
Nutritional Management of Patients with Fatty Acid Oxidation Disorders


Abstract
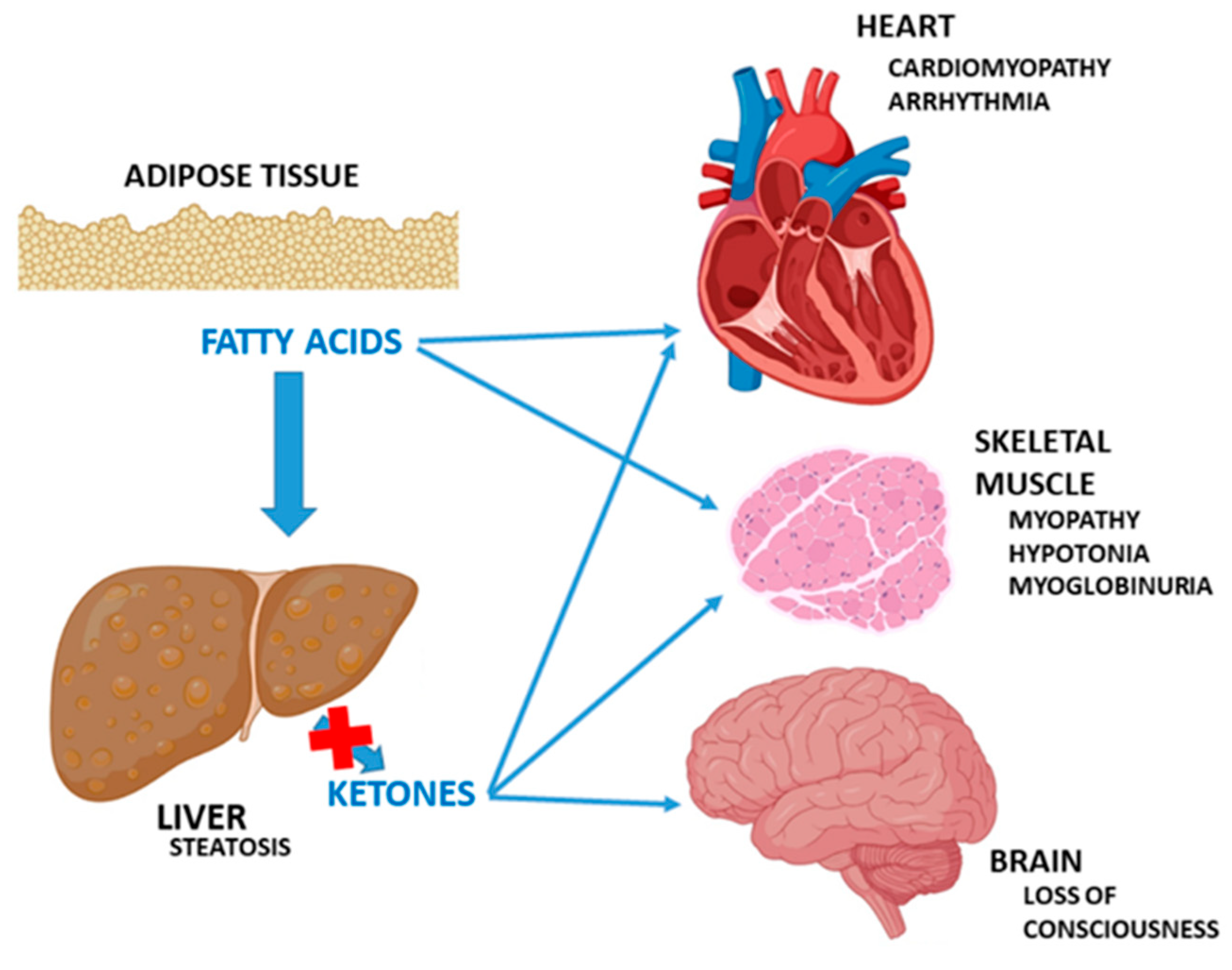
1. Introduction
2. Treatment of Fatty Acid Oxidation Disorders
Dietary Treatment
- For this purpose, frequent meals throughout the day are advised, ensuring a constant supply of glucose. The optimal time between meals is not well established and may vary individually, depending on age, weight, growth and enzyme deficiency.
- Never skip breakfast.
- For children under 1 year, meals should be every 3–4 h.
- It is advisable to have a midnight meal. Cornstarch can be used from 8 months of age when the pancreatic enzymes have the optimal capacity for adequate absorption. This preparation has a large amount of branched glucose chains that are hydrolysed and released slowly, allowing normoglycaemia figures to be maintained for 6–8 h, being more effective than an equivalent intake of glucose every 3 h. Start with 1.0–1.5 g/kg and gradually increase to 1.75–2 g/kg at 2 years of age. If other carbohydrate sources (rice, wheat, oats) are used, their effect may not be as desired due to lower amylopectin content [10].
- In severe cases with cardiomyopathy or feeding difficulties, nocturnal continuous enteral feeding by nasogastric tube or gastrostomy should be considered.
- A.
- LC-FAODs (CPT-I, CPT-II, CACT, VLCAD, LCHAD/MTP):
- Avoidance of fasting (primary measure) (midnight cornstarch or carbohydrates, nocturnal enteral nutrition).
- Long-chain triglyceride (LCT) restriction to 10% of total energy (normalises plasma acylcarnitines), as restrictions below this figure entail a high risk of essential fatty acid deficiency and higher figures increase their potentially toxic metabolites [15,16]. It is very useful to provide the family with a traffic-light-type diet with recommended, limited or in moderation, and non-recommended foods (Table 3) [9].
- Linoleic acid (C18:2n6) and linolenic acid (C18:3n3) intake of 3–4% and 0.5–1%, respectively, of total calories, with a 5/1–10/1 ratio, to avoid the risk of essential fatty acid deficiency. To this end, vegetable oils should be included in the diet as a source of essential fatty acid precursors, within 10% of LCT. Soybean, walnut, canola, flaxseed, wheat germ, sunflower or safflower oils are recommended.
- In patients with MTP complex disorders including long-chain 3-hydroxy- acyl-CoA dehydrogenase (LCHAD) deficiency, there is no consensus on the intake of docoxahexanoic acid (DHA) (C22:6n3). DHA deficiency has been suggested to contribute to the pathogenesis of chorioretinitis, although supplementation does not appear to prevent retinal degeneration [17]. However, several groups [8,17,18,19] advise 65 mg/day in children weighing less than 20 kg, 130 mg/day in children weighing more than 20 kg and 100–200 mg/day in adults. Other groups do not routinely prescribe it, adding walnut oil to the diet [20].
- Medium-chain triglyceride (MCT) supplementation at 10–25% of total energy. The minimum amount to be administered is 10%, as lower amounts do not decrease abnormal metabolites [15,16]. After ingestion, MCT diffuses easily and directly into the venous system and consequently into the tissues, not requiring the carnitine system or long-chain enzymes for its metabolisation in which four molecules of acetyl CoA are produced. These oils contain a mixture of even-chain fatty acids of 8 (octanoate) (mainly), 10 (decanoate) and some 12 (dodecanoate) carbons in length, which vary according to presentation. It has been suggested that the best octanoate/decanoate ratio is 1:3 [16]. MCT suppresses the long-chain FAO, preventing the accumulation of toxic metabolites (long-chain 3-hydroxy fatty acid intermediates), lactate and acylcarnitines [16]. This supplementation can be carried out in several ways.
- ○
- Pure form at doses of 2–3 g/kg/day in the first year of life and 1–1.25 g/kg/day in those older than 1 year.
- ○
- Complete diets with a high MCT content.
- ○
- Modular, virtually fat-free diets such as those with the addition of LCT and MCT.
- Triheptanoin (TH) is a highly purified odd-chain synthetic triglyceride composed of three seven-carbon fatty acids esterified with a glycerol. TH is catabolised in the gut to triheptanoate, which diffuses across membranes to enter cells. Its oxidation leads to two molecules of acetyl CoA and one of propionyl CoA, which through methylmanolyl CoA is irreversibly converted into succinyl-CoA (intermediate of the Krebs cycle), increasing the pool of these (anaplerotic function). This anaplerotic function of TH is what theoretically gives it added value, together with the increase in the production of oxaloacetate necessary for neoglucogenesis (improving glucose production) and the formation of aspartate and citrate.
- Since its first publication in 2002 by Charles R Roe et al. [21], it has been used on a compassionate basis or in clinical trials in both non-responders to MCT therapy and naïve patients with different LC-FAODs (CACT, CPT I, CPT II, MTP, LCHAD, VLCAD) [22,23,24,25]. Different doses (3–4 g/kg/day in children; 1 g/kg/day in adolescents and adults; 20% total energy; 25–35% total energy) have been used, describing improvements in quality of life, cardiac function, muscle function and exercise tolerance, with reductions in mortality, time to hospitalisation for crises, number of episodes of hypoglycaemia, cardiomyopathy and rhabdomyolysis. TH has been approved by the FDA and can be administered from the neonatal period, has a good safety profile and in some patients induces diarrhoea (which improves after fractionation of the dose), abdominal pain or vomiting.
- Total MCT+LCT = 20–35% of total energy.
- Physical exercise: Patients with exercise-induced muscle pain or weakness (rhabdomyolysis) benefit 20 min prior to exercise from MCT at a dose of 0.25–0.5 g/kg of ideal weight for height [8,27] or carbohydrate at a dose of 1 g/kg of ideal weight for height or a combination of both at a lower dose. Rest and rehydration periods should be taken if training is long [18].
- Monitor plasma levels of essential fatty acids and fat-soluble vitamins for risk of deficiency. There may be biochemical deficiency without clinical manifestations.
- B
- Medium- and Short-Chain Deficiencies:
- Maintain regular meals, avoiding prolonged fasting especially in the first 6 months of life [18].
- When age permits, introduce foods containing slow sugars [28].
- At present, dietary modifications are not recommended (except during exacerbations), maintaining a normal lipid intake according to WHO recommendations (30–35% of the total caloric value) [28], limiting saturated fatty acids and prioritising extra virgin olive oil and fats from foods such as avocado, nuts, seeds, etc.
- The infant can be breastfed or take the usual infant formulas, checking their possible coconut oil content.
- MCT is contraindicated in medium- and short-chain disorders and in multiple dehydrogenase deficiency (MADD). Modular diets without MCT can be used, taking great care not to ingest medium-chain fatty acids in the normal diet (formulas with MCT or processed products with MCT—mainly coconut).
- Considering that most short-chain acyl-CoA dehydrogenase deficiency (SCADD) patients diagnosed by newborn screening remain asymptomatic, it is suggested that SCADD is more likely a biochemical entity without clinical correlate, and no treatment or chronic management is necessary.
3. Nutritional Measures in Special Situations
3.1. Preoperative
3.2. Home Measures for Possible Decompensation
4. Emergency Hospital Treatment
5. Pharmacological Treatment
- Anaesthetics
- 2
- Other drugs to avoid:
- Pivalic acid, valproic acid, salicylates and acetaminophen due to carnitine consumption.
- Mildronate for decreasing carnitine synthesis and competing with the carnitine transporter (OCTN2).
- Omeprazole, levofloxacin and various antitumour drugs (etoposide, vinblastine, actinomycin D) due to inhibition of OCTN2.
- Statins for increasing OAG.
- Adrenaline for its lipolytic effect [37].
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.I.; Ferdinandusse, S.; Ijlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef]
- Lindner, M.; Hoffmann, G.F.; Matern, D. Newborn cribado for disorders of fatty-acid oxidation: Experience and recomendations from an expert meeting. J. Inherit. Metab. Dis. 2010, 33, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Bowling, F.; Carpenter, K.; Christodoulou, J.; Cowley, D.; Ellaway, C.; Fletcher, J.; et al. Expanded newborn screening: Outcome in screened and unscreened patients at age 6 years. Pediatrics 2009, 124, e241–e248. [Google Scholar] [CrossRef] [PubMed]
- Peña-Quintana, L. Alteraciones de la β-oxidación de los ácidos grasos y del sistema carnitina. In Diagnóstico y Tratamiento de las Enfermedades Metabólicas Hereditarias, 5th ed.; Couce, M.L., Aldámiz-Echevarría, L., Jiménez, C.G., Lamuño, D.G., Eds.; Ergón: Madrid, Spain, 2022; pp. 753–776. [Google Scholar]
- Longo, N.; di San Filippo, C.A.; Pasquali, M. Disorders of Carnitine Transport and the Carnitine Cycle. Am. J. Med. Genet. Part C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sala, P.; Peña-Quintana, L. Biochemical Markers for the Diagnosis of Mitochondrial Fatty Acid Oxidation Diseases. J. Clin. Med. 2021, 10, 4855. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.L.; MacLeod, E.; Jurecka, A.; Hainline, B. Clinical manifestations and management of fatty acid oxidation disorders. Rev. Endocr. Metab. Disord. 2020, 21, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Lindner, M.; Santer, R.; Grotzke, M.; Baumgartner, M.R.; Boehles, H.; Das, A.; Haase, C.; Hennermann, J.B.; Karall, D.; et al. Treatment recommendations in long-chain fatty acid oxidation defects: Consensus from a workshop. J. Inherit. Metab. Dis. 2009, 32, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Peña Quintana, L.; Meavilla Olivas, S.; Egea Castillo, N. Tratamiento nutricional de los EIM de los lípidos. Trastornos de la β-oxidación de ácidos grasos. In Tratamiento Nutricional de los Errores Innatos del Metabolismo, 4th ed.; Ruiz Pons, M., Sánchez-Valverde Visus, F., Vitoria Miñana, I., Eds.; Ergon: Madrid, Spain, 2024; pp. 99–112. [Google Scholar]
- Seung, D. Amylose in starch: Towards an understanding of biosynthesis, structure and function. New Phytol. 2020, 228, 1490–1504. [Google Scholar] [CrossRef]
- Merritt, J.L., II; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef]
- Vockley, J. Long-chain fatty acid oxidation disorders and current management strategies. Am. J. Manag. Care 2020, 26 (Suppl. S7), S147–S154. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Elizondo, G.; Behrend, A.; Matern, D.; Schoeller, D.A.; Harding, C.O.; Purnell, J.Q. Higher dietary protein intake preserves lean body mass, lowers liver lipid deposition, and maintains metabolic control in participants with long-chain fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2019, 42, 857–869. [Google Scholar] [CrossRef] [PubMed]
- DeLany, J.P.; Horgan, A.; Gregor, A.; Vockley, J.; Harding, C.O.; Gillingham, M.B. Resting and Total Energy Expenditure of Patients with Long-chain Fatty Acid Oxidation Disorders (LC-FAODs). Mol. Genet. Metab. 2023, 138, 107519. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, M.B.; Connor, W.E.; Matern, D.; Rinaldo, P.; Burlingame, T.; Meeuws, K.; Harding, C.O. Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2003, 79, 114–123. [Google Scholar] [CrossRef]
- Jones, P.M.; Butt, Y.; Bennett, M.J. Accumulation of 3-hydroxy-fatty acids in the culture medium of long-chain L-3-hydroxyacyl CoA dehydrogenase (LCHAD) and mitochondrial trifunctional protein-deficient skin fibroblasts: Implications for medium chain triglyceride dietary treatment of LCHAD deficiency. Pediatr. Res. 2003, 53, 783–787. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Weleber, R.G.; Neuringer, M.; Connor, W.E.; Mills, M.; van Calcar, S.; Hoeve, J.V.; Wolff, J.; Harding, C.O. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol. Genet. Metab. 2005, 86, 124–133. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Bastin, J.; Gillingham, M.; Morris, A.; Wijburg, F.; Wilcken, B. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2010, 33, 555–561. [Google Scholar] [CrossRef]
- Harding, C.O.; Gillingham, M.B.; van Calcar, S.C.; Wolff, J.A.; Verhoeve, J.N.; Mills, M.D. Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.M.; Skovby, F.; Vestergaard, H.; Christensen, M.; Christensen, E. Clinical and biochemical monitoring of patients with fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2010, 33, 495–500. [Google Scholar] [CrossRef]
- Roe Ch, R.; Sweetman, L.; Roe, D.S.; David, F.; Brunengraber, H. Treatment of cardiomyopathy and rhabdomyolisis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J. Clin. Investig. 2002, 110, 259–269. [Google Scholar] [CrossRef]
- Roe, C.R.; Brunengraber, H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: Review of 15 years Experience. Mol. Genet. Metab. 2015, 116, 260–268. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Heitner, S.B.; Martin, J.; Rose, S.; Goldstein, A.; El-Gharbawy, A.H.; Deward, S.; Lasarev, M.R.; Pollaro, J.; DeLany, J.P.; et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial. J. Inherit. Metab. Dis. 2017, 40, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Burton, B.; Berry, G.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; Humphrey, R.; et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: Safety and efficacy in children and adults following 24weeks of treatment. Mol. Genet. Metab. 2017, 120, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Burton, B.K.; Berry, G.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Chapman, K.A.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; et al. Triheptanoin for the treatment of long-chain fatty acid oxidation disorders: Final results of an open-label, long-term extension study. J. Inherit. Metab. Dis. 2023, 46, 943–955. [Google Scholar] [CrossRef]
- Van Calcar, S.; Sowa, M.; Rohr, F.; Beazer, J.; Setlock, T.; Weihe, T.; Pendyal, S.; Wallace, L.; Hansen, J.; Stembridge, A.; et al. Nutrition management guideline for VLCAD: An evidence- and consensus-based approach. Mol. Genet. Metab. 2020, 131, 23–37. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Scott, B.; Elliott, D.; Harding, C.O. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol. Genet. Metab. 2006, 89, 58–63. [Google Scholar] [CrossRef]
- Feillet, F.; Ogier, H.; Cheillan, D.; Aquaviva, C.; Labarthe, F.; Baruteau, J.; Chabrol, B.; de Lonlay, P.; Valayanopoulos, V.; Garnotel, R.; et al. Medium-chain acyl-CoA-dehydrogenase (MCAD) deficiency: French consensus for neonatal cribado, diagnosis, and management. Arch Pediatr. 2012, 19, 184–193. [Google Scholar] [CrossRef]
- Nasser, M.; Javaheri, H.; Fedorowicz, Z.; Noorani, Z. Carnitine supplementation for inborn errors of metabolism. Cochrane Database Syst. Rev. 2012, 2015, CD006659. [Google Scholar] [CrossRef]
- Vallance, H.; Koochin, A.; Branov, J.; Rosen-Heath, A.; Bosdet, T.; Wang, Z.; Hazen, S.; Horvath, G. Marked elevation in plasma trimethylamine-N-oxide (TMAO) in patients with mitochondrial disorders treated with oral l-carnitine. Mol. Genet. Metab. Rep. 2018, 15, 130–133. [Google Scholar] [CrossRef]
- Magoulas, P.L.; El-Hattab, A.W. Systemic primary carnitine deficiency: An overview of clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2012, 7, 68. [Google Scholar] [CrossRef]
- Shiraishi, H.; Yamada, K.; Oki, E.; Ishige, M.; Fukao, T.; Hamada, Y.; Sakai, N.; Ochi, F.; Watanabe, A.; Kawakami, S.; et al. Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan; 2nd report QOL survey. Mol. Genet. Metab. Rep. 2019, 20, 100496. [Google Scholar] [CrossRef] [PubMed]
- D’annibale, O.M.; Phua, Y.L.; Land, C.V.; Karunanidhi, A.; Dorenbaum, A.; Mohsen, A.-W.; Vockley, J. Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics. Cells 2022, 11, 2635. [Google Scholar] [CrossRef] [PubMed]
- Storgaard, J.H.; Løkken, N.; Madsen, K.L.; Voermans, N.C.; Laforêt, P.; Nadaj-Pakleza, A.; Tard, C.; van Hall, G.; Vissing, J.; Ørngreen, M.C. No effect of resveratrol on fatty acid oxidation or exercise capacity in patients with fatty acid oxidation disorders: A randomized clinical cross-over trial. J. Inherit. Metab. Dis. 2022, 45, 517–528. [Google Scholar] [CrossRef] [PubMed]
- van Rijt, W.J.; Jager, E.A.; Allersma, D.P.; Zeybek, A.A.; Bhattacharya, K.; Debray, F.-G.; Ellaway, C.J.; Gautschi, M.; Geraghty, M.T.; Gil-Ortega, D.; et al. Efficacy and safety of D,L-3-hydroxybutyrate (D,L-3-HB) treatment in multiple acyl-CoA dehydrogenase deficiency. Genet. Med. 2020, 22, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, J.C.; Visser, G.; Clarke, K.; Ferdinandusse, S.; de Haan, F.H.; Houtkooper, R.H.; Ijlst, L.; Kok, I.L.; Langeveld, M.; van der Pol, W.L.; et al. Nutritional ketosis improves exercise metabolism in patients with very long-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2020, 43, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Pico, M.L.C.; García-Villoria, J.; Hernández, E.M.; Peña-Quintana, L.; Félix, L.R.; Ribes, A. Coordinador: Isidro Vitoria Miñana. Protocolo de diagnóstico y tratamiento de las deficiencias de la β-oxidación mitocondrial de los ácidos grasos. In Protocolos de Diagnóstico y Tratamiento de los Errores Congénitos del Metabolismo, 2nd ed.; Ortega, D.G., Ed.; Ergon: Madrid, Spain, 2017; pp. 43–66. [Google Scholar]


{kind=link}
{kind=link}
|
| Age | Overnight Fasting Hours |
|---|---|
| Newborns | 3 |
| <6 months | 4 |
| 6–12 months | 6–8 |
| >1 year | 8–10 |
| Foods | Advised Every Day (<1.5 g/100 g) | Limited Prescribed Amount (1.5–3 g/100 g) | Not Recommended On Prescription Only (>3 g/100 g) |
|---|---|---|---|
| Cereals and flour | Rice, pasta, flour, semolina, tapioca, barley flour, corn, others White loaf bread Cereal porridge-type cereals Corn, wheat or rice breakfast cereals | Quinoa, barley Italian pasta with egg Sliced bread Wholemeal bread Cereals with fibre Brown rice | Bread rusks Low-fat biscuits Oatmeal, bran and germ cereals Viennese pastries, croissants, biscuits, doughnuts, etc. Muesli, breakfast cereals with nuts, fillings, chocolate chips, etc. |
| Milk and dairy products | Skimmed milk and yoghurts Fresh cheeses and spreads, 0% fat | Milkshakes with semi-skimmed milk Semi-skimmed milk | Whole milk, plain, powdered or condensed milk Whole yoghurts Milk shakes with whole milk Cream Cheeses Cream ice creams |
| Fish, shellfish and molluscs | White fish such as haddock, cod, hake, megrim, sole, monkfish, whiting, others. Cockles, clams, squid, cuttlefish, octopus, Norway lobster, prawns. Yellowfin tuna in its natural state with less than 1g of fat x100 tuna. | Oily fish: anchovies, canned tuna Semi-fatty fish: Sea bream, sea bass, turbot Shellfish (mussels, red shrimp) | Oily fish: salmon, red mullet, sardines, halibut, trout Fried, pre-cooked fish Fish roe Fried squid |
| Meat and poultry | Skinless chicken and turkey breast. Rabbit Cooked low-fat ham. Turkey breast (cold meat). | Lean cuts of horse, pork and veal without visible fat Cooked and serrano ham Sausage loin | Pork, veal Fatty sausages, pâtés, sausages, offal. |
| Eggs | Egg white | - | Whole egg |
| Vegetables and root vegetables | All fresh and frozen | - | Pre-cooked or fried |
| Fruits and nuts | All fresh, except avocado and olives | - | Avocado, olives All nuts and dried fruits |
| Legumes | Lentils, beans, peas, broad beans | Chickpeas | Soya beans Soy bean and tofu drink |
| Oils and fats | MCT oil | On medical prescription | Olive/sunflower/soybean/nut oil, butter, lard, palm and coconut oil, margarines, mayonnaise |
| Sweets and desserts | Homemade jams and pastries made with skimmed milk and egg whites | Egg-free vanilla custard | Chocolates, soluble cocoa, marzipan, nougat Desserts made with whole milk, egg, cream Egg custard |
| Spices and sauces | All kinds of herbs, salt, lemon, homemade vegetable stock. Bechamel sauce with skimmed milk | - | Sauces made with cream, mayonnaise, butter, etc. Bechamel sauce with whole milk |
| Disorders of Medium-Chain Fatty Acid Oxidation (MCAD), and Multiple Dehydrogenase Deficiency (MADD). | Disorders of Long-Chain Fatty Acid Oxidation (LC-FAODs) (CPT1, CPT2, CACT, LCHAD/MTP, VLCAD) | ||||||
|---|---|---|---|---|---|---|---|
| Dextrinomaltose (DTM) | Medium-Chain Triglycerides (MCTs) Added to Dextrinomaltose | ||||||
| Age (Years) | DTM (g/100 mL water) | Kcal/mL | Daily Volume | Calories/Day | MCT (mL/100 mL) | Kcal/mL (DTM + MCT) | Calories/Day (DTM + MCT) |
| 0–1 | 10 | 0.4 | 150–200 mL/kg | 60–80 Kcal/Kg | 2 | 0.57 | 85–114 Kcal/g |
| 1–2 | 15 | 0.6 | 100 mL/kg | 60 Kcal/Kg | 2 | 0.77 | 77 Kcal/Kg |
| 2–6 | 20 | 0.8 | 1200–1500 mL | 960–1200 Kcal | 2 | 0.97 | 1165–1455 Kcal/Kg |
| 6–10 | 20 | 0.8 | 1500–2000 mL | 1200–1600 Kcal | 2 | 0.97 | 1455–1940 Kcal/Kg |
| >10 | 25 | 1 | 2000 mL | 2000 Kcal | 2 | 1.17 | 2340 Kcal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Quintana, L.; Correcher-Medina, P. Nutritional Management of Patients with Fatty Acid Oxidation Disorders. Nutrients 2024, 16, 2707. https://doi.org/10.3390/nu16162707
Peña-Quintana L, Correcher-Medina P. Nutritional Management of Patients with Fatty Acid Oxidation Disorders. Nutrients. 2024; 16(16):2707. https://doi.org/10.3390/nu16162707
Chicago/Turabian StylePeña-Quintana, Luis, and Patricia Correcher-Medina. 2024. "Nutritional Management of Patients with Fatty Acid Oxidation Disorders" Nutrients 16, no. 16: 2707. https://doi.org/10.3390/nu16162707
APA StylePeña-Quintana, L., & Correcher-Medina, P. (2024). Nutritional Management of Patients with Fatty Acid Oxidation Disorders. Nutrients, 16(16), 2707. https://doi.org/10.3390/nu16162707