
Both Maternal High-Fat and Post-Weaning High-Carbohydrate Diets Increase Rates of Spontaneous Hepatocellular Carcinoma in Aged-Mouse Offspring

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
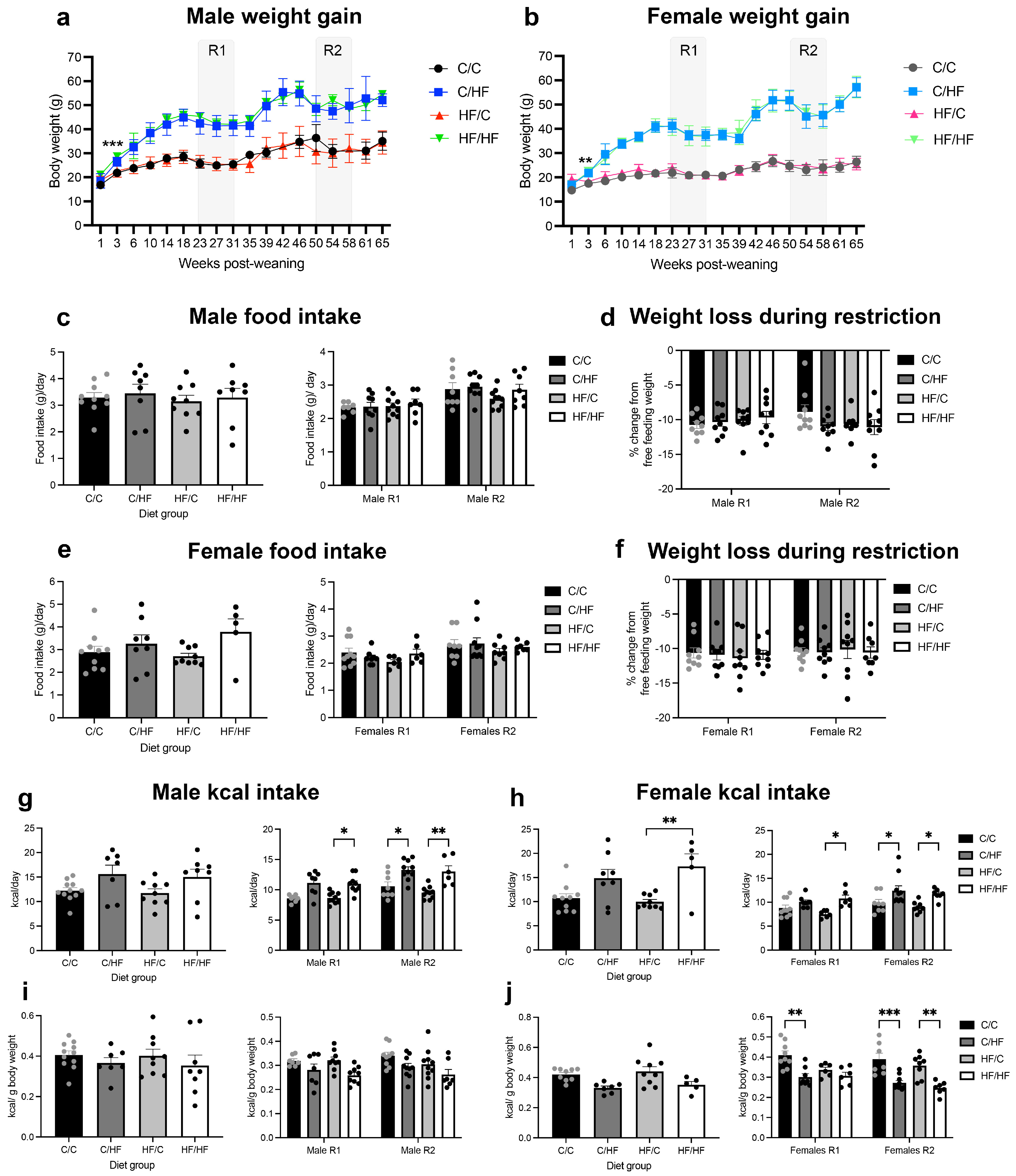
3.1. Postnatal HF Diet Induced Weight Gain, While Postnatal C Diet Increased HCC Incidence
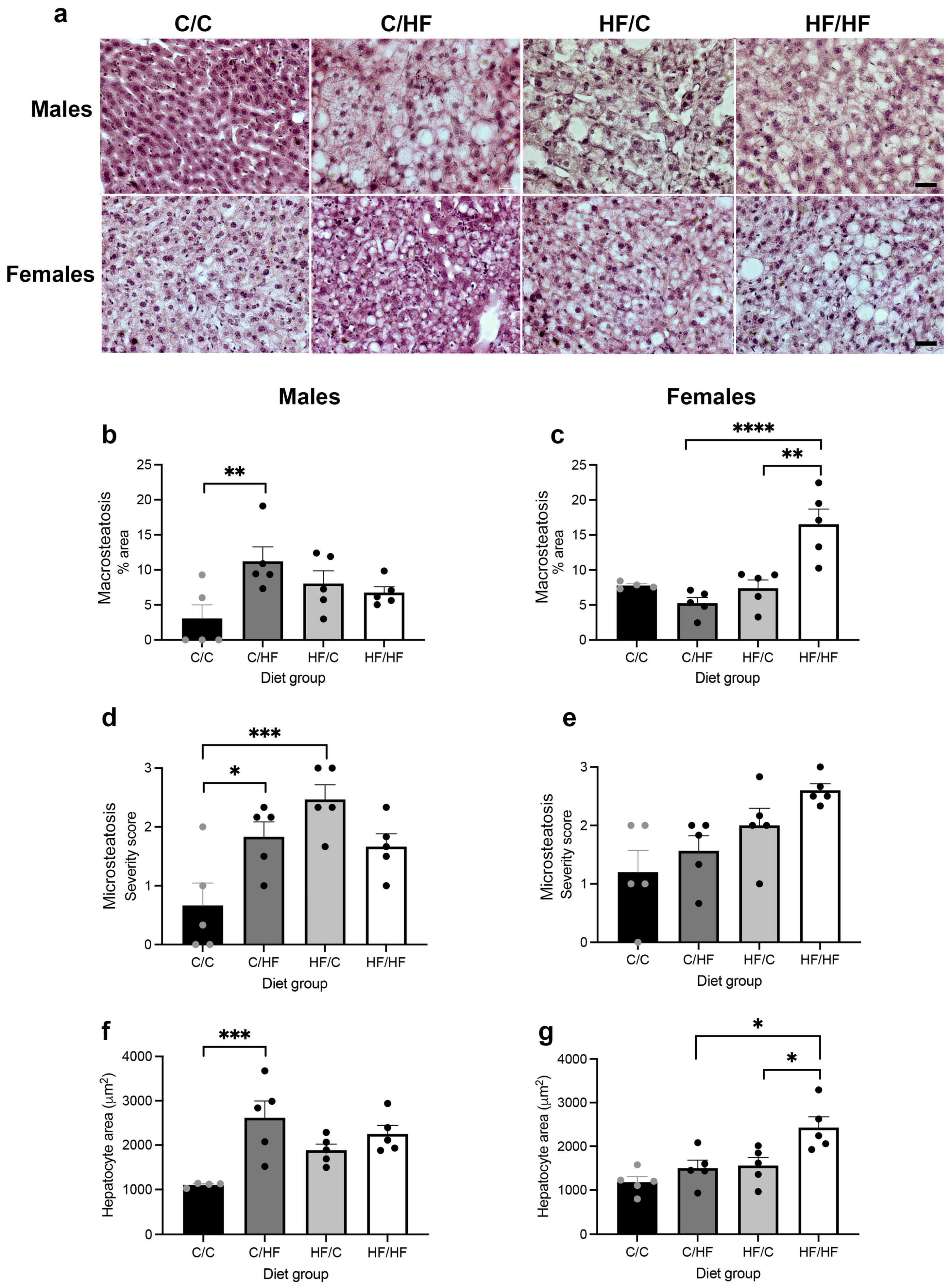
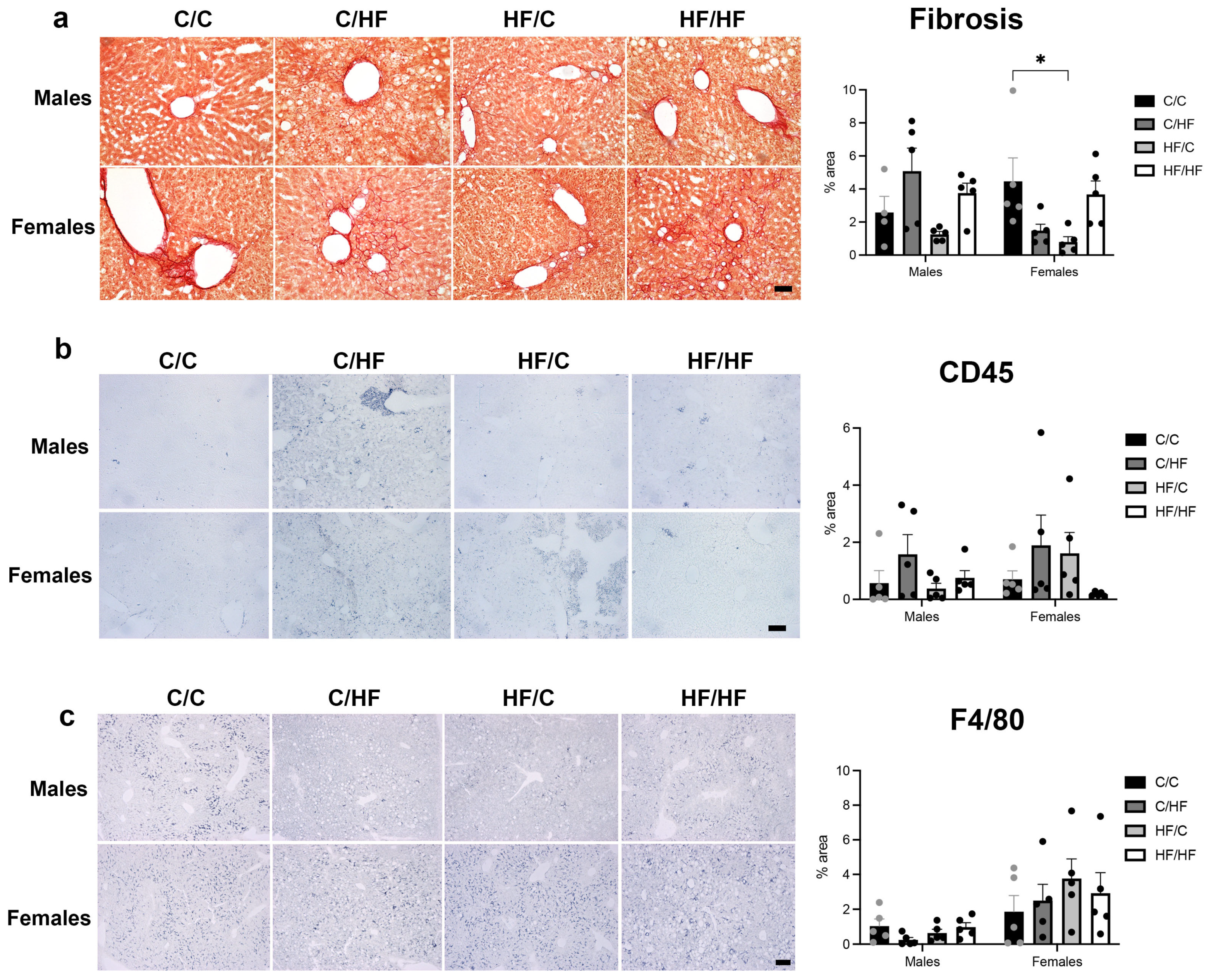
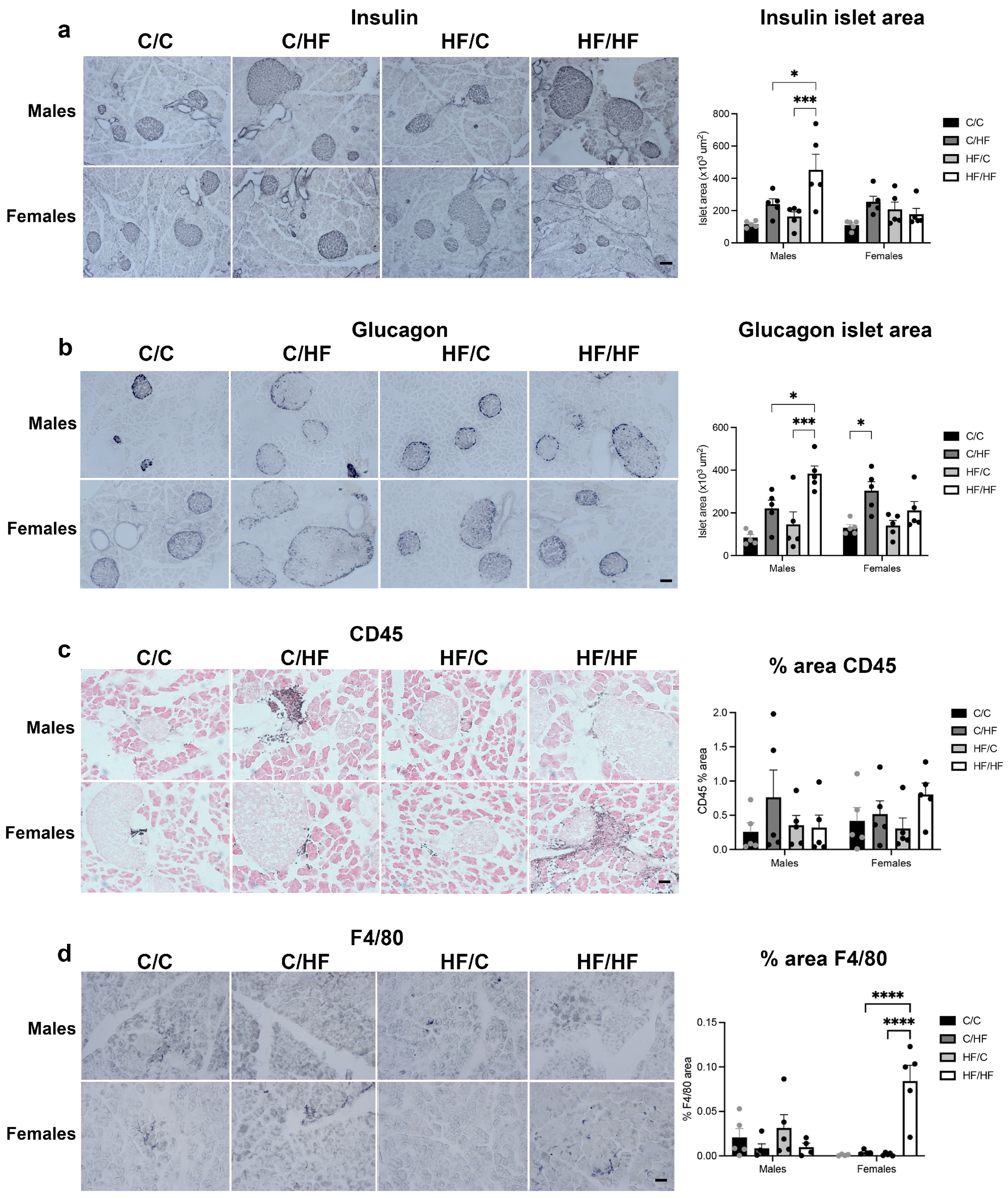
3.2. Measures of MASLD Were Higher in HF-Fed Animals
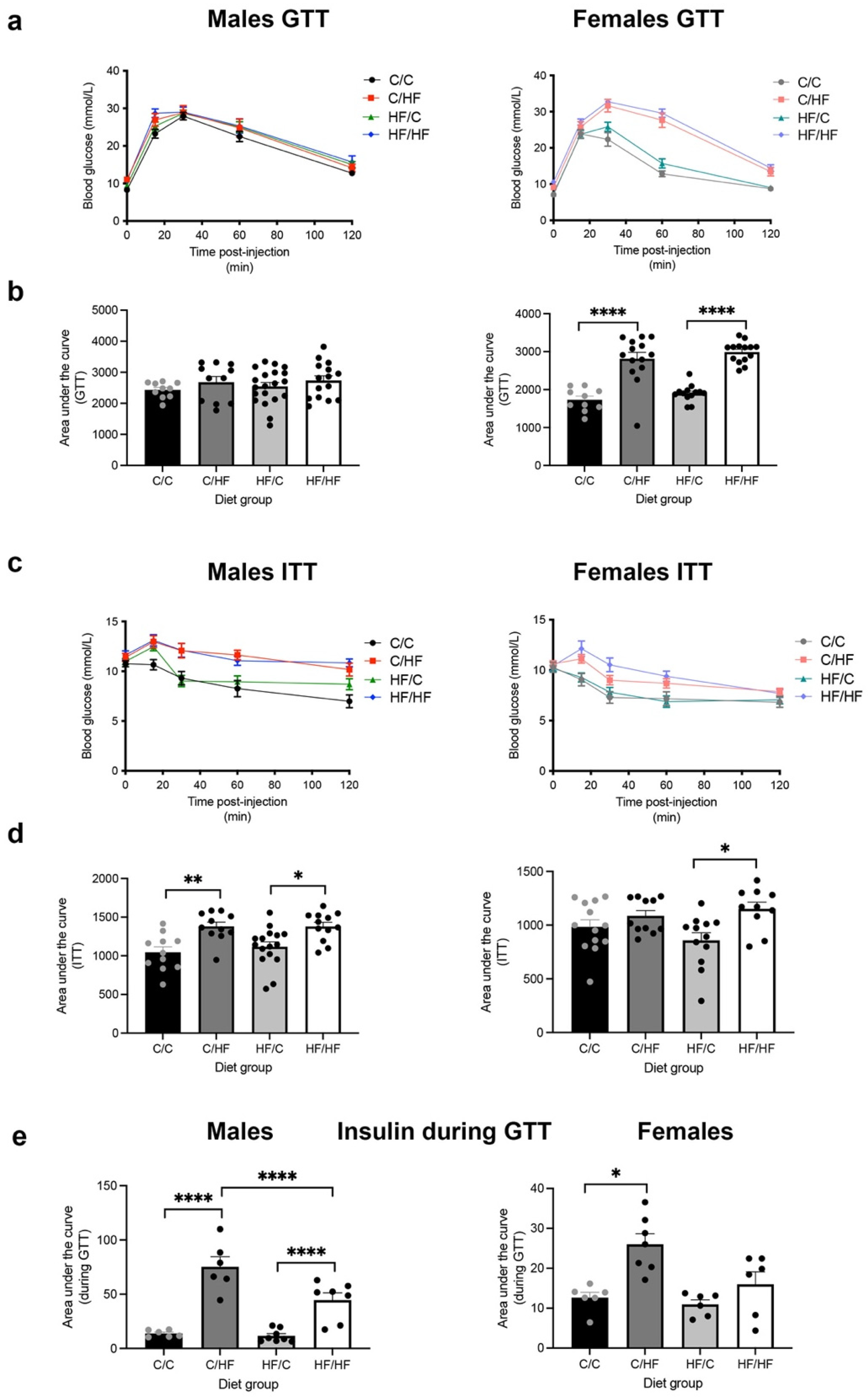
3.3. Postnatal HF, but Not C Diet, Induced Glucose Intolerance and Hyperinsulinemia
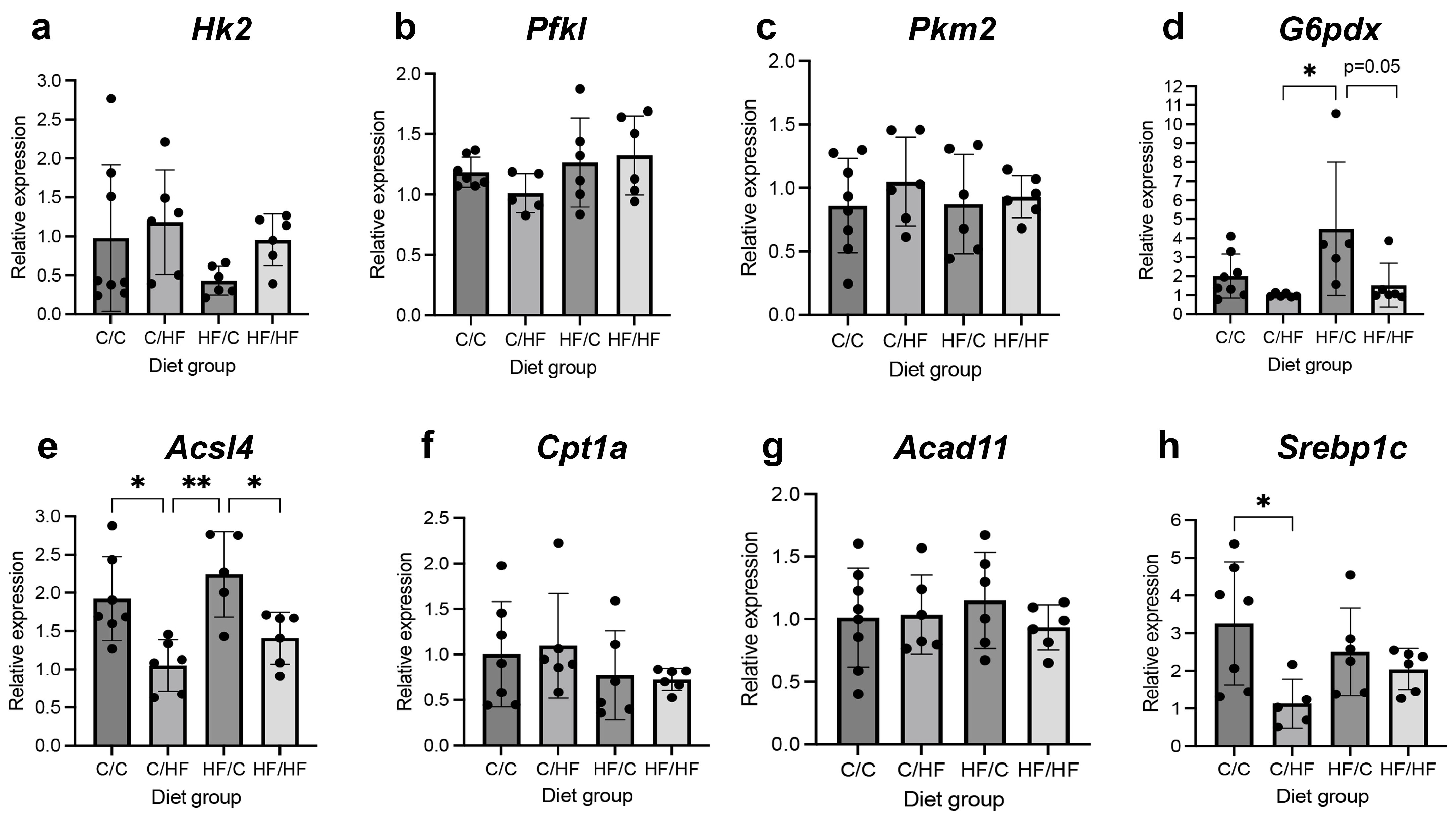
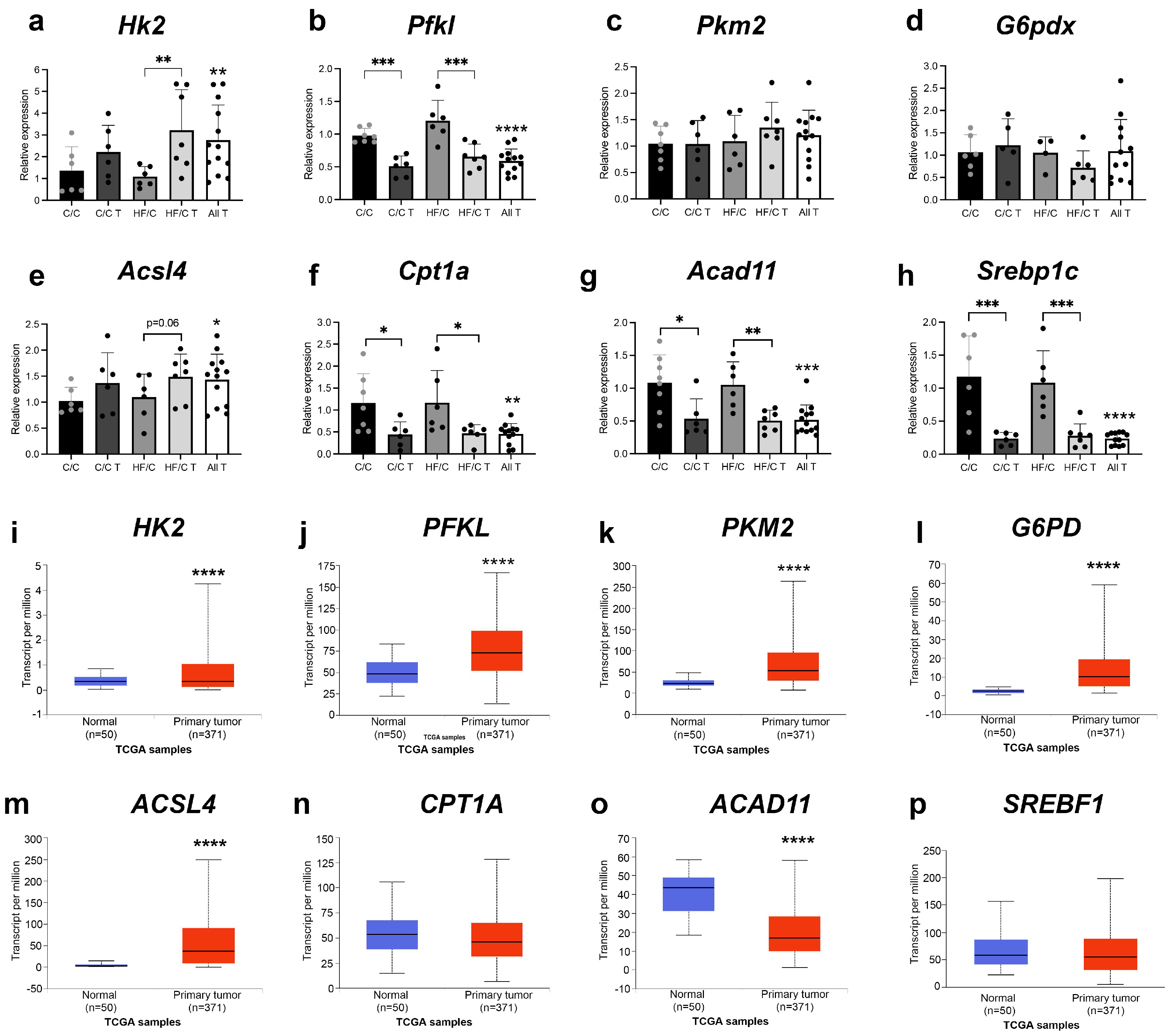
3.4. Markers of Glycolysis and Fatty Acid Oxidation Were Altered in Both Diet Groups and in HCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef]
- Macias, R.I.R.; Monte, M.J.; Serrano, M.A.; Gonzalez-Santiago, J.M.; Martin-Arribas, I.; Simao, A.L.; Castro, R.E.; Gonzalez-Gallego, J.; Mauriz, J.L.; Marin, J.J.G. Impact of aging on primary liver cancer: Epidemiology, pathogenesis and therapeutics. Aging 2021, 13, 23416–23434. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.M.; Wong, L.L.; Hernandez, B.Y.; Ji, J.F.; Jia, W.; Kwee, S.A.; Kalathil, S. Gender differences in hepatocellular cancer: Disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma Res. 2018, 4, 66. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Shah, P.A.; Patil, R.; Harrison, S.A. NAFLD-related hepatocellular carcinoma: The growing challenge. Hepatology 2023, 77, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef]
- Cao, B.; Liu, C.; Zhang, Q.; Dong, Y. Maternal High-Fat Diet Leads to Non-alcoholic Fatty Liver Disease Through Upregulating Hepatic SCD1 Expression in Neonate Rats. Front. Nutr. 2020, 7, 581723. [Google Scholar] [CrossRef]
- Hagstrom, H.; Simon, T.G.; Roelstraete, B.; Stephansson, O.; Soderling, J.; Ludvigsson, J.F. Maternal obesity increases the risk and severity of NAFLD in offspring. J. Hepatol. 2021, 75, 1042–1048. [Google Scholar] [CrossRef]
- Moeckli, B.; Delaune, V.; Prados, J.; Tihy, M.; Peloso, A.; Oldani, G.; Delmi, T.; Slits, F.; Gex, Q.; Rubbia-Brandt, L.; et al. Impact of Maternal Obesity on Liver Disease in the Offspring: A Comprehensive Transcriptomic Analysis and Confirmation of Results in a Murine Model. Biomedicines 2022, 10, 294. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Q.; Zhang, Y.; Geng, M.; Wei, Y.; Liu, Y.; Liu, S.; Petersen, R.B.; Yue, J.; Huang, K.; et al. Multigenerational maternal obesity increases the incidence of HCC in offspring via miR-27a-3p. J. Hepatol. 2020, 73, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, D.J.M.; Feitosa de Oliveira, K.G.; Pereira, I.C.; do Nascimento, G.T.M.; Barrense, C.O.; Martins, J.A.; Pereira Rego, B.M.; Oliveira da Silva, T.E.; Carneiro da Silva, F.C.; Torres-Leal, F.L. Dietary restriction and hepatic cancer: Systematic review and meta-analysis of animal studies. Crit. Rev. Oncol. Hematol. 2024, 196, 104264. [Google Scholar] [CrossRef]
- Lagopoulos, L.; Sunahara, G.I.; Wurzner, H.; Dombrowsky, I.; Stalder, R. The effects of alternating dietary restriction and ad libitum feeding of mice on the development of diethylnitrosamine-induced liver tumours and its correlation to insulinaemia. Carcinogenesis 1991, 12, 311–315. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, W.; Simon, T.G.; Smith-Warner, S.A.; Fung, T.T.; Sui, J.; Chong, D.; VoPham, T.; Meyerhardt, J.A.; Wen, D.; et al. Dietary Patterns and Risk of Hepatocellular Carcinoma among U.S. Men and Women. Hepatology 2019, 70, 577–586. [Google Scholar] [CrossRef]
- Vogtmann, E.; Li, H.L.; Shu, X.O.; Chow, W.H.; Ji, B.T.; Cai, H.; Gao, J.; Zhang, W.; Gao, Y.T.; Zheng, W.; et al. Dietary glycemic load, glycemic index, and carbohydrates on the risk of primary liver cancer among Chinese women and men. Ann. Oncol. 2013, 24, 238–244. [Google Scholar] [CrossRef]
- Koh, W.P.; Dan, Y.Y.; Goh, G.B.; Jin, A.; Wang, R.; Yuan, J.M. Dietary fatty acids and risk of hepatocellular carcinoma in the Singapore Chinese health study. Liver Int. 2016, 36, 893–901. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.; Liu, X.; Wu, X.; Xu, Q.; Qu, J.; Li, X.; Zhu, Y.; Wen, L.; Wang, J. High-Carbohydrate Diet Consumption Poses a More Severe Liver Cholesterol Deposition than a High-Fat and High-Calorie Diet in Mice. Int. J. Mol. Sci. 2023, 24, 14700. [Google Scholar] [CrossRef]
- Tessitore, A.; Mastroiaco, V.; Vetuschi, A.; Sferra, R.; Pompili, S.; Cicciarelli, G.; Barnabei, R.; Capece, D.; Zazzeroni, F.; Capalbo, C.; et al. Development of hepatocellular cancer induced by long term low fat-high carbohydrate diet in a NAFLD/NASH mouse model. Oncotarget 2017, 8, 53482–53494. [Google Scholar] [CrossRef]
- Healy, M.E.; Chow, J.D.; Byrne, F.L.; Breen, D.S.; Leitinger, N.; Li, C.; Lackner, C.; Caldwell, S.H.; Hoehn, K.L. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J. Hepatol. 2015, 62, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Healy, M.E.; Lahiri, S.; Hargett, S.R.; Chow, J.D.; Byrne, F.L.; Breen, D.S.; Kenwood, B.M.; Taddeo, E.P.; Lackner, C.; Caldwell, S.H.; et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci. Rep. 2016, 6, 22292. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Lin, J.Z.; Yang, X.B.; Sang, X.T. Aberrant lipid metabolism in hepatocellular carcinoma cells as well as immune microenvironment: A review. Cell Prolif. 2020, 53, e12772. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Heucke, N.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhutter, H.G. Characterization of Lipid and Lipid Droplet Metabolism in Human HCC. Cells 2019, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef]
- Peng, H.; Xu, H.; Wu, J.; Li, J.; Zhou, Y.; Ding, Z.; Siwko, S.K.; Yuan, X.; Schalinske, K.L.; Alpini, G.; et al. Maternal high-fat diet disrupted one-carbon metabolism in offspring, contributing to nonalcoholic fatty liver disease. Liver Int. 2021, 41, 1305–1319. [Google Scholar] [CrossRef]
- Hafner, H.; Mulcahy, M.C.; Carlson, Z.; Hartley, P.; Sun, H.; Westerhoff, M.; Qi, N.; Bridges, D.; Gregg, B. Lactational High Fat Diet in Mice Causes Insulin Resistance and NAFLD in Male Offspring Which Is Partially Rescued by Maternal Metformin Treatment. Front. Nutr. 2021, 8, 759690. [Google Scholar] [CrossRef]
- Contu, L.; Heath, C.J.; Hawkes, C.A. Appetitive Motivation and Associated Neurobiology Change Differentially across the Life Course of Mouse Offspring Exposed to Peri- and Postnatal High Fat Feeding. Nutrients 2022, 14, 5161. [Google Scholar] [CrossRef] [PubMed]
- Contu, L.; Nizari, S.; Heath, C.J.; Hawkes, C.A. Pre- and Post-natal High Fat Feeding Differentially Affects the Structure and Integrity of the Neurovascular Unit of 16-Month Old Male and Female Mice. Front. Neurosci. 2019, 13, 1045. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Dragani, T.A.; Manenti, G.; Gariboldi, M.; De Gregorio, L.; Pierotti, M.A. Genetics of liver tumor susceptibility in mice. Toxicol. Lett. 1995, 82–83, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Chettouh, H.; Lequoy, M.; Fartoux, L.; Vigouroux, C.; Desbois-Mouthon, C. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int. 2015, 35, 2203–2217. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lin, X.; Wang, G. Targeting SREBP-1-Mediated Lipogenesis as Potential Strategies for Cancer. Front. Oncol. 2022, 12, 952371. [Google Scholar] [CrossRef]
- Pan, F.; Lin, X.; Hao, L.; Wang, T.; Song, H.; Wang, R. The Critical Role of Ferroptosis in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2022, 10, 882571. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Ghai, A.; Deng, Y.; Li, S.; Zhang, R.; Egbulefu, C.; Liang, G.; Achilefu, S.; Ye, J. Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases. Mol. Cell 2023, 83, 3931–3939.e5. [Google Scholar] [CrossRef]
- Gregorio, B.M.; Souza-Mello, V.; Carvalho, J.J.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Maternal high-fat intake predisposes nonalcoholic fatty liver disease in C57BL/6 offspring. Am. J. Obstet. Gynecol. 2010, 203, 495.e1–495.e8. [Google Scholar] [CrossRef]
- Thompson, M.D. Developmental Programming of NAFLD by Parental Obesity. Hepatol. Commun. 2020, 4, 1392–1403. [Google Scholar] [CrossRef]
- Takiyama, T.; Sera, T.; Nakamura, M.; Hoshino, M.; Uesugi, K.; Horike, S.I.; Meguro-Horike, M.; Bessho, R.; Takiyama, Y.; Kitsunai, H.; et al. A maternal high-fat diet induces fetal origins of NASH-HCC in mice. Sci. Rep. 2022, 12, 13136. [Google Scholar] [CrossRef]
- Venugopal, S.; Dhanoa, R.K.; Selvamani, T.Y.; Shoukrie, S.I.; Zahra, A.; Malla, J.; Selvaraj, R.; Hamouda, R.K.; Mohammed, L. Does Type 2 Diabetes Increase the Risk of Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease Patients? A Systematic Review. Cureus 2023, 15, e36079. [Google Scholar] [CrossRef]
- Savva, C.; Helguero, L.A.; Gonzalez-Granillo, M.; Melo, T.; Couto, D.; Angelin, B.; Domingues, M.R.; Li, X.; Kutter, C.; Korach-Andre, M. Molecular programming modulates hepatic lipid metabolism and adult metabolic risk in the offspring of obese mothers in a sex-specific manner. Commun. Biol. 2022, 5, 1057. [Google Scholar] [CrossRef]
- Duan, X.Y.; Pan, Q.; Yan, S.Y.; Ding, W.J.; Fan, J.G.; Qiao, L. High-saturate-fat diet delays initiation of diethylnitrosamine-induced hepatocellular carcinoma. BMC Gastroenterol. 2014, 14, 195. [Google Scholar] [CrossRef]
- Lan, Y.; Jin, C.; Kumar, P.; Yu, X.; Lenahan, C.; Sheng, J. Ketogenic Diets and Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 879205. [Google Scholar] [CrossRef] [PubMed]
- Bertram, H.C.; Larsen, L.B.; Chen, X.; Jeppesen, P.B. Impact of high-fat and high-carbohydrate diets on liver metabolism studied in a rat model with a systems biology approach. J. Agric. Food Chem. 2012, 60, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg. Nutr. 2015, 4, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Dewdney, B.; Roberts, A.; Qiao, L.; George, J.; Hebbard, L. A Sweet Connection? Fructose’s Role in Hepatocellular Carcinoma. Biomolecules 2020, 10, 496. [Google Scholar] [CrossRef]
- Minehira, K.; Young, S.G.; Villanueva, C.J.; Yetukuri, L.; Oresic, M.; Hellerstein, M.K.; Farese, R.V., Jr.; Horton, J.D.; Preitner, F.; Thorens, B.; et al. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J. Lipid Res. 2008, 49, 2038–2044. [Google Scholar] [CrossRef]
- Cho, Y.; Cho, E.J.; Yoo, J.J.; Chang, Y.; Chung, G.E.; Jeong, S.M.; Park, S.H.; Han, K.; Shin, D.W.; Yu, S.J. Association between Lipid Profiles and the Incidence of Hepatocellular Carcinoma: A Nationwide Population-Based Study. Cancers 2021, 13, 1599. [Google Scholar] [CrossRef]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Pulliam, C.F.; Patel, A.; Del Piero, F.; Watanabe, T.T.N.; Wankhade, U.D.; Shankar, K.; Hicks, C.; Ronis, M.J. Liver tumorigenesis is promoted by a high saturated fat diet specifically in male mice and is associated with hepatic expression of the proto-oncogene Agap2 and enrichment of the intestinal microbiome with Coprococcus. Carcinogenesis 2019, 40, 349–359. [Google Scholar] [CrossRef]
- Jia, B.; Li, J.; Song, Y.; Luo, C. ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries. Int. J. Mol. Sci. 2023, 24, 21. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Tang, D.; Kroemer, G.; Ren, J. Ferroptosis in hepatocellular carcinoma: Mechanisms and targeted therapy. Br. J. Cancer 2023, 128, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Legrand, P.; Rioux, V. The complex and important cellular and metabolic functions of saturated fatty acids. Lipids 2010, 45, 941–946. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Li, C.; Yang, W.; Zhang, J.; Zheng, X.; Yao, Y.; Tu, K.; Liu, Q. SREBP-1 has a prognostic role and contributes to invasion and metastasis in human hepatocellular carcinoma. Int. J. Mol. Sci. 2014, 15, 7124–7138. [Google Scholar] [CrossRef]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Lu, Y.; Wu, W.; Zhang, Y.; Yang, B.; Wu, H.; Peng, C.; et al. ACSL4 promotes hepatocellular carcinoma progression via c-Myc stability mediated by ERK/FBW7/c-Myc axis. Oncogenesis 2020, 9, 42. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Gumaa, K.A.; McLean, P. Effect of insulin and diet on the steady state concentrations of intermediates of the pentose phosphate pathway of glucose metabolism in liver. FEBS Lett. 1968, 1, 227–229. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holt, D.; Contu, L.; Wood, A.; Chadwick, H.; Alborelli, I.; Insilla, A.C.; Crea, F.; Hawkes, C.A. Both Maternal High-Fat and Post-Weaning High-Carbohydrate Diets Increase Rates of Spontaneous Hepatocellular Carcinoma in Aged-Mouse Offspring. Nutrients 2024, 16, 2805. https://doi.org/10.3390/nu16162805
Holt D, Contu L, Wood A, Chadwick H, Alborelli I, Insilla AC, Crea F, Hawkes CA. Both Maternal High-Fat and Post-Weaning High-Carbohydrate Diets Increase Rates of Spontaneous Hepatocellular Carcinoma in Aged-Mouse Offspring. Nutrients. 2024; 16(16):2805. https://doi.org/10.3390/nu16162805
Chicago/Turabian StyleHolt, Daniel, Laura Contu, Alice Wood, Hannah Chadwick, Ilaria Alborelli, Andrea Cacciato Insilla, Francesco Crea, and Cheryl A. Hawkes. 2024. "Both Maternal High-Fat and Post-Weaning High-Carbohydrate Diets Increase Rates of Spontaneous Hepatocellular Carcinoma in Aged-Mouse Offspring" Nutrients 16, no. 16: 2805. https://doi.org/10.3390/nu16162805
APA StyleHolt, D., Contu, L., Wood, A., Chadwick, H., Alborelli, I., Insilla, A. C., Crea, F., & Hawkes, C. A. (2024). Both Maternal High-Fat and Post-Weaning High-Carbohydrate Diets Increase Rates of Spontaneous Hepatocellular Carcinoma in Aged-Mouse Offspring. Nutrients, 16(16), 2805. https://doi.org/10.3390/nu16162805