
Role of Folate in Liver Diseases
{kind=link}
{kind=link}
Abstract
1. Introduction
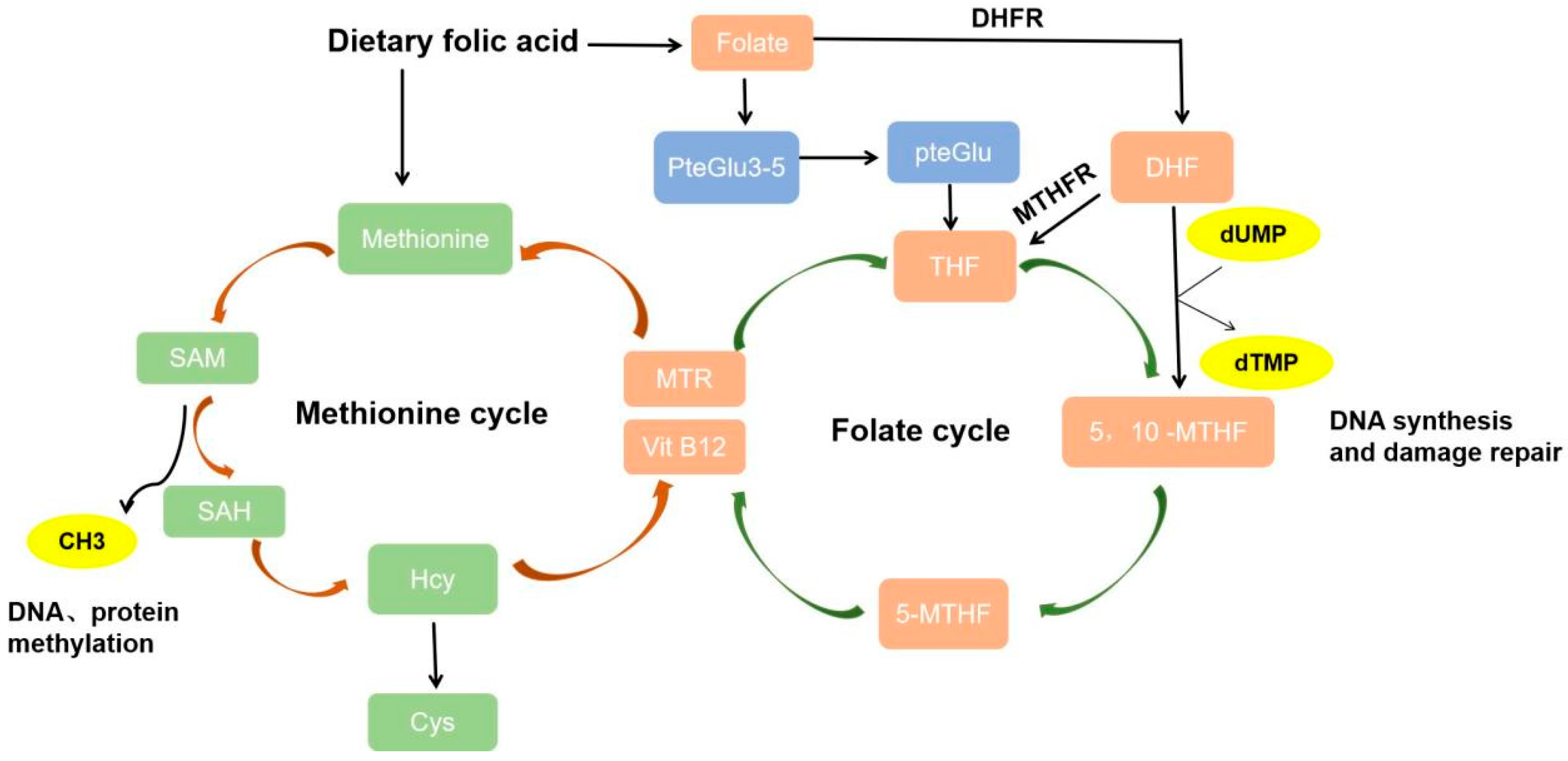
2. Folate Metabolism
2.1. Folate Metabolic Process
2.2. Folate Mediates One-Carbon Metabolism and Promotes DNA Synthesis
2.3. Folate Mediates the Methionine Cycle
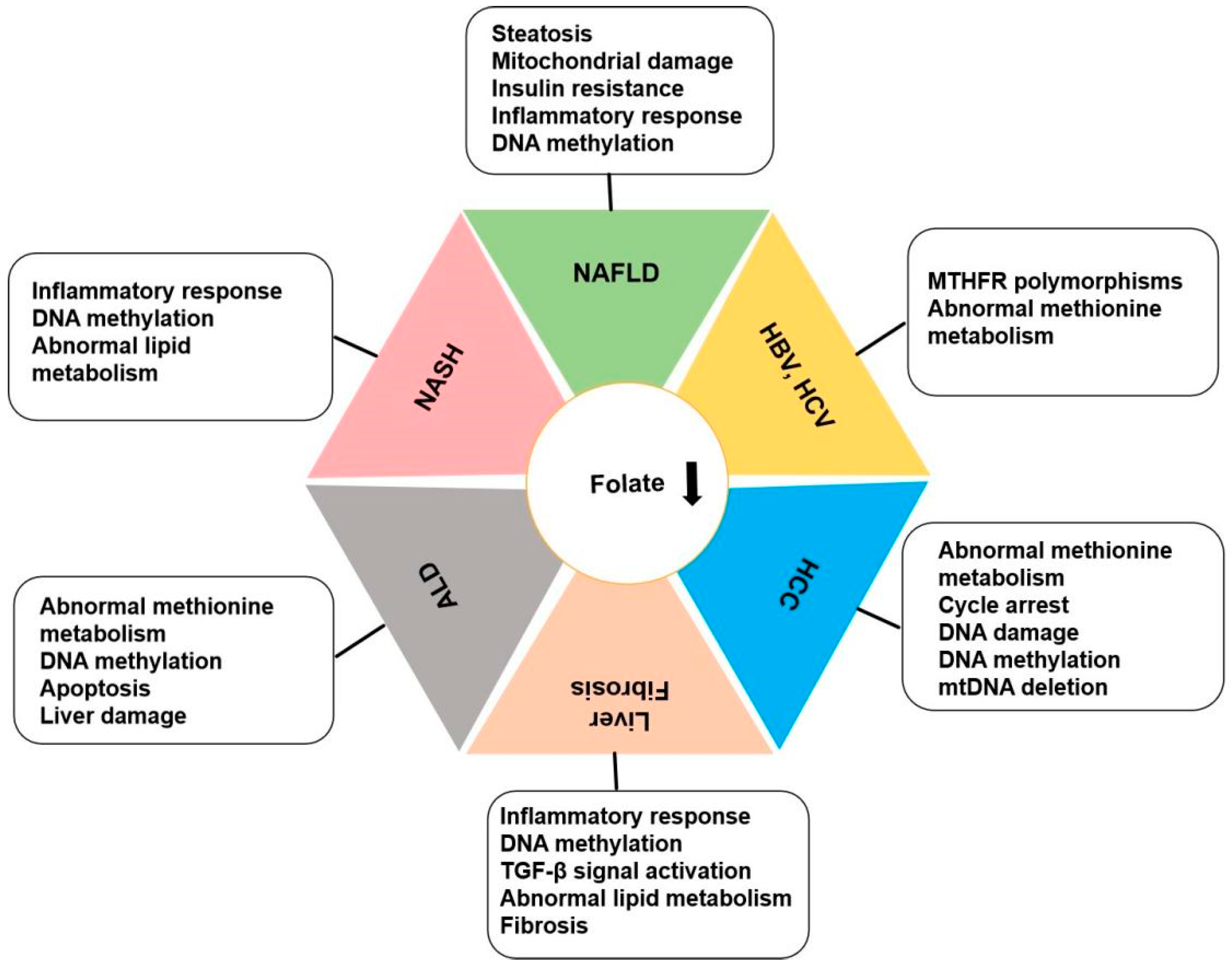
3. Folate and Liver Diseases
3.1. Folate and NAFLD, NASH
3.1.1. Folate Mediates One-Carbon Metabolism to Regulate NAFLD Progression
3.1.2. Folate Mediates DNA Methylation to Regulate NAFLD Progression
3.1.3. The Role of Folate Receptors in NAFLD
3.2. Folate and Alcoholic Liver Disease
3.2.1. Folate Mediates Methionine Metabolism in ALD
3.2.2. Folate Mediates DNA Methylation in ALD
3.3. Folate and Liver Fibrosis and Cirrhosis
3.3.1. Folate Mediates DNA Methylation in Liver Fibrosis
3.3.2. Folate Receptors in Liver Fibrosis
3.3.3. Folate Mediates 1C Metabolism in Liver Fibrosis
3.4. Folate and Chronic Viral Hepatitis
3.4.1. Role of Folate Metabolism in Chronic Viral Hepatitis
3.4.2. Role of Folate in Mediating Methionine Metabolism in Chronic Hepatitis
3.5. Folate and Liver Cancer: Hepatocellular Carcinoma
3.5.1. Folate Is Involved in the Regulation of HCC
3.5.2. Folate Mediates the Methionine Cycle in HCC
3.5.3. Folate Mediates DNA Methylation in HCC
3.5.4. Role of Folate Receptors in HCC
3.5.5. Other Mechanisms
3.6. Therapeutic Manipulation of Folate for Liver Disease Treatment and Management
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wills, L. Treatment of “pernicious anaemia of pregnancy” and “tropical anaemia” with special reference to yeast extract as a curative agent 1931. Natl. Med. J. India 2013, 26, 117–122. [Google Scholar] [PubMed]
- Welch, A.D. Folic acid: Discovery and the exciting first decade. Perspect. Biol. Med. 1983, 27, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, J.; Liao, M.; Yang, Y.; Wang, Y.; Yuan, Y.; Ouyang, L. Folate-mediated one-carbon metabolism: A targeting strategy in cancer therapy. Drug Discov. Today 2021, 26, 817–825. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Ly, A.; Hoyt, L.; Crowell, J.; Kim, Y.I. Folate and DNA methylation. Antioxid. Redox Signal 2012, 17, 302–326. [Google Scholar] [CrossRef] [PubMed]
- Rampersaud, G.C.; Kauwell, G.P.; Hutson, A.D.; Cerda, J.J.; Bailey, L.B. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am. J. Clin. Nutr. 2000, 72, 998–1003. [Google Scholar] [CrossRef]
- Kang, S.S.; Wong, P.W.; Norusis, M. Homocysteinemia due to folate deficiency. Metabolism 1987, 36, 458–462. [Google Scholar] [CrossRef]
- Green, R.; Dwyre, D.M. Evaluation of Macrocytic Anemias. Semin. Hematol. 2015, 52, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Sung, B.; Kim, N.D. Roles of folate in skeletal muscle cell development and functions. Arch. Pharm. Res. 2019, 42, 319–325. [Google Scholar] [CrossRef]
- Medici, V.; Halsted, C.H. Folate, alcohol, and liver disease. Mol. Nutr. Food Res. 2013, 57, 596–606. [Google Scholar] [CrossRef]
- Malaguarnera, G.; Catania, V.E.; Bertino, G.; Drago, F.; Madeddu, R.; Bonfiglio, C.; Malaguarnera, M. Serum Folate deficiency in HCV related Hepatocellular Carcinoma. Sci. Rep. 2022, 12, 5025. [Google Scholar] [CrossRef] [PubMed]
- Youssry, S.; Kamel, M.A. Effect of folate supplementation on immunological and autophagy markers in experimental nonalcoholic fatty liver disease. Eur. Cytokine Netw. 2019, 30, 135–143. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.P.; Kelly, K.B.; Al Rajabi, A.; Jacobs, R.L. Novel insights on interactions between folate and lipid metabolism. Biofactors 2014, 40, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Castano-Moreno, E.; Castillo, V.; Penailillo, R.; Llanos, M.N.; Valenzuela, R.; Ronco, A.M. Fatty acid and lipid metabolism in liver of pregnant mice and their offspring is influenced by unbalanced folates/vitamin B12 diets. Prostaglandins Leukot. Essent. Fat. Acids 2020, 154, 102057. [Google Scholar] [CrossRef]
- Mejos, K.K.; Kim, H.W.; Lim, E.M.; Chang, N. Effects of parental folate deficiency on the folate content, global DNA methylation, and expressions of FRalpha, IGF-2 and IGF-1R in the postnatal rat liver. Nutr. Res. Pract. 2013, 7, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, M.; Mahroum, N.; Bragazzi, N.L.; Shalaata, K.; Yavne, Y.; Adawi, M.; Amital, H.; Watad, A. Folate and B12 Levels Correlate with Histological Severity in NASH Patients. Nutrients 2018, 10, 440. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.K.; Luo, J.; Li, X.J.; Liao, W.Z.; Hu, Y.Q.; Guo, X.G. Serum folate associated with nonalcoholic fatty liver disease and advanced hepatic fibrosis. Sci. Rep. 2023, 13, 12933. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, Q.; Yang, L.; Zhang, R.; Gao, L.; Han, X.; Ji, L.; Zou, X. The Association between Non-Alcoholic Fatty Liver Disease (NAFLD) and Advanced Fibrosis with Serological Vitamin B12 Markers: Results from the NHANES 1999–2004. Nutrients 2022, 14, 1224. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Rasanen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietilainen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571. [Google Scholar] [CrossRef]
- Sid, V.; Siow, Y.L.; O, K. Role of folate in nonalcoholic fatty liver disease. Can. J. Physiol. Pharmacol. 2017, 95, 1141–1148. [Google Scholar] [CrossRef]
- Schalinske, K.L.; Steele, R.D. Methotrexate alters carbon flow through the hepatic folate-dependent one-carbon pool in rats. Carcinogenesis 1996, 17, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O‘Brien, P.J. Mitochondrial function and toxicity: Role of B vitamins on the one-carbon transfer pathways. Chem. Biol. Interact. 2006, 163, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Goh, C.E.; Demmer, R.T.; Whitcomb, B.W.; Du, P.; Liu, Z. Association between Serum Folate and Insulin Resistance among U.S. Nondiabetic Adults. Sci. Rep. 2017, 7, 9187. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.E.; Mikael, L.G.; Leung, K.Y.; Levesque, N.; Deng, L.; Wu, Q.; Malysheva, O.V.; Best, A.; Caudill, M.A.; Greene, N.D.; et al. High folic acid consumption leads to pseudo-MTHFR deficiency, altered lipid metabolism, and liver injury in mice. Am. J. Clin. Nutr. 2015, 101, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, M.; Wang, R.; Fan, X.; Tang, T.; Li, P.; Zhou, X.; Qi, K. Suppression of high-fat-diet-induced obesity in mice by dietary folic acid supplementation is linked to changes in gut microbiota. Eur. J. Nutr. 2022, 61, 2015–2031. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wu, Y.; Peng, H.; Cai, X.; Hu, Z.; Lin, X.; Peng, X.E. Genome-wide DNA methylation profiling in nonalcoholic fatty liver reveals predictive aberrant methylation in PRKCE and SEC14L3 promoters. Dig. Liver Dis. 2022, 54, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.A.; Mavis, A.M.; Jima, D.; Maguire, R.; Bashir, M.; Hyun, J.; Cabezas, M.N.; Parish, A.; Niedzwiecki, D.; Diehl, A.M.; et al. Alterations in DNA methylation associate with fatty liver and metabolic abnormalities in a multi-ethnic cohort of pre-teenage children. Epigenetics-Us 2022, 17, 1446–1461. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; von Schonfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Zhou, B.; Yang, J.; Lu, Y.; Mao, F.; Song, Y. Whole-genome DNA methylation and gene expression profiling in the livers of mice with nonalcoholic steatohepatitis. Life Sci. 2023, 329, 121951. [Google Scholar] [CrossRef]
- Lake, A.D.; Hardwick, R.N.; Leamon, C.P.; Low, P.S.; Cherrington, N.J. Folate receptor-beta expression as a diagnostic target in human & rodent nonalcoholic steatohepatitis. Toxicol. Appl. Pharmacol. 2019, 368, 49–54. [Google Scholar]
- Halsted, C.H.; Villanueva, J.A.; Devlin, A.M.; Chandler, C.J. Metabolic interactions of alcohol and folate. J. Nutr. 2002, 132, 2367S–2372S. [Google Scholar] [CrossRef]
- McClain, C.; Barve, S.; Joshi-Barve, S.; Song, Z.; Deaciuc, I.; Chen, T.; Hill, D. Dysregulated cytokine metabolism, altered hepatic methionine metabolism and proteasome dysfunction in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2005, 29, 180S–188S. [Google Scholar] [CrossRef]
- Halsted, C.H.; Villanueva, J.A.; Devlin, A.M. Folate deficiency, methionine metabolism, and alcoholic liver disease. Alcohol 2002, 27, 169–172. [Google Scholar] [CrossRef]
- Halsted, C.H. B-Vitamin dependent methionine metabolism and alcoholic liver disease. Clin. Chem. Lab. Med. 2013, 51, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, Y.A.; Cubero, F.J.; Hu, W.; Hao, F.; Haas, U.; Ramadori, P.; Gassler, N.; Hoss, M.; Strnad, P.; Zimmermann, H.W.; et al. Enhanced expression of c-myc in hepatocytes promotes initiation and progression of alcoholic liver disease. J. Hepatol. 2016, 64, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Chen, K.D.; Lo, M.H.; Cai, X.Y.; Chang, L.S.; Kuo, Y.H.; Huang, W.D.; Kuo, H.C. Decreased DNA methyltransferases expression is associated with coronary artery lesion formation in Kawasaki disease. Int. J. Med. Sci. 2019, 16, 576–582. [Google Scholar] [CrossRef]
- Chater-Diehl, E.J.; Laufer, B.I.; Castellani, C.A.; Alberry, B.L.; Singh, S.M. Alteration of Gene Expression, DNA Methylation, and Histone Methylation in Free Radical Scavenging Networks in Adult Mouse Hippocampus following Fetal Alcohol Exposure. PLoS ONE 2016, 11, e0154836. [Google Scholar] [CrossRef]
- Hao, F.; Cubero, F.J.; Ramadori, P.; Liao, L.; Haas, U.; Lambertz, D.; Sonntag, R.; Bangen, J.M.; Gassler, N.; Hoss, M.; et al. Inhibition of Caspase-8 does not protect from alcohol-induced liver apoptosis but alleviates alcoholic hepatic steatosis in mice. Cell Death Dis. 2017, 8, e3152. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; Thomes, P.G.; Orlicky, D.J.; Osna, N.A.; French, S.W.; Tuma, D.J. Increased methylation demand exacerbates ethanol-induced liver injury. Exp. Mol. Pathol. 2014, 97, 49–56. [Google Scholar] [CrossRef]
- Zhao, H.; Gao, H.; Zhang, Y.; Lan, T.; Wang, J.; Zhao, H.; Zhang, H.; Xue, M.; Liang, H. Folic Acid Protects against Ethanol-Induced Hepatic Mitophagy Imbalance by ROS Scavenging and Attenuating the Elevated Hcy Levels. J. Agric. Food Chem. 2023, 71, 14276–14288. [Google Scholar] [CrossRef]
- Page, A.; Paoli, P.; Moran Salvador, E.; White, S.; French, J.; Mann, J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J. Hepatol. 2016, 64, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Kicheva, A.; Ribeiro, A.; Blassberg, R.; Page, K.M.; Barnes, C.P.; Briscoe, J. Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat. Commun. 2015, 6, 6709. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Oakley, F.; Akiboye, F.; Elsharkawy, A.; Thorne, A.W.; Mann, D.A. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: Implications for wound healing and fibrogenesis. Cell Death Differ. 2007, 14, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zheng, B.; Xu, S.; Zhao, Z.; Liu, W.; Wang, T.; Yuan, M.; Sun, X.; Tan, Y.; Xu, Q.; et al. Mitochondrial folate metabolism-mediated alpha-linolenic acid exhaustion masks liver fibrosis resolution. J. Biol. Chem. 2023, 299, 104909. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.; Rico, M.C.; Merali, C.; Barrero, C.A.; Perez-Leal, O.; Mischley, V.; Karanicolas, J.; Friedman, S.L.; Merali, S. Secreted folate receptor gamma drives fibrogenesis in metabolic dysfunction-associated steatohepatitis by amplifying TGFbeta signaling in hepatic stellate cells. Sci. Transl. Med. 2023, 15, eade2966. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Singh, B.K.; Zhou, J.; Tikno, K.; Widjaja, A.; Sandireddy, R.; Arul, K.; Abdul Ghani, S.A.B.; Bee, G.G.B.; Wong, K.A.; et al. Vitamin B(12) and folate decrease inflammation and fibrosis in NASH by preventing syntaxin 17 homocysteinylation. J. Hepatol. 2022, 77, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Liu, W.H.; Wang, Z.H.; Yin, M.C. Vitamins B status and antioxidative defense in patients with chronic hepatitis B or hepatitis C virus infection. Eur. J. Nutr. 2011, 50, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Qiao, K.; Zhang, S.; Trieu, C.; Dai, Q.; Huo, Z.; Du, Y.; Lu, W.; Hou, W. Genetic Polymorphism of MTHFR C677T Influences Susceptibility to HBV-Related Hepatocellular Carcinoma in a Chinese Population: A Case-Control Study. Clin. Lab. 2017, 63, 787–795. [Google Scholar] [CrossRef] [PubMed]
- ElDeeb, M.K.; Ghazal, A.A.; Metwally, D.E.; Elghlied, L.A. Possible roles of methylenetetrahydrofolate reductase polymorphism and folate status in patients with early hepatitis C virus genotype 4. Arab. J. Gastroenterol. 2021, 22, 121–126. [Google Scholar] [CrossRef]
- Jiao, X.; Luo, Y.; Yang, B.; Jing, L.; Li, Y.; Liu, C.; Jing, X.; Wang, F.; Wang, Y.; Du, Z.; et al. The MTHFR C677T mutation is not a risk factor recognized for HBV-related HCC in a population with a high prevalence of this genetic marker. Infect. Genet. Evol. 2017, 49, 66–72. [Google Scholar] [CrossRef]
- Li, Z.; Wang, F.; Liang, B.; Su, Y.; Sun, S.; Xia, S.; Shao, J.; Zhang, Z.; Hong, M.; Zhang, F.; et al. Methionine metabolism in chronic liver diseases: An update on molecular mechanism and therapeutic implication. Signal Transduct. Target. Ther. 2020, 5, 280. [Google Scholar] [CrossRef] [PubMed]
- Key, T.J.; Bradbury, K.E.; Perez-Cornago, A.; Sinha, R.; Tsilidis, K.K.; Tsugane, S. Diet, nutrition, and cancer risk: What do we know and what is the way forward? BMJ 2020, 368, m511. [Google Scholar] [CrossRef] [PubMed]
- Biselli, M.; Reggidori, N.; Iavarone, M.; Renzulli, M.; Lani, L.; Granito, A.; Piscaglia, F.; Lorenzini, S.; Alimenti, E.; Vara, G.; et al. Impact of Sarcopenia on the Survival of Patients with Hepatocellular Carcinoma Treated with Sorafenib. Cancers 2024, 16, 1080. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Chanarin, I.; Slavin, G.; Levi, A.J. Folate deficiency in the alcoholic--its relationship to clinical and haematological abnormalities, liver disease and folate stores. Br. J. Haematol. 1975, 29, 469–478. [Google Scholar] [CrossRef]
- Jiang, Y.; Cao, H.; Chen, X.; Yu, G.; Song, C.; Duan, H.; Tian, F.; Wan, H.; Shen, J. Associations of serum folate and vitamin C levels with metabolic dysfunction-associated fatty liver disease in US adults: A nationwide cross-sectional study. Front. Public Health 2022, 10, 1022928. [Google Scholar] [CrossRef]
- Areekul, S.; Hitanant, S.; Panatampon, P.; Chantachum, Y. Serum vitamin B12 and folate levels, vitamin B12 and folic acid binding proteins in patients with primary carcinoma of the liver. Southeast. Asian J. Trop. Med. Public Health 1977, 8, 519–524. [Google Scholar] [PubMed]
- Cui, L.H.; Quan, Z.Y.; Piao, J.M.; Zhang, T.T.; Jiang, M.H.; Shin, M.H.; Choi, J.S. Plasma Folate and Vitamin B12 Levels in Patients with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2016, 17, 1032. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.S.; Lin, C.Y.; Wu, M.Y.; Lu, C.L.; Huang, R.F. Relationship between folate status and tumour progression in patients with hepatocellular carcinoma. Br. J. Nutr. 2008, 100, 596–602. [Google Scholar] [CrossRef]
- Fang, A.P.; Liu, Z.Y.; Liao, G.C.; Chen, P.Y.; Wang, X.Y.; Zhang, D.M.; Luo, Y.; Long, J.A.; Zhong, R.H.; Zhou, Z.G.; et al. Serum folate concentrations at diagnosis are associated with hepatocellular carcinoma survival in the Guangdong Liver Cancer Cohort study. Br. J. Nutr. 2019, 121, 1376–1388. [Google Scholar] [CrossRef]
- Persson, E.C.; Schwartz, L.M.; Park, Y.; Trabert, B.; Hollenbeck, A.R.; Graubard, B.I.; Freedman, N.D.; McGlynn, K.A. Alcohol consumption, folate intake, hepatocellular carcinoma, and liver disease mortality. Cancer Epidemiol. Biomark. Prev. 2013, 22, 415–421. [Google Scholar] [CrossRef]
- Li, J.T.; Yang, H.; Lei, M.Z.; Zhu, W.P.; Su, Y.; Li, K.Y.; Zhu, W.Y.; Wang, J.; Zhang, L.; Qu, J.; et al. Dietary folate drives methionine metabolism to promote cancer development by stabilizing MAT IIA. Signal Transduct. Target. Ther. 2022, 7, 192. [Google Scholar] [CrossRef]
- Ho, C.T.; Shang, H.S.; Chang, J.B.; Liu, J.J.; Liu, T.Z. Folate deficiency-triggered redox pathways confer drug resistance in hepatocellular carcinoma. Oncotarget 2015, 6, 26104–26118. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Ali, T.; Negi, I.; Das, A.; Duseja, A.; Kaur, J. Dietary modulations of folic acid affect the development of diethylnitrosamine induced hepatocellular carcinoma in a rat model. J. Mol. Histol. 2021, 52, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, P.; Mi, W.; Li, L.; Anderson, N.M.; Lesner, N.P.; Burrows, M.; Plesset, J.; Majer, A.; Wang, G.; et al. Blocking methionine catabolism induces senescence and confers vulnerability to GSK3 inhibition in liver cancer. Nat. Cancer 2024, 5, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Peng, J.X.; Hao, X.Y.; Cai, J.P.; Liang, L.J.; Zhai, J.M.; Zhang, K.S.; Lai, J.M.; Yin, X.Y. DNA methylation profiling identifies EYA4 gene as a prognostic molecular marker in hepatocellular carcinoma. Ann. Surg. Oncol. 2014, 21, 3891–3899. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Cheng, X.; Gong, M.; Chen, X.; Yin, F.; Lai, K. Hypermethylation and Expression Silencing of PDCD4 Gene in Hepatocellular Carcinoma: A Consort Study. Medicine (Baltimore) 2016, 95, e2729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, J.; Huang, T.; Duan, S.; Dai, D.; Jiang, D.; Sui, X.; Li, D.; Chen, Y.; Ding, F.; et al. Meta-analysis of DNA methylation biomarkers in hepatocellular carcinoma. Oncotarget 2016, 7, 81255–81267. [Google Scholar] [CrossRef]
- Chen, G.; Fan, X.; Li, Y.; He, L.; Wang, S.; Dai, Y.; Bin, C.; Zhou, D.; Lin, H. Promoter aberrant methylation status of ADRA1A is associated with hepatocellular carcinoma. Epigenetics-Us 2020, 15, 684–701. [Google Scholar] [CrossRef]
- Ling, Y.H.; Liu, M.N.; Yin, Y.X.; Zhou, Z.G.; Chen, J.W.; Wei, W.; Yun, J.P.; Xie, D.; Guo, R.P.; Cai, M.Y. Integrated genetic and epigenetic analysis reveals DNA repair alterations in multifocal hepatocellular carcinoma. Signal Transduct. Target. Ther. 2023, 8, 244. [Google Scholar] [CrossRef]
- Sharma, R.; Ali, T.; Kaur, J. Tumor suppressor genes are differentially regulated with dietary folate modulations in a rat model of hepatocellular carcinoma. Mol. Cell Biochem. 2021, 476, 385–399. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.; Serna, S.; Yang, Z.; Delso, I.; Taleb, V.; Hicks, T.; Artschwager, R.; Vakhrushev, S.Y.; Clausen, H.; Angulo, J.; et al. FUT8-Directed Core Fucosylation of N-glycans Is Regulated by the Glycan Structure and Protein Environment. ACS Catal. 2021, 11, 9052–9065. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, T.; Guan, J.; Liu, X.; Wang, J.; Jin, P.; Hou, S.; Lu, W.; Qian, J.; Wang, W.; et al. Interrogation of Folic Acid-Functionalized Nanomedicines: The Regulatory Roles of Plasma Proteins Reexamined. ACS Nano 2020, 14, 14779–14789. [Google Scholar] [CrossRef]
- Koirala, N.; Das, D.; Fayazzadeh, E.; Sen, S.; McClain, A.; Puskas, J.E.; Drazba, J.A.; McLennan, G. Folic acid conjugated polymeric drug delivery vehicle for targeted cancer detection in hepatocellular carcinoma. J. Biomed. Mater. Res. A 2019, 107, 2522–2535. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Wang, X.; Ma, Q.; Li, J.; Li, M.; Huang, Y.; Liang, W.; Su, D.; Guo, R. Triformyl cholic acid and folic acid functionalized magnetic graphene oxide nanocomposites: Multiple-targeted dual-modal synergistic chemotherapy/photothermal therapy for liver cancer. J. Inorg. Biochem. 2021, 223, 111558. [Google Scholar] [CrossRef] [PubMed]
- Guariento, A.H.; Furtado, K.S.; de Conti, A.; Campos, A.; Purgatto, E.; Carrilho, J.; Shinohara, E.M.; Tryndyak, V.; Han, T.; Fuscoe, J.C.; et al. Transcriptomic responses provide a new mechanistic basis for the chemopreventive effects of folic acid and tributyrin in rat liver carcinogenesis. Int. J. Cancer 2014, 135, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Kuo, C.S.; Lin, C.Y.; Lu, C.L.; Syu Huang, R.F. Lymphocytic mitochondrial DNA deletions, biochemical folate status and hepatocellular carcinoma susceptibility in a case-control study. Br. J. Nutr. 2009, 102, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Min, H. Folic acid supplementation prevents high fructose-induced non-alcoholic fatty liver disease by activating the AMPK and LKB1 signaling pathways. Nutr. Res. Pract. 2020, 14, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Molaqanbari, M.R.; Zarringol, S.; Talari, H.R.; Taghizadeh, M.; Bahmani, F.; Mohtashamian, A.; Ebrahimzadeh, A.; Sharifi, N. Effects of Folic Acid Supplementation on Liver Enzymes, Lipid Profile, and Insulin Resistance in Patients with Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Adv. Biomed. Res. 2023, 12, 103. [Google Scholar]
- Sid, V.; Shang, Y.; Siow, Y.L.; Hewage, S.M.; House, J.D.; O, K. Folic Acid Supplementation Attenuates Chronic Hepatic Inflammation in High-Fat Diet Fed Mice. Lipids 2018, 53, 709–716. [Google Scholar] [CrossRef]
- Zhao, H.; Guo, P.; Zuo, Y.; Wang, Y.; Zhao, H.; Lan, T.; Xue, M.; Zhang, H.; Liang, H. Folic acid intervention changes liver Foxp3 methylation and ameliorates the damage caused by Th17/Treg imbalance after long-term alcohol exposure. Food Funct. 2022, 13, 5262–5274. [Google Scholar] [CrossRef]
- Lu, H.; Cao, W.; Zhang, L.; Yang, L.; Bi, X.; Lin, Y.; Deng, W.; Jiang, T.; Sun, F.; Zeng, Z.; et al. Effects of hepatitis B virus infection and strategies for preventing mother-to-child transmission on maternal and fetal T-cell immunity. Front. Immunol. 2023, 14, 1122048. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lian, J.; Yi, L.; Yao, T.; Feng, S.; Wang, B.; Li, J.; Wang, S.; Feng, Y. Folic acid supplementation in pregnant women with hepatitis B surface antigen improves infant hepatitis B surface antibody mediated by infant IL-4. Br. J. Nutr. 2023, 129, 1812–1819. [Google Scholar] [CrossRef]
- Huang, M.; Cheng, L.; Mo, S.; Ru, H.; Mo, X.; Yan, L. Evaluation of colorectal cancer liver metastases based on liquid biopsy combined with folate receptor- Positive circulating tumor cells and HSP90. Front. Oncol. 2022, 12, 912016. [Google Scholar] [CrossRef]
- Chen, Q.; Meng, X.; McQuade, P.; Rubins, D.; Lin, S.A.; Zeng, Z.; Haley, H.; Miller, P.; Gonzalez Trotter, D.; Low, P.S. Folate-PEG-NOTA-Al(18)F: A New Folate Based Radiotracer for PET Imaging of Folate Receptor-Positive Tumors. Mol. Pharm. 2017, 14, 4353–4361. [Google Scholar] [CrossRef] [PubMed]
- Radford, L.L.; Fernandez, S.; Beacham, R.; El Sayed, R.; Farkas, R.; Benesova, M.; Muller, C.; Lapi, S.E. New (55)Co-labeled Albumin-Binding Folate Derivatives as Potential PET Agents for Folate Receptor Imaging. Pharmaceuticals 2019, 12, 166. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Wang, D.; Wang, X.; Mei, J.; Gong, Q. Role of Folate in Liver Diseases. Nutrients 2024, 16, 1872. https://doi.org/10.3390/nu16121872
Yang M, Wang D, Wang X, Mei J, Gong Q. Role of Folate in Liver Diseases. Nutrients. 2024; 16(12):1872. https://doi.org/10.3390/nu16121872
Chicago/Turabian StyleYang, Minlan, Dingye Wang, Xiyuan Wang, Jie Mei, and Quan Gong. 2024. "Role of Folate in Liver Diseases" Nutrients 16, no. 12: 1872. https://doi.org/10.3390/nu16121872
APA StyleYang, M., Wang, D., Wang, X., Mei, J., & Gong, Q. (2024). Role of Folate in Liver Diseases. Nutrients, 16(12), 1872. https://doi.org/10.3390/nu16121872