickle Cell Disease
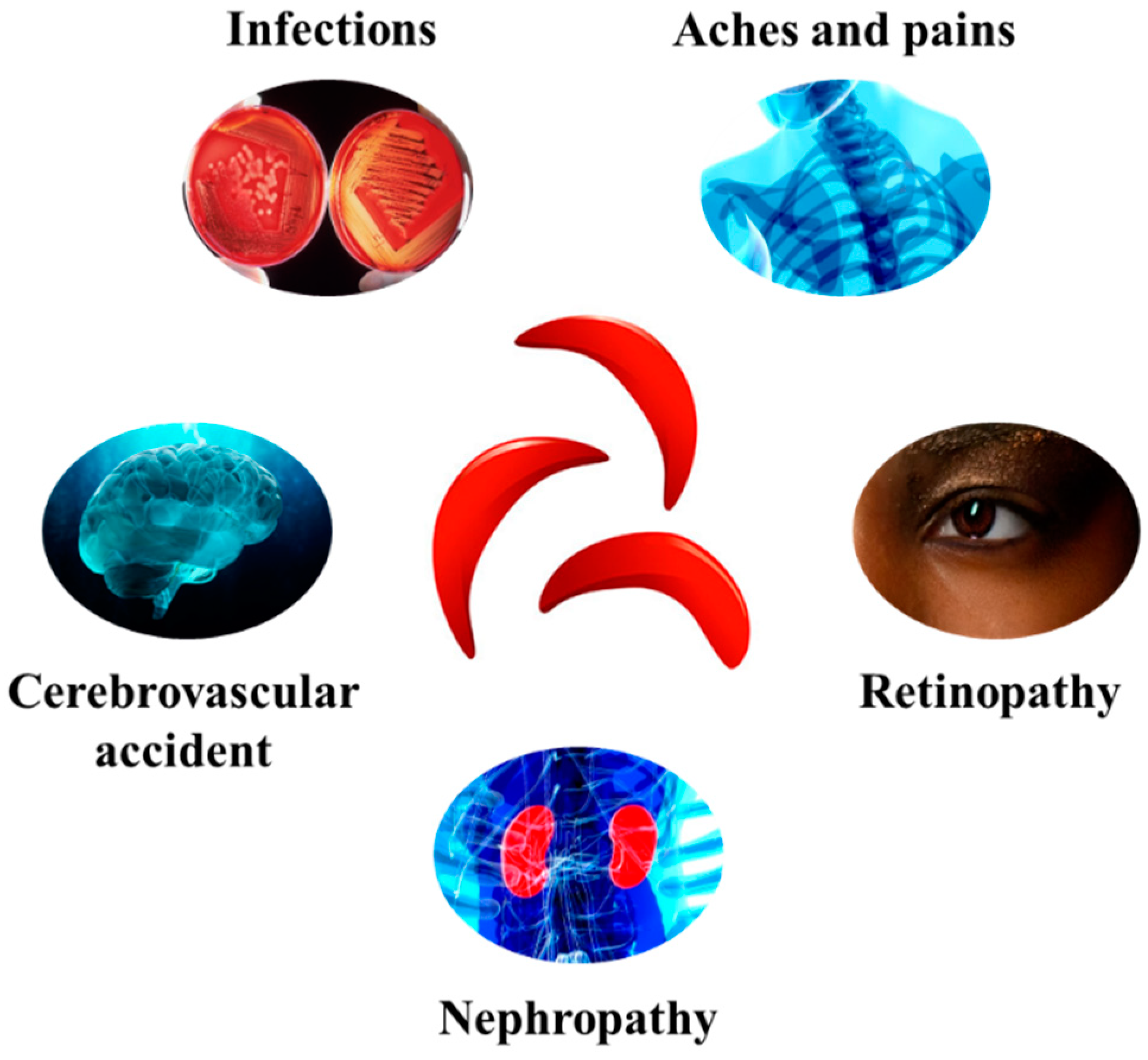
1.2. Sickle Cell Disease Physiopathology
1.3. Sickle Cell Disease Diagnosis
2. The Advent of New Technologies
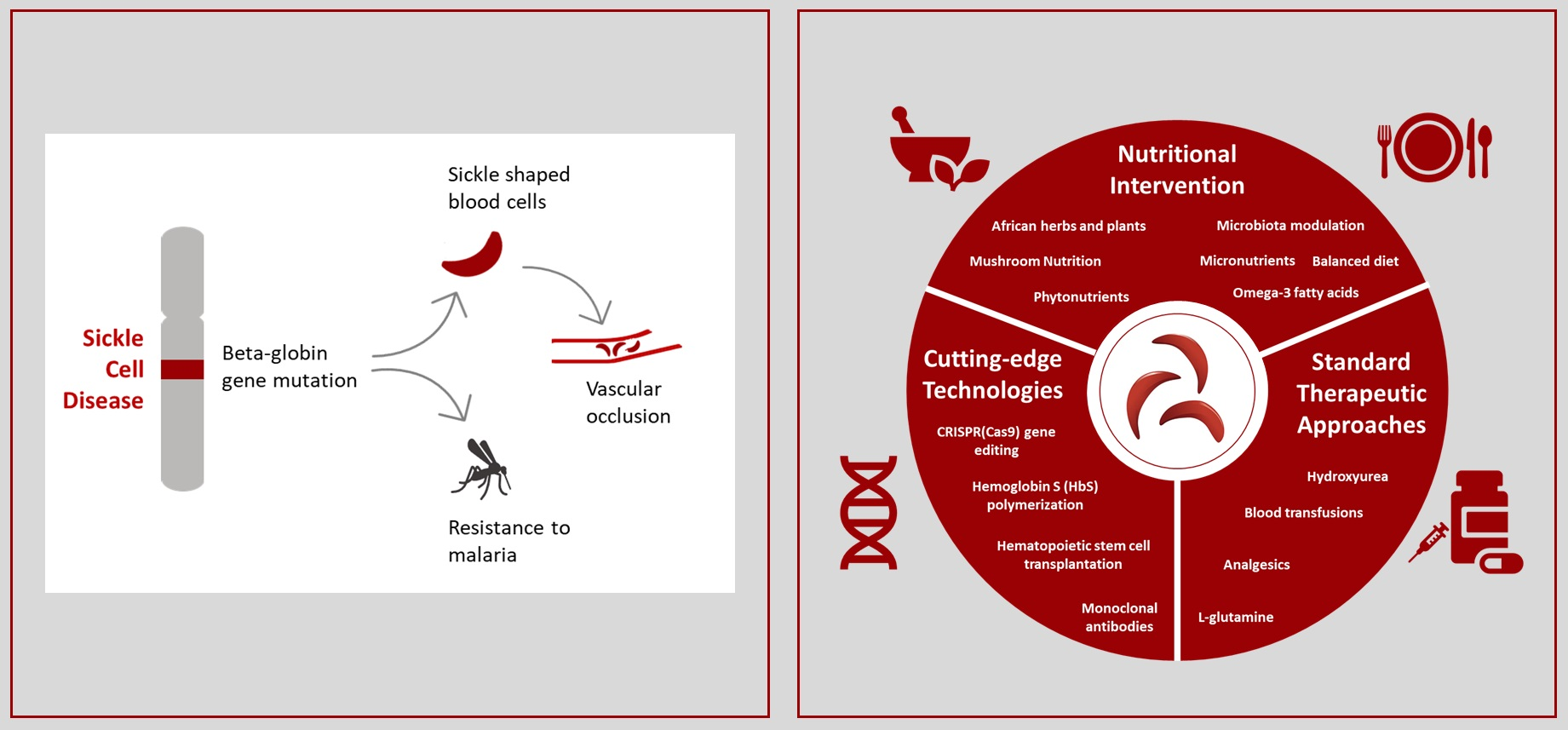
3. Current Treatments of Sickle Cell Disease
Recent Advanced Therapeutic Approaches for Sickle Cell Disease
4. Influence of Gut Microbiome in Sickle Cell Disease
5. Nutritional Perspectives in Sickle Cell Disease
5.1. African Plant Resources in Sickle Cell Disease
5.2. Mushroom Nutritional Prospects
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Elendu, C.; Amaechi, D.C.; Alakwe-Ojimba, C.E.; Elendu, T.C.; Elendu, R.C.; Ayabazu, C.P.; Aina, T.O.; Aborisade, O.; Adenikinju, J.S. Understanding Sickle Cell Disease: Causes, Symptoms, and Treatment Options. Medicine 2023, 102, E35237. [Google Scholar] [CrossRef] [PubMed]
- Tebbi, C.K. Sickle Cell Disease, a Review. Hemato 2022, 3, 341–366. [Google Scholar] [CrossRef]
- Quinn, C.T. Minireview: Clinical Severity in Sickle Cell Disease: The Challenges of Definition and Prognostication. Exp. Biol. Med. 2016, 241, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Ershler, W.B.; De Castro, L.M.; Pakbaz, Z.; Moynahan, A.; Weycker, D.; Delea, T.E.; Agodoa, I.; Cong, Z. Hemoglobin and End-Organ Damage in Individuals with Sickle Cell Disease. Curr. Ther. Res. 2023, 98, 100696. [Google Scholar] [CrossRef] [PubMed]
- Inusa, B.P.D.; Hsu, L.L.; Kohli, N.; Patel, A.; Ominu-Evbota, K.; Anie, K.A.; Atoyebi, W. Sickle Cell Disease—Genetics, Pathophysiology, Clinical Presentation and Treatment. Int. J. Neonatal Screen. 2019, 5, 20. [Google Scholar] [CrossRef]
- Kargutkar, N.; Sawant-Mulay, M.; Hariharan, P.; Chandrakala, S.; Nadkarni, A. Role of MicroRNA in Hydroxyurea Mediated HbF Induction in Sickle Cell Anaemia Patients. Sci. Rep. 2023, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harb. Perspect. Med. 2013, 3, a011858. [Google Scholar] [CrossRef] [PubMed]
- Aldakeel, S.A.; Ghanem, N.Z.; Al-Amodi, A.M.; Osman, A.K.; Al Asoom, L.I.; Ahmed, N.R.; Almandil, N.B.; Akhtar, M.S.; Azeez, S.A.; Borgio, J.F. Identification of Seven Novel Variants in the β-Globin Gene in Transfusion-Dependent and Normal Patients. Arch. Med. Sci. 2020, 16, 453–459. [Google Scholar] [CrossRef]
- Egesa, W.I.; Nakalema, G.; Waibi, W.M.; Turyasiima, M.; Amuje, E.; Kiconco, G.; Odoch, S.; Kumbakulu, P.K.; Abdirashid, S.; Asiimwe, D. Sickle Cell Disease in Children and Adolescents: A Review of the Historical, Clinical, and Public Health Perspective of Sub-Saharan Africa and Beyond. Int. J. Pediatr. 2022, 2022, 3885979. [Google Scholar] [CrossRef]
- Uçucu, S.; Karabıyık, T.; Azik, F. Difficulties in the Diagnosis of HbS/Beta Thalassemia: Really a Mild Disease? J. Med. Biochem. 2022, 41, 32–39. [Google Scholar] [CrossRef]
- Liu, Y.; Su, S.; Shayo, S.; Bao, W.; Pal, M.; Dou, K.; Shi, P.A.; Aygun, B.; Campbell-Lee, S.; Lobo, C.A.; et al. Hemolysis Dictates Monocyte Differentiation via Two Distinct Pathways in Sickle Cell Disease Vaso-Occlusion. J. Clin. Investig. 2023, 133, e172087. [Google Scholar] [CrossRef] [PubMed]
- Nader, E.; Romana, M.; Connes, P. The Red Blood Cell—Inflammation Vicious Circle in Sickle Cell Disease. Front. Immunol. 2020, 11, 517556. [Google Scholar] [CrossRef] [PubMed]
- Mangla, A.; Ehsan, M.; Agarwal, N.; Maruvada, S. Sickle Cell Anemia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Venugopal, A.; Chandran, M.; Eruppakotte, N.; Kizhakkillach, S.; Breezevilla, S.C.; Vellingiri, B. Monogenic Diseases in India. Mutat. Res. Mutat. Res. 2018, 776, 23–31. [Google Scholar] [CrossRef]
- Mehta, A.B.; Hoffbrand, A.V. Hemolytic Anaemias V Inherited Defects of Haemoglobin—Sickle Cell Disease. In Haematology at a Glance; Blackwell Science: Malden, MA, USA, 2000; ISBN 9780632047932. [Google Scholar]
- Stuart, M.J.; Nagel, R.L. Sickle-Cell Disease. Lancet 2004, 364, 1343–1360. [Google Scholar] [CrossRef]
- Habara, A.; Steinberg, M.H. Minireview: Genetic Basis of Heterogeneity and Severity in Sickle Cell Disease. Exp. Biol. Med. 2016, 241, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Van Ness, S.; Gordeuk, V.; Nouraie, S.M.; Nekhai, S.; Gladwin, M.; Steinberg, M.H.; Sebastiani, P. Biomarker Signatures of Sickle Cell Disease Severity. Blood Cells Mol. Dis. 2018, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Njoku, F.; Zhang, X.; Shah, B.N.; Machado, R.F.; Han, J.; Saraf, S.L.; Gordeuk, V.R. Biomarkers of Clinical Severity in Treated and Untreated Sickle Cell Disease: A Comparison by Genotypes of a Single Center Cohort and African Americans in the NHANES Study. Br. J. Haematol. 2021, 194, 767–778. [Google Scholar] [CrossRef]
- Takaoka, K.; Cyril, A.C.; Jinesh, S.; Radhakrishnan, R. Mechanisms of Pain in Sickle Cell Disease. Br. J. Pain 2020, 15, 213–220. [Google Scholar] [CrossRef]
- Sadler, K.E.; Atkinson, S.N.; Ehlers, V.L.; Waltz, T.B.; Hayward, M.; García, D.M.R.; Salzman, N.H.; Stucky, C.L.; Brandow, A.M. Gut Microbiota and Metabolites Drive Chronic Sickle Cell Disease Pain. bioRxiv 2023. [Google Scholar] [CrossRef]
- Matthie, N.; Ross, D.; Sinha, C.; Khemani, K.; Bakshi, N.; Krishnamurti, L. A Qualitative Study of Chronic Pain and Self-Management in Adults with Sickle Cell Disease. J. Natl. Med. Assoc. 2019, 111, 158–168. [Google Scholar] [CrossRef]
- Osunkwo, I.; O’Connor, H.F.; Saah, E. Optimizing the Management of Chronic Pain in Sickle Cell Disease. Hematology 2020, 2020, 562–569. [Google Scholar] [CrossRef]
- Thomson, A.M.; McHugh, T.A.; Oron, A.P.; Teply, C.; Lonberg, N.; Vilchis Tella, V.; Wilner, L.B.; Fuller, K.; Hagins, H.; Aboagye, R.G.; et al. Global, Regional, and National Prevalence and Mortality Burden of Sickle Cell Disease, 2000–2021: A Systematic Analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023, 10, e585–e599. [Google Scholar] [CrossRef]
- Grosse, S.D.; Odame, I.; Atrash, H.K.; Amendah, D.D.; Piel, F.B.; Williams, T.N. Sickle Cell Disease in Africa: A Neglected Cause of Early Childhood Mortality. Am. J. Prev. Med. 2011, 41, S398–S405. [Google Scholar] [CrossRef]
- Delgadinho, M.; Ginete, C.; Santos, B.; Mendes, J.; Miranda, A.; Vasconcelos, J.; Brito, M. Microbial Gut Evaluation in an Angolan Paediatric Population with Sickle Cell Disease. J. Cell. Mol. Med. 2022, 26, 5360–5368. [Google Scholar] [CrossRef]
- Ranque, B.; Kitenge, R.; Ndiaye, D.D.; Ba, M.D.; Adjoumani, L.; Traore, H.; Coulibaly, C.; Guindo, A.; Boidy, K.; Mbuyi, D.; et al. Estimating the Risk of Child Mortality Attributable to Sickle Cell Anaemia in Sub-Saharan Africa: A Retrospective, Multicentre, Case-Control Study. Lancet Haematol. 2022, 9, e208–e216. [Google Scholar] [CrossRef]
- Ali Hazzazi, A.; Ageeli, M.H.; Alfaqih, A.M.; Ali Jaafari, A.; Malhan, H.M.; Bakkar, M.M. Epidemiology and Characteristics of Sickle Cell Patients Admitted to Hospitals in Jazan Region, Saudi Arabia. J. Appl. Hematol. 2020, 11, 10–14. [Google Scholar] [CrossRef]
- Arji, E.E.; Eze, U.J.; Ezenwaka, G.O.; Kennedy, N. Evidence-Based Interventions for Reducing Sickle Cell Disease-Associated Morbidity and Mortality in Sub-Saharan Africa: A Scoping Review. SAGE Open Med. 2023, 11. [Google Scholar] [CrossRef]
- Piel, F.B.; Rees, D.C.; DeBaun, M.R.; Nnodu, O.; Ranque, B.; Thompson, A.A.; Ware, R.E.; Abboud, M.R.; Abraham, A.; Ambrose, E.E.; et al. Defining Global Strategies to Improve Outcomes in Sickle Cell Disease: A Lancet Haematology Commission. Lancet Haematol. 2023, 10, e633–e686. [Google Scholar] [CrossRef]
- Sedrak, A.; Kondamudi, N.P. Sickle Cell Disease. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global Burden of Sickle Cell Anaemia in Children under Five, 2010–2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef]
- Ansong, D.; Akoto, A.O.; Ocloo, D.; Ohene-Frempong, K. Sickle Cell Disease: Management Options and Challenges in Developing Countries. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013062. [Google Scholar] [CrossRef]
- Colombatti, R.; Hegemann, I.; Medici, M.; Birkegård, C. Systematic Literature Review Shows Gaps in Data on Global Prevalence and Birth Prevalence of Sickle Cell Disease and Sickle Cell Trait: Call for Action to Scale up and Harmonize Data Collection. J. Clin. Med. 2023, 12, 5538. [Google Scholar] [CrossRef]
- Adigwe, O.P.; Onoja, S.O.; Onavbavba, G. A Critical Review of Sickle Cell Disease Burden and Challenges in Sub-Saharan Africa. J. Blood Med. 2023, 14, 367–376. [Google Scholar] [CrossRef]
- Piel, F.B.; Patil, A.P.; Howes, R.E.; Nyangiri, O.A.; Gething, P.W.; Dewi, M.; Temperley, W.H.; Williams, T.N.; Weatherall, D.J.; Hay, S.I. Global Epidemiology of Sickle Haemoglobin in Neonates: A Contemporary Geostatistical Model-Based Map and Population Estimates. Lancet 2013, 381, 142–151. [Google Scholar] [CrossRef]
- Nieuwenhuis, F.; Wolf, B.; Bomba, A.; Graaf, P. De Haematological Study in Cabo Delgado Province, Mozambique; Sickle Cell Trait and G6PD Deficiency. Trop. Geogr. Med. 1986, 38, 183–187. [Google Scholar]
- Galatas, B.; Mabote, L.; Simone, W.; Matambisso, G.; Nhamussua, L.; Mañú-Pereira, M.D.M.; Menéndez, C.; Saute, F.; Macete, E.; Bassat, Q.; et al. Heterogeneity of G6PD Deficiency Prevalence in Mozambique: A School-Based Cross-Sectional Survey in Three Different Regions CIBS-25/013 CIBS IRB00002657 IRB. Malar. J. 2017, 16, 36. [Google Scholar] [CrossRef]
- Williams, T.N.; Mwangi, T.W.; Wambua, S.; Alexander, N.D.; Kortok, M.; Snow, R.W.; Marsh, K. Sickle Cell Trait and the Risk of Plasmodium Falciparum Malaria and Other Childhood Diseases. J. Infect. Dis. 2005, 192, 178–186. [Google Scholar] [CrossRef]
- Saelens, J.W.; Petersen, J.E.V.; Freedman, E.; Moseley, R.C.; Konaté, D.; Diakité, S.A.S.; Traoré, K.; Vance, N.; Fairhurst, R.M.; Diakité, M.; et al. Impact of Sickle Cell Trait Hemoglobin on the Intraerythrocytic Transcriptional Program of Plasmodium Falciparum. mSphere 2021, 6, e0075521. [Google Scholar] [CrossRef]
- Ngou, C.M.; Bayibéki, A.N.; Abate, L.; Makinde, O.S.; Feufack-Donfack, L.B.; Sarah-Matio, E.M.; Bouopda-Tuedom, A.G.; Taconet, P.; Moiroux, N.; Awono-Ambéné, P.H.; et al. Influence of the Sickle Cell Trait on Plasmodium Falciparum Infectivity from Naturally Infected Gametocyte Carriers. BMC Infect. Dis. 2023, 23, 317. [Google Scholar] [CrossRef]
- Sesethu, G.; Nombalentle, M.; Yamkela, M.; Anelisa, M.; Makumire, S.; Mkwetshana, N.; Govender, K.K.; Makhoba, X.H. In Silico Evaluation of Heat Shock Proteins Reveals an Interplay with Polyamines as a Survival Strategy for the Plasmodium falciparum. INNOSC Theranostics Pharmacol. Sci. 2023, 1228. [Google Scholar] [CrossRef]
- Aolymat, I.; Hatmal, M.M.; Olaimat, A.N. The Emerging Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis. Pathophysiology 2023, 30, 63–82. [Google Scholar] [CrossRef]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef]
- Menon, A.V.; Liu, J.; Tsai, H.P.; Zeng, L.; Yang, S.; Asnani, A.; Kim, J. Excess Heme Upregulates Heme Oxygenase 1 and Promotes Cardiac Ferroptosis in Mice with Sickle Cell Disease. Blood 2022, 139, 936–941. [Google Scholar] [CrossRef]
- Rutter, M. Gene–Environment Interdependence. Dev. Sci. 2007, 10, 12–18. [Google Scholar] [CrossRef]
- Bodaghi, A.; Fattahi, N.; Ramazani, A. Biomarkers: Promising and Valuable Tools towards Diagnosis, Prognosis and Treatment of COVID-19 and Other Diseases. Heliyon 2023, 9, e13323. [Google Scholar] [CrossRef]
- Kavanagh, P.L.; Fasipe, T.A.; Wun, T. Sickle Cell Disease: A Review. JAMA 2022, 328, 57–68. [Google Scholar] [CrossRef]
- Matter, B.; Seiler, C.L.; Murphy, K.; Ming, X.; Zhao, J.; Lindgren, B.; Jones, R.; Tretyakova, N. Mapping Three Guanine Oxidation Products along DNA Following Exposure to Three Types of Reactive Oxygen Species. Free Radic. Biol. Med. 2018, 121, 180–189. [Google Scholar] [CrossRef]
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Populations and Dynamics of Guanine Radicals in DNA Strands—Direct versus Indirect Generation. Molecules 2019, 24, 2347. [Google Scholar] [CrossRef]
- Sesti-Costa, R.; Costa, F.F.; Conran, N. Role of Macrophages in Sickle Cell Disease Erythrophagocytosis and Erythropoiesis. Int. J. Mol. Sci. 2023, 24, 6333. [Google Scholar] [CrossRef]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARγ/PGC1α Signaling as a Potential Therapeutic Target for Mitochondrial Biogenesis in Neurodegenerative Disorders. Pharmacol. Ther. 2021, 219, 107705. [Google Scholar] [CrossRef]
- Mihaylov, S.R.; Castelli, L.M.; Lin, Y.H.; Gül, A.; Soni, N.; Hastings, C.; Flynn, H.R.; Păun, O.; Dickman, M.J.; Snijders, A.P.; et al. The Master Energy Homeostasis Regulator PGC-1α Exhibits an MRNA Nuclear Export Function. Nat. Commun. 2023, 14, 5496. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Martinez, G.; Mayoral-Andrade, G.; Tomanek-Chalkley, A.; Zepeda-Orozco, D.; Allen, B.G.; Mapuskar, K.A.; Vasquez-Martinez, G.; Mayoral-Andrade, G.; Tomanek-Chalkley, A.; Zepeda-Orozco, D.; et al. Mitochondrial Oxidative Metabolism: An Emerging Therapeutic Target to Improve CKD Outcomes. Biomedicines 2023, 11, 1573. [Google Scholar] [CrossRef]
- Williams, T.N.; Thein, S.L. Sickle Cell Anemia and Its Phenotypes. Annu. Rev. Genom. Hum. Genet. 2018, 19, 113–147. [Google Scholar] [CrossRef]
- Arishi, W.A.; Al-hadrami, H.A.; Zourob, M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines 2021, 12, 519. [Google Scholar] [CrossRef]
- Ontario Health (Quality). Carrier Screening Programs for Cystic Fibrosis, Fragile X Syndrome, Hemoglobinopathies and Thalassemia, and Spinal Muscular Atrophy: A Health Technology Assessment. Ont. Health Technol. Assess. Ser. 2023, 23, 1–398. [Google Scholar]
- Yenilmez, E.D.; Tuli, A. New Perspectives in Prenatal Diagnosis of Sickle Cell Anemia. In Sickle Cell Disease Pain and Common Chronic Complications; Inusa, B.P.D., Ed.; IntechOpen: London, UK, 2016; ISBN 978-953-51-2767-3. [Google Scholar]
- Sato, M.; Miyoshi, K.; Nagao, Y.; Nishi, Y.; Ohtsuka, M.; Nakamura, S.; Sakurai, T.; Watanabe, S. The Combinational Use of CRISPR/Cas9-Based Gene Editing and Targeted Toxin Technology Enables Efficient Biallelic Knockout of the α-1,3-Galactosyltransferase Gene in Porcine Embryonic Fibroblasts. Xenotransplantation 2014, 21, 291–300. [Google Scholar] [CrossRef]
- Gostimskaya, I. CRISPR–Cas9: A History of Its Discovery and Ethical Considerations of Its Use in Genome Editing. Biochem. 2022, 87, 777–788. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C. In the Business of Base Editors: Evolution from Bench to Bedside. PLoS Biol. 2023, 21, e3002071. [Google Scholar] [CrossRef]
- Bhokisham, N.; Laudermilch, E.; Traeger, L.L.; Bonilla, T.D.; Ruiz-Estevez, M.; Becker, J.R. CRISPR-Cas System: The Current and Emerging Translational Landscape. Cells 2023, 12, 1103. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Li, R.; Wang, Q.; She, K.; Lu, F.; Yang, Y. CRISPR/Cas Systems Usher in a New Era of Disease Treatment and Diagnosis. Mol. Biomed. 2022, 3, 31. [Google Scholar] [CrossRef]
- Rees, H.A.; Minella, A.C.; Burnett, C.A.; Komor, A.C.; Gaudelli, N.M. CRISPR-Derived Genome Editing Therapies: Progress from Bench to Bedside. Mol. Ther. 2021, 29, 3125–3139. [Google Scholar] [CrossRef] [PubMed]
- Raguram, A.; Banskota, S.; Liu, D.R. Therapeutic in Vivo Delivery of Gene Editing Agents. Cell 2022, 185, 2806–2827. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene Editing and CRISPR in the Clinic: Current and Future Perspectives. Biosci. Rep. 2020, 40, BSR20200127. [Google Scholar] [CrossRef]
- Wei, T.; Cheng, Q.; Min, Y.L.; Olson, E.N.; Siegwart, D.J. Systemic Nanoparticle Delivery of CRISPR-Cas9 Ribonucleoproteins for Effective Tissue Specific Genome Editing. Nat. Commun. 2020, 11, 3232. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, J.; Liang, L.; Liu, R. Multiplex Gene Editing and Regulation Techniques Based on CRISPR/Cas System. Sheng Wu Gong Cheng Xue Bao 2023, 39, 2449–2465. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.L.; Pereira, E.R.; de Oliveira, C.S.; Ferreira, E.D.S.; Menon, E.T.N.; Diniz, S.N.; Pezuk, J.A. MicroRNAs: Understanding Their Role in Gene Expression and Cancer. Einstein 2021, 19, eRB5996. [Google Scholar] [CrossRef]
- Kim, T.; Croce, C.M. MicroRNA: Trends in Clinical Trials of Cancer Diagnosis and Therapy Strategies. Exp. Mol. Med. 2023, 55, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167. [Google Scholar] [CrossRef]
- Rosolen, D.; Nunes-Souza, E.; Marchi, R.; Tofolo, M.V.; Antunes, V.C.; Berti, F.C.B.; Fonseca, A.S.; Cavalli, L.R. MiRNAs Action and Impact on Mitochondria Function, Metabolic Reprogramming and Chemoresistance of Cancer Cells: A Systematic Review. Biomedicines 2023, 11, 693. [Google Scholar] [CrossRef]
- Suriya Muthukumaran, N.; Velusamy, P.; Akino Mercy, C.S.; Langford, D.; Natarajaseenivasan, K.; Shanmughapriya, S. MicroRNAs as Regulators of Cancer Cell Energy Metabolism. J. Pers. Med. 2022, 12, 1329. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Frimpong, E.; Ababio, G.K.; Dzudzor, B.; Ekem, I.; Gyan, B.; Sodzi-Tettey, N.A.; Antwi, D.A. Sickle Cell Disease: Reappraisal of the Role of Foetal Haemoglobin Levels in the Frequency of Vaso-Occlusive Crisis. Ghana Med. J. 2015, 49, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Cyrus, C.; Vatte, C.; Al-Nafie, A.; Chathoth, S.; Akhtar, M.S.; Darwish, M.; Almohazey, D.; AlDubayan, S.H.; Steinberg, M.H.; Al-Ali, A. MiRNA Expression Associated with HbF in Saudi Sickle Cell Anemia. Medicina 2022, 58, 1470. [Google Scholar] [CrossRef]
- Starlard-Davenport, A.; Gu, Q.; Pace, B.S. Targeting Genetic Modifiers of HBG Gene Expression in Sickle Cell Disease: The MiRNA Option. Mol. Diagn. Ther. 2022, 26, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Cyrus, C. The Role of MiRNAs as Therapeutic Tools in Sickle Cell Disease. Medicina 2021, 57, 1106. [Google Scholar] [CrossRef]
- Li-Thiao-Te, V.; Uettwiller, F.; Quartier, P.; Lacaille, F.; Bader-Meunier, B.; Brousse, V.; de Montalembert, M. Coexistent Sickle-Cell Anemia and Autoimmune Disease in Eight Children: Pitfalls and Challenges. Pediatr. Rheumatol. 2018, 16, 5. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Q.; Zhang, R.; Dai, X.; Chen, W.; Xing, D. Circulating MicroRNAs: Biomarkers of Disease. Clin. Chim. Acta 2021, 516, 46–54. [Google Scholar] [CrossRef]
- Bou-Fakhredin, R.; De Franceschi, L.; Motta, I.; Cappellini, M.D.; Taher, A.T. Pharmacological Induction of Fetal Hemoglobin in β-Thalassemia and Sickle Cell Disease: An Updated Perspective. Pharmaceuticals 2022, 15, 753. [Google Scholar] [CrossRef]
- Hassan Murad, M.; Liem, R.I.; Lang, E.S.; Akl, E.A.; Meerpohl, J.J.; DeBaun, M.R.; Tisdale, J.F.; Brandow, A.M.; Lanzkron, S.M.; Chou, S.T.; et al. 2019 Sickle Cell Disease Guidelines by the American Society of Hematology: Methodology, Challenges, and Innovations. Blood Adv. 2019, 3, 3945–3950. [Google Scholar] [CrossRef]
- Brandow, A.M.; Carroll, C.P.; Creary, S.; Edwards-Elliott, R.; Glassberg, J.; Hurley, R.W.; Kutlar, A.; Seisa, M.; Stinson, J.; Strouse, J.J.; et al. American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Management of Acute and Chronic Pain. Blood Adv. 2020, 4, 2656–2701. [Google Scholar] [CrossRef]
- FDA. FDA Approved L-Glutamine Powder for the Treatment of Sickle Cell Disease. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approved-l-glutamine-powder-treatment-sickle-cell-disease (accessed on 31 December 2023).
- Ali, M.A.; Ahmad, A.; Chaudry, H.; Aiman, W.; Aamir, S.; Anwar, M.Y.; Khan, A. Efficacy and Safety of Recently Approved Drugs for Sickle Cell Disease: A Review of Clinical Trials. Exp. Hematol. 2020, 92, 11–18.e1. [Google Scholar] [CrossRef]
- Ware, R.E. Optimizing Hydroxyurea Therapy for Sickle Cell Anemia. Hematology 2015, 2015, 436–443. [Google Scholar] [CrossRef]
- EMA. Oxbryta: EPAR—Medicine Overview; EMA/131392/2022; EMA: Amsterdam, The Netherlands, 2022.
- Eaton, W.A.; Bunn, H.F. Treating Sickle Cell Disease by Targeting HbS Polymerization. Blood 2017, 129, 2719–2726. [Google Scholar] [CrossRef] [PubMed]
- Darshana, T.; Rees, D.; Premawardhena, A. Hydroxyurea and Blood Transfusion Therapy for Sickle Cell Disease in South Asia: Inconsistent Treatment of a Neglected Disease. Orphanet J. Rare Dis. 2021, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Matos, C.; Comont, T.; Castex, M.P.; Lafaurie, M.; Walter, O.; Moulis, G.; Dion, J.; Cougoul, P. Risk of Vaso-Occlusive Episodes in Patients with Sickle Cell Disease Exposed to Systemic Corticosteroids: A Comprehensive Review. Expert Rev. Hematol. 2022, 15, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J.; Wun, T.; McCavit, T.L.; De Castro, L.M.; Krishnamurti, L.; Lanzkron, S.; Hsu, L.L.; Smith, W.R.; Rhee, S.; Magnani, J.L.; et al. Randomized Phase 2 Study of GMI-1070 in SCD: Reduction in Time to Resolution of Vaso-Occlusive Events and Decreased Opioid Use. Blood 2015, 125, 2656–2664. [Google Scholar] [CrossRef]
- Doss, J.F.; Jonassaint, J.C.; Garrett, M.E.; Ashley-Koch, A.E.; Telen, M.J.; Chi, J.T. Phase 1 Study of a Sulforaphane-Containing Broccoli Sprout Homogenate for Sickle Cell Disease. PLoS ONE 2016, 11, e0152895. [Google Scholar] [CrossRef]
- Khaddour, K.; Hana, C.K.; Mewawalla, P. Hematopoietic Stem Cell Transplantation. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chou, S.T.; Alsawas, M.; Fasano, R.M.; Field, J.J.; Hendrickson, J.E.; Howard, J.; Kameka, M.; Kwiatkowski, J.L.; Pirenne, F.; Shi, P.A.; et al. American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Transfusion Support. Blood Adv. 2020, 4, 327–355. [Google Scholar] [CrossRef]
- Han, H.; Hensch, L.; Tubman, V.N. Indications for Transfusion in the Management of Sickle Cell Disease. Hematology 2021, 2021, 696–703. [Google Scholar] [CrossRef]
- Rankine-Mullings, A.E.; Nevitt, S.J. Hydroxyurea (Hydroxycarbamide) for Sickle Cell Disease. Cochrane Database Syst. Rev. 2022, 2022, CD002202. [Google Scholar] [CrossRef]
- Inam, Z.; Tisdale, J.F.; Leonard, A. Outcomes and Long-Term Effects of Hematopoietic Stem Cell Transplant in Sickle Cell Disease. Expert Rev. Hematol. 2023, 16, 879–903. [Google Scholar] [CrossRef]
- Bhalla, N.; Bhargav, A.; Yadav, S.K.; Singh, A.K. Allogeneic Hematopoietic Stem Cell Transplantation to Cure Sickle Cell Disease: A Review. Front. Med. 2023, 10, 1036939. [Google Scholar] [CrossRef]
- Sadaf, A.; Quinn, C.T. L-Glutamine for Sickle Cell Disease: Knight or Pawn? Exp. Biol. Med. 2020, 245, 146–154. [Google Scholar] [CrossRef]
- Yenamandra, A.; Marjoncu, D. Voxelotor: A Hemoglobin S Polymerization Inhibitor for the Treatment of Sickle Cell Disease. J. Adv. Pract. Oncol. 2020, 11, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Karki, N.R.; Saunders, K.; Kutlar, A. A Critical Evaluation of Crizanlizumab for the Treatment of Sickle Cell Disease. Expert Rev. Hematol. 2022, 15, 5–13. [Google Scholar] [CrossRef]
- Gov. UK. MHRA Authorises World-First Gene Therapy That Aims to Cure Sickle-Cell Disease and Transfusion-Dependent β-Thalassemia. Available online: https://www.gov.uk/government/news/mhra-authorises-world-first-gene-therapy-that-aims-to-cure-sickle-cell-disease-and-transfusion-dependent-thalassemia (accessed on 31 December 2023).
- De Souza, D.C.; Hebert, N.; Esrick, E.B.; Ciuculescu, M.F.; Archer, N.M.; Armant, M.; Audureau, É.; Brendel, C.; Di Caprio, G.; Galactéros, F.; et al. Genetic Reversal of the Globin Switch Concurrently Modulates Both Fetal and Sickle Hemoglobin and Reduces Red Cell Sickling. Nat. Commun. 2023, 14, 5850. [Google Scholar] [CrossRef] [PubMed]
- The National Heart, Lung, and Blood Institute (NHLBI). The Management of Sickle Cell Disease; NIH Publication: Bethesda, MD, USA, 2002.
- Wang, W.C. Sickle Cell Anemia and Other Sickling Syndromes. In Wintrobe’s Clinical Hematology; Greer, J.P., Foerster, J., Rodgers, G.M., Paraskevas, F., Glader, B., Arber, D.A., Means, R.T., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2009; ISBN 978-0781765077. [Google Scholar]
- Ratko, T.A.; Belinson, S.E.; Brown, H.M.; Noorani, H.Z.; Chopra, R.D.; Marbella, A.; Samson, D.J.; Bonnell, C.J.; Ziegler, K.M.; Aronson, N. Hematopoietic Stem-Cell Transplantation in the Pediatric Population; AHRO: Rockville, MD, USA, 2012. [Google Scholar]
- Gee, B.E. Biologic Complexity in Sickle Cell Disease: Implications for Developing Targeted Therapeutics. Sci. World J. 2013, 2013, 694146. [Google Scholar] [CrossRef]
- ABScience. Available online: https://www.ab-science.com/news-and-media/press-releases/ (accessed on 31 December 2023).
- Conran, N.; Belcher, J.D. Inflammation in Sickle Cell Disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Mei, J.X.; Yu, G.; Lei, L.; Zhang, W.H.; Liu, K.; Chen, X.L.; Kołat, D.; Yang, K.; Hu, J.K. Role of the Gut Microbiota in Anticancer Therapy: From Molecular Mechanisms to Clinical Applications. Signal Transduct. Target. Ther. 2023, 8, 201. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Cabrera-Mulero, A.; Tinahones, A.; Bandera, B.; Moreno-Indias, I.; Macías-González, M.; Tinahones, F.J. Keto Microbiota: A Powerful Contributor to Host Disease Recovery. Rev. Endocr. Metab. Disord. 2019, 20, 415–425. [Google Scholar] [CrossRef]
- Roy, S.; Nag, S.; Saini, A.; Choudhury, L. Association of Human Gut Microbiota with Rare Diseases: A Close Peep Through. Intractable Rare Dis. Res. 2022, 11, 52–62. [Google Scholar] [CrossRef]
- Brim, H.; Taylor, J.; Abbas, M.; Vilmenay, K.; Daremipouran, M.; Varma, S.; Lee, E.; Pace, B.; Song-Naba, W.L.; Gupta, K.; et al. The Gut Microbiome in Sickle Cell Disease: Characterization and Potential Implications. PLoS ONE 2021, 16, e0255956. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Zhang, H.; Sparks, J.B.; Karyala, S.V.; Settlage, R.; Luo, X.M. Host Adaptive Immunity Alters Gut Microbiota. ISME J. 2014, 9, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Dike, C.R.; Hanson, C.; Davies, H.D.; Obaro, S.; Yu, F.; Harper, J.; Grace, H.; Lebensburger, J.; Raulji, C.; Ma, J.; et al. The Relationship between Nutrition, Gut Dysbiosis, and Pediatric Sickle Cell Pain Outcomes: A Pilot Study. Pediatr. Blood Cancer 2023, 70, e30397. [Google Scholar] [CrossRef]
- Mallott, E.K.; Sitarik, A.R.; Leve, L.D.; Cioffi, C.; Camargo, C.A.; Hasegawa, K.; Bordenstein, S.R. Human Microbiome Variation Associated with Race and Ethnicity Emerges as Early as 3 Months of Age. PLoS Biol. 2023, 21, e3002230. [Google Scholar] [CrossRef]
- Qin, Y.; Havulinna, A.S.; Liu, Y.; Jousilahti, P.; Ritchie, S.C.; Tokolyi, A.; Sanders, J.G.; Valsta, L.; Brożyńska, M.; Zhu, Q.; et al. Combined Effects of Host Genetics and Diet on Human Gut Microbiota and Incident Disease in a Single Population Cohort. Nat. Genet. 2022, 54, 134–142. [Google Scholar] [CrossRef]
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to Host Microbiome Symbiosis in Health and Disease. Mucosal Immunol. 2021, 14, 547–554. [Google Scholar] [CrossRef]
- Mutalub, Y.B.; Abdulwahab, M.; Mohammed, A.; Yahkub, A.M.; AL-Mhanna, S.B.; Yusof, W.; Tang, S.P.; Rasool, A.H.G.; Mokhtar, S.S. Gut Microbiota Modulation as a Novel Therapeutic Strategy in Cardiometabolic Diseases. Foods 2022, 11, 2575. [Google Scholar] [CrossRef] [PubMed]
- Huda, M.N.; Salvador, A.C.; Barrington, W.T.; Gacasan, C.A.; D’Souza, E.M.; Deus Ramirez, L.; Threadgill, D.W.; Bennett, B.J. Gut Microbiota and Host Genetics Modulate the Effect of Diverse Diet Patterns on Metabolic Health. Front. Nutr. 2022, 9, 896348. [Google Scholar] [CrossRef] [PubMed]
- Lajqi, T.; Pöschl, J.; Frommhold, D.; Hudalla, H. The Role of Microbiota in Neutrophil Regulation and Adaptation in Newborns. Front. Immunol. 2020, 11, 568685. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Turpin, W.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Kevans, D.; Smith, M.I.; Guttman, D.S.; Griffiths, A.; Panaccione, R.; Otley, A.; et al. Association of Host Genome with Intestinal Microbial Composition in a Large Healthy Cohort. Nat. Genet. 2016, 48, 1413–1417. [Google Scholar] [CrossRef]
- Xu, F.; Fu, Y.; Sun, T.Y.; Jiang, Z.; Miao, Z.; Shuai, M.; Gou, W.; Ling, C.W.; Yang, J.; Wang, J.; et al. The Interplay between Host Genetics and the Gut Microbiome Reveals Common and Distinct Microbiome Features for Complex Human Diseases. Microbiome 2020, 8, 145. [Google Scholar] [CrossRef]
- Quan, Y.; Zhang, K.X.; Zhang, H.Y. The Gut Microbiota Links Disease to Human Genome Evolution. Trends Genet. 2023, 39, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Blouin, M.J.; Beauchemin, H.; Wright, A.; De Paepe, M.; Soretie, M.; Bleau, A.M.; Nakamoto, B.; Ou, C.N.; Stamatoyannopoulos, G.; Trudel, M. Genetic Correction of Sickle Cell Disease: Insights Using Transgenic Mouse Models. Nat. Med. 2000, 6, 177–182. [Google Scholar] [CrossRef]
- Beuzard, Y. Mouse Models of Sickle Cell Disease. Transfus. Clin. Biol. 2008, 15, 7–11. [Google Scholar] [CrossRef]
- Pawliuk, R.; Westerman, K.A.; Fabry, M.E.; Payen, E.; Tighe, R.; Bouhassira, E.E.; Acharya, S.A.; Ellis, J.; London, I.M.; Eaves, C.J.; et al. Correction of Sickle Cell Disease in Transgenic Mouse Models by Gene Therapy. Science 2001, 294, 2368–2371. [Google Scholar] [CrossRef]
- Hugenholtz, F.; de Vos, W.M. Mouse Models for Human Intestinal Microbiota Research: A Critical Evaluation. Cell. Mol. Life Sci. 2017, 75, 149–160. [Google Scholar] [CrossRef]
- Cahana, I.; Iraqi, F.A. Impact of Host Genetics on Gut Microbiome: Take-Home Lessons from Human and Mouse Studies. Anim. Model. Exp. Med. 2020, 3, 229–236. [Google Scholar] [CrossRef]
- Samakoglu, S.; Lisowski, L.; Budak-Alpdogan, T.; Usachenko, Y.; Acuto, S.; Di Marzo, R.; Maggio, A.; Zhu, P.; Tisdale, J.F.; Riviere, I.; et al. A Genetic Strategy to Treat Sickle Cell Anemia by Coregulating Globin Transgene Expression and RNA Interference. Nat. Biotechnol. 2005, 24, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.V.; Sellak, H.; Sawan, M.A.; Joseph, G.; Darby, T.M.; VanInsberghe, D.; Naudin, C.R.; Archer, D.R.; Jones, R.M.; Taylor, W.R. Intestinal Barrier Dysfunction in Murine Sickle Cell Disease Is Associated with Small Intestine Neutrophilic Inflammation, Oxidative Stress, and Dysbiosis. FASEB BioAdvances 2023, 5, 199–210. [Google Scholar] [CrossRef]
- Forcados, G.E.; Muhammad, A.; Oladipo, O.O.; Makama, S.; Meseko, C.A. Metabolic Implications of Oxidative Stress and Inflammatory Process in SARS-CoV-2 Pathogenesis: Therapeutic Potential of Natural Antioxidants. Front. Cell. Infect. Microbiol. 2021, 11, 654813. [Google Scholar] [CrossRef]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Ren, H.; Ghebremeskel, K.; Okpala, I.; Lee, A.; Ibegbulam, O.; Crawford, M. Patients with Sickle Cell Disease Have Reduced Blood Antioxidant Protection. Int. J. Vitam. Nutr. Res. 2013, 78, 139–147. [Google Scholar] [CrossRef]
- Setty, B.N.Y.; Betal, S.G.; Miller, R.E.; Brown, D.S.; Meier, M.; Cahill, M.; Lerner, N.B.; Apollonsky, N.; Stuart, M.J. Relationship of Omega-3 Fatty Acids DHA and EPA with the Inflammatory Biomarker Hs-CRP in Children with Sickle Cell Anemia. Prostaglandins Leukot. Essent. Fat. Acids 2019, 146, 11–18. [Google Scholar] [CrossRef]
- Daak, A.A.; Lopez-Toledano, M.A.; Heeney, M.M. Biochemical and Therapeutic Effects of Omega-3 Fatty Acids in Sickle Cell Disease. Complement. Ther. Med. 2020, 52, 102482. [Google Scholar] [CrossRef]
- Pittman, D.D.; Hines, P.C.; Beidler, D.; Rybin, D.; Frelinger, A.L.; Michelson, A.D.; Liu, K.; Gao, X.; White, J.; Zaidi, A.U.; et al. Evaluation of Longitudinal Pain Study in Sickle Cell Disease (ELIPSIS) by Patient-Reported Outcomes, Actigraphy, and Biomarkers. Blood 2021, 137, 2010–2020. [Google Scholar] [CrossRef]
- Sadler, K.E.; Mogil, J.S.; Stucky, C.L. Innovations and Advances in Modelling and Measuring Pain in Animals. Nat. Rev. Neurosci. 2021, 23, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Sadler, K.; Ehlers, V.; Brandow, A.; Stucky, C. Sickle Cell Disease Associated Changes in the Gut Microbiome Contribute to Persistent Pain. J. Pain 2022, 23, 7. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Dutta, D.; Aujla, A.; Knoll, B.M.; Lim, S.H. Intestinal Pathophysiological and Microbial Changes in Sickle Cell Disease: Potential Targets for Therapeutic Intervention. Br. J. Haematol. 2020, 188, 488–493. [Google Scholar] [CrossRef]
- Umeakunne, K.; Hibbert, J.M. Nutrition in Sickle Cell Disease: Recent Insights. Nutr. Diet. Suppl. 2019, 11, 9–17. [Google Scholar] [CrossRef]
- Reber, E.; Gomes, F.; Vasiloglou, M.F.; Schuetz, P.; Stanga, Z. Nutritional Risk Screening and Assessment. J. Clin. Med. 2019, 8, 1065. [Google Scholar] [CrossRef] [PubMed]
- Ohemeng, A.; Nartey, E.B.; Quaidoo, E.; Ansong, R.S.; Asiedu, M.S. Knowledge and Nutrition-Related Practices among Caregivers of Adolescents with Sickle Cell Disease in the Greater Accra Region of Ghana. BMC Public Health 2023, 23, 434. [Google Scholar] [CrossRef]
- Boadu, I.; Ohemeng, A.; Renner, L.A. Dietary Intakes and Nutritional Status of Children with Sickle Cell Disease at the Princess Marie Louise Hospital, Accra—A Survey. BMC Nutr. 2018, 4, 33. [Google Scholar] [CrossRef]
- Fernandes, T.H.; Bell, V. The Imprecision of Micronutrient Requirement Values: The Example of Vitamin D. J. Food Sci. 2023, 89, 51–63. [Google Scholar] [CrossRef]
- Zemel, B.S.; Kawchak, D.A.; Fung, E.B.; Ohene-Frempong, K.; Stallings, V.A. Effect of Zinc Supplementation on Growth and Body Composition in Children with Sickle Cell Disease. Am. J. Clin. Nutr. 2002, 75, 300–307. [Google Scholar] [CrossRef]
- Hibbert, J.M.; Hsu, L.L.; Bhathena, S.J.; Irune, I.; Sarfo, B.; Creary, M.S.; Gee, B.E.; Mohamed, A.I.; Buchanan, I.D.; Al-Mahmoud, A.; et al. Proinflammatory Cytokines and the Hypermetabolism of Children with Sickle Cell Disease. Exp. Biol. Med. 2005, 230, 68–74. [Google Scholar] [CrossRef]
- McCormick, M.; Osei-Anto, H.A.; Martinez, R.M. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; National Academies Press: Washington, DC, USA, 2020. [Google Scholar]
- Dutta, D.; Methe, B.; Amar, S.; Morris, A.; Lim, S.H. Intestinal Injury and Gut Permeability in Sickle Cell Disease. J. Transl. Med. 2019, 17, 183. [Google Scholar] [CrossRef] [PubMed]
- Haroun, E.; Kumar, P.A.; Saba, L.; Kassab, J.; Ghimire, K.; Dutta, D.; Lim, S.H. Intestinal Barrier Functions in Hematologic and Oncologic Diseases. J. Transl. Med. 2023, 21, 233. [Google Scholar] [CrossRef] [PubMed]
- Hyacinth, H.I.; Gee, B.E.; Hibbert, J.M. The Role of Nutrition in Sickle Cell Disease. Nutr. Metab. Insights 2010, 3. [Google Scholar] [CrossRef]
- Da Guarda, C.C.; Yahouédéhou, S.C.M.A.; Santiago, R.P.; Neres, J.S.D.S.; Fernandes, C.F.D.L.; Aleluia, M.M.; Figueiredo, C.V.B.; Fiuza, L.M.; Carvalho, S.P.; Oliveira, R.M.D.; et al. Sickle Cell Disease: A Distinction of Two Most Frequent Genotypes (HbSS and HbSC). PLoS ONE 2020, 15, e0228399. [Google Scholar] [CrossRef]
- Soe, H.H.K.; Abas, A.B.L.; Than, N.N.; Ni, H.; Singh, J.; Said, A.R.B.M.; Osunkwo, I. Vitamin D Supplementation for Sickle Cell Disease. Cochrane Database Syst. Rev. 2020, 5, CD010858. [Google Scholar] [CrossRef] [PubMed]
- Cairo, C.; Webb, T.J. Effective Barriers: The Role of NKT Cells and Innate Lymphoid Cells in the Gut. J. Immunol. 2022, 208, 235–246. [Google Scholar] [CrossRef]
- Yin, R.; Kuo, H.C.; Hudlikar, R.; Sargsyan, D.; Li, S.; Wang, L.; Wu, R.; Kong, A.N. Gut Microbiota, Dietary Phytochemicals, and Benefits to Human Health. Curr. Pharmacol. Rep. 2019, 5, 332–344. [Google Scholar] [CrossRef]
- Townsend, J.R.; Kirby, T.O.; Marshall, T.M.; Church, D.D.; Jajtner, A.R.; Esposito, R. Foundational Nutrition: Implications for Human Health. Nutrients 2023, 15, 2837. [Google Scholar] [CrossRef]
- Teixeira, T.V.; Da Silva, A.C.F.; Rodrigues, C.d.S.C.; Brito, F.d.S.B.; Canella, D.S.; Citelli, M. Food Consumption of People with Sickle Cell Anemia in a Middle-Income Country. Nutrients 2023, 15, 1478. [Google Scholar] [CrossRef]
- Al-Saqladi, A.W.M.; Cipolotti, R.; Fijnvandraat, K.; Brabin, B.J. Growth and Nutritional Status of Children with Homozygous Sickle Cell Disease. Ann. Trop. Paediatr. 2008, 28, 165–189. [Google Scholar] [CrossRef] [PubMed]
- Bello-Manga, H.; DeBaun, M.R.; Kassim, A.A. Epidemiology and Treatment of Relative Anemia in Children with Sickle Cell Disease in Sub-Saharan Africa. Expert Rev. Hematol. 2016, 9, 1031–1042. [Google Scholar] [CrossRef]
- Nartey, E.B.; Spector, J.; Adu-Afarwuah, S.; Jones, C.L.; Jackson, A.; Ohemeng, A.; Shah, R.; Koryo-Dabrah, A.; Kuma, A.B.A.; Hyacinth, H.I.; et al. Nutritional Perspectives on Sickle Cell Disease in Africa: A Systematic Review. BMC Nutr. 2021, 7, 9. [Google Scholar] [CrossRef]
- Sahu, T.; Pande, B.; Verma, H.K.; Bhaskar, L.V.K.S.; Sinha, M.; Sinha, R.; Rao, P.V. Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective. Thalass. Rep. 2023, 13, 206–229. [Google Scholar] [CrossRef]
- Kamal, S.; Naghib, M.M.; Zahrani, J.A.; Hassan, H.; Moawad, K.; Arrahman, O. The Influence of Nutrition on Disease Severity and Health-Related Quality of Life in Adults with Sickle Cell Disease: A Prospective Longitudinal Study. Mediterr. J. Hematol. Infect. Dis. 2021, 13, e2021007. [Google Scholar] [CrossRef] [PubMed]
- UNICEF. Fed to Fail? The Crisis of Children’s Diets in Early Life. 2021 Child Nutrition Report; UN Children’s Fund: New York, NY, USA, 2021. [Google Scholar]
- Cox, S.E.; Makani, J.; Fulford, A.J.; Komba, A.N.; Soka, D.; Williams, T.N.; Newton, C.R.; Marsh, K.; Prentice, A.M. Nutritional Status, Hospitalization and Mortality among Patients with Sickle Cell Anemia in Tanzania. Haematologica 2011, 96, 948–953. [Google Scholar] [CrossRef]
- Danton, O.; Somboro, A.; Fofana, B.; Diallo, D.; Sidibé, L.; Rubat-Coudert, C.; Marchand, F.; Eschalier, A.; Ducki, S.; Chalard, P. Ethnopharmacological Survey of Plants Used in the Traditional Treatment of Pain Conditions in Mali. J. Herb. Med. 2019, 17–18, 100271. [Google Scholar] [CrossRef]
- Delesderrier, E.; Curioni, C.; Omena, J.; Macedo, C.R.; Cople-Rodrigues, C.; Citelli, M. Antioxidant Nutrients and Hemolysis in Sickle Cell Disease. Clin. Chim. Acta 2020, 510, 381–390. [Google Scholar] [CrossRef]
- Darbari, D.S.; Sheehan, V.A.; Ballas, S.K. The Vaso-Occlusive Pain Crisis in Sickle Cell Disease: Definition, Pathophysiology, and Management. Eur. J. Haematol. 2020, 105, 237–246. [Google Scholar] [CrossRef]
- Reed, J.D.; Redding-Lallinger, R.; Orringer, E.P. Nutrition and Sickle Cell Disease. Am. J. Hematol. 1987, 24, 441–455. [Google Scholar] [CrossRef]
- Dixit, R.; Nettem, S.; Madan, S.S.; Soe, H.H.K.; Abas, A.B.L.; Vance, L.D.; Stover, P.J. Folate Supplementation in People with Sickle Cell Disease. Cochrane Database Syst. Rev. 2018, 2018, CD011130. [Google Scholar] [CrossRef]
- Arruda, M.M.; Mecabo, G.; Rodrigues, C.A.; Matsuda, S.S.; Rabelo, I.B.; Figueiredo, M.S. Antioxidant Vitamins C and E Supplementation Increases Markers of Haemolysis in Sickle Cell Anaemia Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Br. J. Haematol. 2013, 160, 688–700. [Google Scholar] [CrossRef]
- Onalo, R.; Cooper, P.; Cilliers, A.; Vorster, B.C.; Uche, N.A.; Oluseyi, O.O.; Onalo, V.D.; Zubairu, Y.; Ayodele-Kehinde, A.U.; Damilare, O.M.; et al. Randomized Control Trial of Oral Arginine Therapy for Children with Sickle Cell Anemia Hospitalized for Pain in Nigeria. Am. J. Hematol. 2021, 96, 89–97. [Google Scholar] [CrossRef]
- Sadeghi, A.; Taherifard, E.; Dehdari Ebrahimi, N.; Rafiei, E.; Hadianfard, F.; Taherifard, E. Effects of L-Arginine Supplementation in Patients with Sickle Cell Disease: A Systematic Review and Meta-Analysis of Clinical Trials. Health Sci. Reports 2023, 6, e1167. [Google Scholar] [CrossRef]
- Arribas-López, E.; Zand, N.; Ojo, O.; Snowden, M.J.; Kochhar, T. The Effect of Amino Acids on Wound Healing: A Systematic Review and Meta-Analysis on Arginine and Glutamine. Nutrients 2021, 13, 2498. [Google Scholar] [CrossRef] [PubMed]
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative Stress, Antioxidant Capacity, Biomolecule Damage, and Inflammation Symptoms of Sickle Cell Disease in Children. Hematology 2019, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the Role of SOD2 in Sickle Cell Disease. Blood Adv. 2019, 3, 2679–2687. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Enomoto, T.M.; Isichei, C.; Vanderjagt, D.J.; Fry, D.E.; Glew, R.H. Decreased Polyunsaturated Fatty Acids in Sickle Cell Anaemia. J. Trop. Pediatr. 1998, 44, 28–34. [Google Scholar] [CrossRef]
- Lanza, V.; Greco, V.; Bocchieri, E.; Sciuto, S.; Inturri, R.; Messina, L.; Vaccaro, S.; Bellia, F.; Rizzarelli, E. Synergistic Effect of L-Carnosine and Hyaluronic Acid in Their Covalent Conjugates on the Antioxidant Abilities and the Mutual Defense against Enzymatic Degradation. Antioxidants 2022, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative Profile of Patients with Sickle Cell Disease. Med. Sci. 2019, 7, 17. [Google Scholar] [CrossRef]
- Djordjević, V.V.; Kostić, J.; Krivokapić, Ž.; Krtinić, D.; Ranković, M.; Petković, M.; Ćosić, V. Decreased Activity of Erythrocyte Catalase and Glutathione Peroxidase in Patients with Schizophrenia. Medicina 2022, 58, 1491. [Google Scholar] [CrossRef]
- WHO. Special Issue 14—African Traditional Medicine Day. The African Health Monitor; 2010. Available online: https://www.afro.who.int/sites/default/files/2017-06/ahm-special-issue-14.pdf (accessed on 31 December 2023).
- Mahomoodally, M.F. Traditional Medicines in Africa: An Appraisal of Ten Potent African Medicinal Plants. Evid.-Based Complement. Altern. Med. 2013, 2013, 617459. [Google Scholar] [CrossRef]
- WHO. Sickle-Cell Anaemia; WHO: Geneva, Switzerland, 2006.
- Mothibe, M.E.; Sibanda, M. African Traditional Medicine: South African Perspective; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Tluway, F.; Makani, J. Sickle Cell Disease in Africa: An Overview of the Integrated Approach to Health, Research, Education and Advocacy in Tanzania, 2004–2016. Br. J. Haematol. 2017, 177, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Baharlooeian, M.; Terasawa, M.; Shimada, Y.; Nishimura, N. Beneficial Effects of Seaweed-Derived Components on Metabolic Syndrome via Gut Microbiota Modulation. Front. Nutr. 2023, 10, 1173225. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.T.; Hernandez, A.G.; Ware, R.E. Sickle Cell Anemia in Sub-Saharan Africa: Advancing the Clinical Paradigm through Partnerships and Research. Blood 2017, 129, 155–161. [Google Scholar] [CrossRef]
- Ameh, S.; Obodozie, O.O.; Afolabi, E.K.; Oyedele, E.O.; Ache, T.; Onanuga, C.; Ibe, M.C.; Inyang, U. Some Basic Requirements for Preparing an Antisickling Herbal Medicine—NIPRISAN®. African J. Pharm. Pharmacol. 2009, 3, 259–264. [Google Scholar]
- Kitadi, J.M.; Mazasa, P.P.; Tshibangu, D.S.T.; Kasali, F.M.; Tshilanda, D.D.; Ngbolua, K.T.N.; Mpiana, P.T. Ethnopharmacological Survey and Antisickling Activity of Plants Used in the Management of Sickle Cell Disease in Kikwit City, DR Congo. Evid.-Based Complement. Altern. Med. 2020, 2020, 1346493. [Google Scholar] [CrossRef]
- Adejumo, O.E.; Kolapo, A.L.; Folarin, A.O. Moringa oleifera Lam. (Moringaceae) Grown in Nigeria: In Vitro Antisickling Activity on Deoxygenated Erythrocyte Cells. J. Pharm. Bioallied Sci. 2012, 4, 118–122. [Google Scholar] [CrossRef]
- Dafaalla, E.A.A.; Humeida, A.A.K.; Dafaalla, E.A.A.; Humeida, A.A.K. The Effect of Fixed Oil Extracts of Nigella Sativa on Sickle Cells: An in-Vitro Study in Khartoum State -Sudan. World J. Adv. Res. Rev. 2021, 10, 317–321. [Google Scholar] [CrossRef]
- Awor, S.; Bongomin, F.; Kaggwa, M.M.; Pebalo, F.P.; Musoke, D. Prevalence of Use of Herbal Medicines for the Treatment of Sickle Cell Disease in Africa: A Systematic Review and Meta-Analysis. J. Herb. Med. 2023, 42, 100735. [Google Scholar] [CrossRef]
- Mohamed, A.S.; Abd El Dayem, O.Y.; El Shamy, A.M.; El Sakhawy, F.S.; El Gedaily, R.A. Comparative Antisickling and Antioxidant Activities of Pseudobombax Ellipticum Cultivars in Relation to Their Metabolite Profiling Using LC/MS. RSC Adv. 2023, 13, 21327–21335. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 552535. [Google Scholar] [CrossRef]
- Vamanu, E.; Dinu, L.D.; Pelinescu, D.R.; Gatea, F. Therapeutic Properties of Edible Mushrooms and Herbal Teas in Gut Microbiota Modulation. Microorganisms 2021, 9, 1262. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Acebo, L.; López de las Hazas, M.C.; Margollés, A.; Dávalos, A.; García-Ruiz, A. Eating MicroRNAs: Pharmacological Opportunities for Cross-Kingdom Regulation and Implications in Host Gene and Gut Microbiota Modulation. Br. J. Pharmacol. 2021, 178, 2218–2245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, F.; Zhang, J.; Wang, W.; Li, L.; Yan, J. Modulatory Effects of Polysaccharides from Plants, Marine Algae and Edible Mushrooms on Gut Microbiota and Related Health Benefits: A Review. Int. J. Biol. Macromol. 2022, 204, 169–192. [Google Scholar] [CrossRef]
- Aboderin, F.I.; Oduola, T.; Davison, G.M.; Oguntibeju, O.O. A Review of the Relationship between the Immune Response, Inflammation, Oxidative Stress, and the Pathogenesis of Sickle Cell Anaemia. Biomedicines 2023, 11, 2413. [Google Scholar] [CrossRef]
- Oguntibeju, O. The Interplay among Immune Response, Inflammation, Oxida-Tive Stress and Sickle Cell Anaemia Pathogenesis. J. Forensic Med. 2023, 8, 1–2. [Google Scholar] [CrossRef]
- Fernandes, T.; Garrine, C.; Ferrão, J.; Bell, V.; Varzakas, T. Mushroom Nutrition as Preventative Healthcare in Sub-Saharan Africa. Appl. Sci. 2021, 11, 4221. [Google Scholar] [CrossRef]
- Mshelia Halilu, E. Cultivation and Conservation of African Medicinal Plants for Pharmaceutical Research and Socio-Economic Development; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Patel, D.K.; Dutta, S.D.; Ganguly, K.; Cho, S.-J.; Lim, K.-T.; Patel, D.K.; Dutta, S.D.; Ganguly, K.; Cho, S.-J.; Lim, K.-T. Mushroom-Derived Bioactive Molecules as Immunotherapeutic Agents: A Review. Molecules 2021, 26, 1359. [Google Scholar] [CrossRef] [PubMed]
- Łysakowska, P.; Sobota, A.; Wirkijowska, A. Medicinal Mushrooms: Their Bioactive Components, Nutritional Value and Application in Functional Food Production—A Review. Molecules 2023, 28, 5393. [Google Scholar] [CrossRef] [PubMed]
- El-Maradny, Y.A.; El-Fakharany, E.M.; Abu-Serie, M.M.; Hashish, M.H.; Selim, H.S. Lectins Purified from Medicinal and Edible Mushrooms: Insights into Their Antiviral Activity against Pathogenic Viruses. Int. J. Biol. Macromol. 2021, 179, 239–258. [Google Scholar] [CrossRef]
- Das, A.K.; Asif, M.; Hasan, G.M.M.A. A Comparative Study of Fatty Acid Compositions of Three Cultivated Edible Mushroom Species of Bangladesh. J. Agric. Food Res. 2023, 12, 100620. [Google Scholar] [CrossRef]
- Liuzzi, G.M.; Petraglia, T.; Latronico, T.; Crescenzi, A.; Rossano, R. Antioxidant Compounds from Edible Mushrooms as Potential Candidates for Treating Age-Related Neurodegenerative Diseases. Nutrients 2023, 15, 1913. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Wang, Z.; Wang, B.; Hou, B.; Cheng, J.; Bai, T.; Zhang, Y.; Wang, W.; Yan, L.; Zhang, J. Expanding the Application of Tryptophan: Industrial Biomanufacturing of Tryptophan Derivatives. Front. Microbiol. 2023, 14, 1099098. [Google Scholar] [CrossRef]
- Assemie, A.; Abaya, G. The Effect of Edible Mushroom on Health and Their Biochemistry. Int. J. Microbiol. 2022, 2022, 8744788. [Google Scholar] [CrossRef]
- Muszyńska, B.; Grzywacz-Kisielewska, A.; Kała, K.; Gdula-Argasińska, J. Anti-Inflammatory Properties of Edible Mushrooms: A Review. Food Chem. 2018, 243, 373–381. [Google Scholar] [CrossRef]
- Martinez-Medina, G.A.; Chávez-González, M.L.; Verma, D.K.; Prado-Barragán, L.A.; Martínez-Hernández, J.L.; Flores-Gallegos, A.C.; Thakur, M.; Srivastav, P.P.; Aguilar, C.N. Bio-Funcional Components in Mushrooms, a Health Opportunity: Ergothionine and Huitlacohe as Recent Trends. J. Funct. Foods 2021, 77, 104326. [Google Scholar] [CrossRef]
- Cuevas-Cianca, S.I.; Romero-Castillo, C.; Gálvez-Romero, J.L.; Juárez, Z.N.; Hernández, L.R. Antioxidant and Anti-Inflammatory Compounds from Edible Plants with Anti-Cancer Activity and Their Potential Use as Drugs. Molecules 2023, 28, 1488. [Google Scholar] [CrossRef]
- Kozarski, M.; Klaus, A.; Jakovljevic, D.; Todorovic, N.; Vunduk, J.; Petrović, P.; Niksic, M.; Vrvic, M.M.; Van Griensven, L. Antioxidants of Edible Mushrooms. Molecules 2015, 20, 19489–19525. [Google Scholar] [CrossRef] [PubMed]
- Lam-Sidun, D.; Peters, K.M.; Borradaile, N.M. Mushroom-Derived Medicine? Preclinical Studies Suggest Potential Benefits of Ergothioneine for Cardiometabolic Health. Int. J. Mol. Sci. 2021, 22, 3246. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, C.; Esposito, S.; Iacoviello, L. Edible Mushrooms and Beta-Glucans: Impact on Human Health. Nutrients 2021, 13, 2195. [Google Scholar] [CrossRef] [PubMed]
- Timm, T.G.; Costa, T.M.; Alberton, M.D.; Helm, C.V.; Tavares, L.B.B. Mushroom β-Glucans: Application and Innovation for Food Industry and Immunotherapy. Appl. Microbiol. Biotechnol. 2023, 107, 5035–5049. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Sharma, K.K. Fungal Secondary Metabolites in Food and Pharmaceuticals in the Era of Multi-Omics. Appl. Microbiol. Biotechnol. 2022, 106, 3465–3488. [Google Scholar] [CrossRef]
- Nguyen, T.A.N.; Higa, T.; Shiina, A.; Utami, Y.D.; Hiruma, K. Exploring the Roles of Fungal-Derived Secondary Metabolites in Plant-Fungal Interactions. Physiol. Mol. Plant Pathol. 2023, 125, 102021. [Google Scholar] [CrossRef]
- Gupta, S.; Chaturvedi, P.; Kulkarni, M.G.; Van Staden, J. A Critical Review on Exploiting the Pharmaceutical Potential of Plant Endophytic Fungi. Biotechnol. Adv. 2020, 39, 107462. [Google Scholar] [CrossRef]
- Zhao, J.; Hu, Y.; Qian, C.; Hussain, M.; Liu, S.; Zhang, A.; He, R.; Sun, P. The Interaction between Mushroom Polysaccharides and Gut Microbiota and Their Effect on Human Health: A Review. Biology 2023, 12, 122. [Google Scholar] [CrossRef]
- Rani, A.; Saini, K.C.; Bast, F.; Mehariya, S.; Bhatia, S.K.; Lavecchia, R.; Zuorro, A. Microorganisms: A Potential Source of Bioactive Molecules for Antioxidant Applications. Molecules 2021, 26, 1142. [Google Scholar] [CrossRef]
- Hu, W.; Liang, K.; Zhu, H.; Zhao, C.; Hu, H.; Yin, S. Ferroptosis and Its Role in Chronic Diseases. Cells 2022, 11, 2040. [Google Scholar] [CrossRef]
- Tang, H.M.; Cheung, P.C.K. Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins 2019, 11, 492. [Google Scholar] [CrossRef] [PubMed]
- Llabani, E.; Hicklin, R.W.; Lee, H.Y.; Motika, S.E.; Crawford, L.A.; Weerapana, E.; Hergenrother, P.J. Diverse Compounds from Pleuromutilin Lead to a Thioredoxin Inhibitor and Inducer of Ferroptosis. Nat. Chem. 2019, 11, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zennadi, R.; Pantaleo, A.; Secchi, C.; Orecchioni, M. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants 2021, 10, 1608. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, S.; Canabady-Rochelle, L.; Gaucher, C. Nature-Inspired Bioactive Compounds: A Promising Approach for Ferroptosis-Linked Human Diseases? Molecules 2023, 28, 2636. [Google Scholar] [CrossRef] [PubMed]
- Ohiri, R.C.; Essien, E.B. Stabilizing Ability and Anti-Sickling Potentials of Ganoderma Lucidum Decoction Extract on Human HbS Erythrocytes Membrane. Am. J. Biomed. Sci 2018, 10, 202–210. [Google Scholar] [CrossRef]
- Yu, T.; Wu, Q.; Liang, B.; Wang, J.; Wu, D.; Shang, X. The Current State and Future Prospects of Auricularia Auricula’s Polysaccharide Processing Technology Portfolio. Molecules 2023, 28, 582. [Google Scholar] [CrossRef]
- Ohiri, R.C.; Odey, O.P. Human Hemoglobin S Erythrocyte-Stabilizing and Antisickling Potential of Extract of Wood Ear Mushroom, Auricularia Auricula (Agaricomycetes). Int. J. Med. Mushrooms 2022, 24, 25–34. [Google Scholar] [CrossRef]
- Gravina, A.G.; Pellegrino, R.; Auletta, S.; Palladino, G.; Brandimarte, G.; D’Onofrio, R.; Arboretto, G.; Imperio, G.; Ventura, A.; Cipullo, M.; et al. Hericium Erinaceus, a Medicinal Fungus with a Centuries-Old History: Evidence in Gastrointestinal Diseases. World J. Gastroenterol. 2023, 29, 3048–3065. [Google Scholar] [CrossRef]
- Szućko-Kociuba, I.; Trzeciak-Ryczek, A.; Kupnicka, P.; Chlubek, D. Neurotrophic and Neuroprotective Effects of Hericium Erinaceus. Int. J. Mol. Sci. 2023, 24, 15960. [Google Scholar] [CrossRef]
- Anchang, K.Y.; Oko, A.; Richard, S. Toxicological and Positive Changes in Hematological Pattern of Albino Rats Treated with Termitomyces Titanicus: Potential Valorization of an Indigenous Practice in the Treatment of Oris Cancrum (Noma) in Nigeria. J. Pathol. Res. Rev. Rep. 2020, 106, 3. [Google Scholar] [CrossRef]
- Paloi, S.; Kumla, J.; Paloi, B.P.; Srinuanpan, S.; Hoijang, S.; Karunarathna, S.C.; Acharya, K.; Suwannarach, N.; Lumyong, S. Termite Mushrooms (Termitomyces), a Potential Source of Nutrients and Bioactive Compounds Exhibiting Human Health Benefits: A Review. J. Fungi 2023, 9, 112. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Intervention | Outcome |
|---|---|
| Blood transfusion | Reduce the burden of sickled cells [94,95] |
| Hydroxyurea | Increase fetal hemoglobin (HbF) to stop polymers forming in the sickle hemoglobin [96] |
| Hematopoietic stem cell transplantation | Reverse the sickle phenotype [97,98] |
| L-glutamine | Antioxidant effects [99] |
| Hemoglobin S (HbS) polymerization inhibitors | Prevent HbS polymerization [100] |
| Monoclonal antibody (crizanlizumab) | Reduce selectin-mediated adhesion [101] |
| Gene editing therapy (Casgevy™) | Editing faulty gene in a patient’s bone marrow stem cells [102] |
| Nutritional Intervention | Outcome |
|---|---|
| Microbiota modulation | Reverse established microbial dysbiosis [127,128,129] |
| Omega-3 fatty acids | Improve VOC rate, markers of inflammation, adhesion, and hemolysis [146] |
| Plant | |
|---|---|
| Alchornea cordifolia | Traditional use as “blood tonic” [193] |
| Ceiba pentandra | Traditional use as “blood tonic” [193] |
| Moringa oleifera | Antiurolithiatic properties [195] |
| Nigella sativa | Antioxidant properties [196] |
| Mushroom | Outcome |
|---|---|
| Ganoderma lucidum | Decrease in hemoglobin polymerization rate [233] |
| Auricularia auricular | Free radical scavenging activity [235] |
| Hericium erinaceus | Regulation of heat shock proteins (HSP70) [237] |
| Termitomyces | Increase hemoglobin levels and white blood cells [238] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bell, V.; Varzakas, T.; Psaltopoulou, T.; Fernandes, T. Sickle Cell Disease Update: New Treatments and Challenging Nutritional Interventions. Nutrients 2024, 16, 258. https://doi.org/10.3390/nu16020258
Bell V, Varzakas T, Psaltopoulou T, Fernandes T. Sickle Cell Disease Update: New Treatments and Challenging Nutritional Interventions. Nutrients. 2024; 16(2):258. https://doi.org/10.3390/nu16020258
Chicago/Turabian StyleBell, Victoria, Theodoros Varzakas, Theodora Psaltopoulou, and Tito Fernandes. 2024. "Sickle Cell Disease Update: New Treatments and Challenging Nutritional Interventions" Nutrients 16, no. 2: 258. https://doi.org/10.3390/nu16020258
APA StyleBell, V., Varzakas, T., Psaltopoulou, T., & Fernandes, T. (2024). Sickle Cell Disease Update: New Treatments and Challenging Nutritional Interventions. Nutrients, 16(2), 258. https://doi.org/10.3390/nu16020258