
Zinc and Central Nervous System Disorders
Abstract
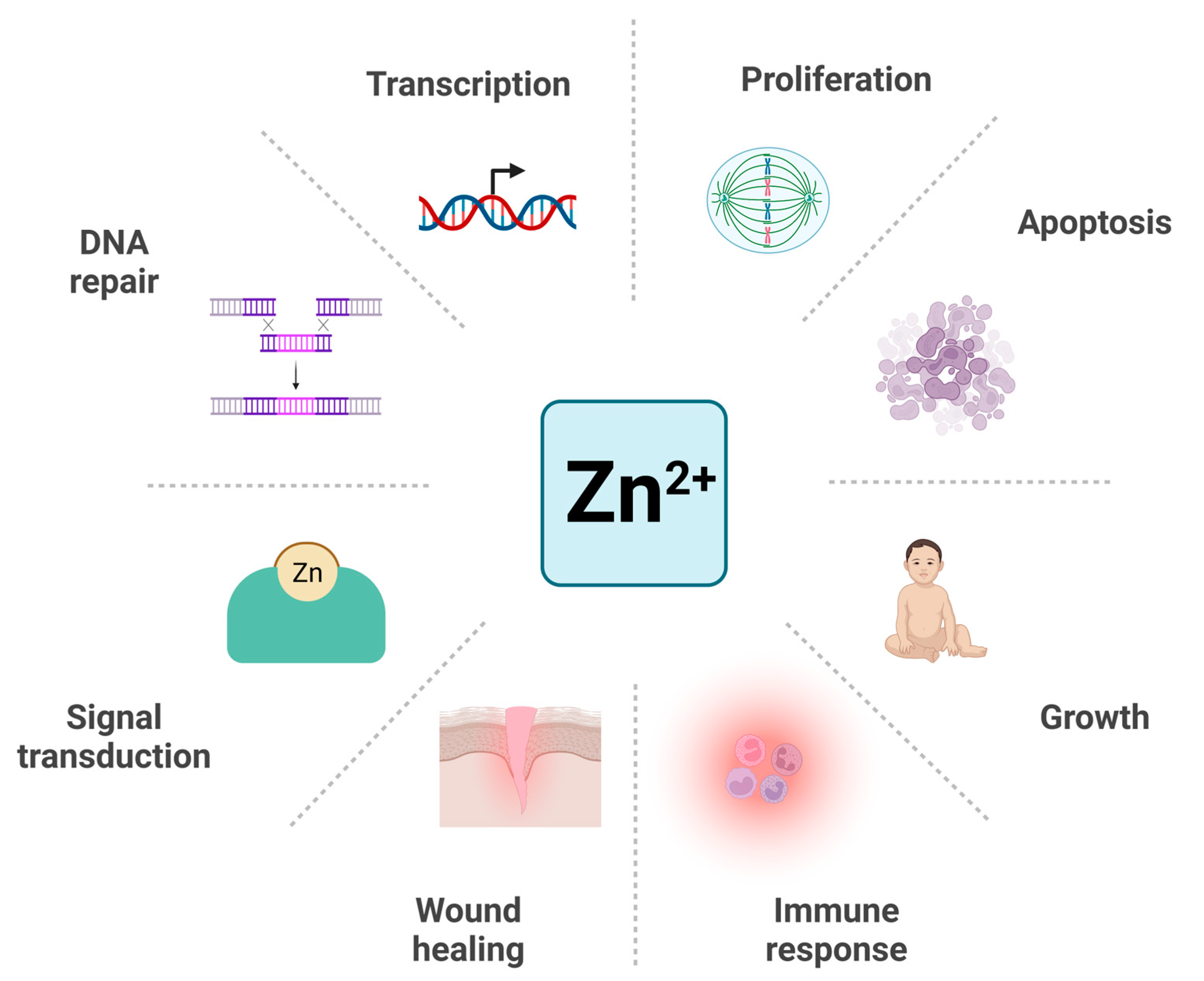
1. Introduction
2. Zn2+ in the CNS
3. Zn2+ and CNS Diseases
3.1. Zn2+ and Alzheimer’s Disease
3.2. Zn2+ and Depression
3.3. Zn2+ and Parkinson’s Disease
3.4. Zn2+ and Multiple Sclerosis
3.5. Zn2+ and Schizophrenia
3.6. Zn2+ and Epilepsy
3.7. Zn2+ and Traumatic Brain Injury
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [CrossRef] [PubMed]
- Rink, L.; Haase, H. Zinc homeostasis and immunity. Trends Immunol. 2007, 28, 1–4. [Google Scholar] [CrossRef] [PubMed]
- O’Halloran, T.V. Transition metals in control of gene expression. Science 1993, 261, 715–725. [Google Scholar] [CrossRef]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef]
- Hambidge, M. Human zinc deficiency. J. Nutr. 2000, 130, 1344S–1349S. [Google Scholar] [CrossRef]
- Sandstead, H.H. Subclinical zinc deficiency impairs human brain function. J. Trace Elem. Med. Biol. 2012, 26, 70–73. [Google Scholar] [CrossRef]
- Kawahara, M.; Tanaka, K.I.; Kato-Negishi, M. Zinc, Carnosine, and Neurodegenerative Diseases. Nutrients 2018, 10, 147. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kinoshita, M.; Shimada, S.; Kawamura, T. Zinc and Skin Disorders. Nutrients 2018, 10, 199. [Google Scholar] [CrossRef]
- Wastney, M.E.; Aamodt, R.L.; Rumble, W.F.; Henkin, R.I. Kinetic analysis of zinc metabolism and its regulation in normal humans. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1986, 251, R398–R408. [Google Scholar] [CrossRef]
- Solomons, N.W. Update on zinc biology. Ann. Nutr. Metab. 2013, 62 (Suppl. 1), 8–17. [Google Scholar] [CrossRef]
- World Health Organization. Zinc Supplementation and Growth in Children: Biological, Behavioural and Contextual Rationale. Available online: http://www.who.int/elena/bbc/zinc_stunting/en/ (accessed on 15 November 2022).
- Heintschel, M.; Heuberger, R. The Potential Role of Zinc Supplementation on Pressure Injury Healing in Older Adults: A Review of the Literature. Wounds-Compend. Clin. Res. Pract. 2017, 29, 56–61. [Google Scholar]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef]
- Krall, R.F.; Tzounopoulos, T.; Aizenman, E. The Function and Regulation of Zinc in the Brain. Neuroscience 2021, 457, 235–258. [Google Scholar] [CrossRef]
- Nuttall, J.R.; Oteiza, P.I. Zinc and the aging brain. Genes Nutr. 2014, 9, 379. [Google Scholar] [CrossRef]
- Koh, J.Y. Zinc and disease of the brain. Mol. Neurobiol. 2001, 24, 99–106. [Google Scholar] [CrossRef]
- Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 2013, 5, 33. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, A.; Singh, K.; Avasthi, K.; Kim, J.J. Neurobiology of zinc and its role in neurogenesis. Eur. J. Nutr. 2021, 60, 55–64. [Google Scholar] [CrossRef]
- Smart, T.G.; Hosie, A.M.; Miller, P.S. Zn2+ ions: Modulators of excitatory and inhibitory synaptic activity. Neuroscientist 2004, 10, 432–442. [Google Scholar] [CrossRef]
- Westbrook, G.L.; Mayer, M.L. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature 1987, 328, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Yin, H.Z.; Carriedo, S.G.; Rao, S.S.; Weiss, J.H. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc. Natl. Acad. Sci. USA 1999, 96, 2414–2419. [Google Scholar] [CrossRef] [PubMed]
- Kodirov, S.A.; Takizawa, S.; Joseph, J.; Kandel, E.R.; Shumyatsky, G.P.; Bolshakov, V.Y. Synaptically released zinc gates long-term potentiation in fear conditioning pathways. Proc. Natl. Acad. Sci. USA 2006, 103, 15218–15223. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Nakamura, M.; Fujii, H.; Tamano, H. Synaptic Zn2+ homeostasis and its significance. Metallomics 2013, 5, 417–423. [Google Scholar] [CrossRef]
- Patrick, W.H.; Dyck, R.H. Signaling by Synaptic Zinc is Required for Whisker-Mediated, Fine Texture Discrimination. Neuroscience 2018, 369, 242–247. [Google Scholar] [CrossRef]
- Barr, C.A.; Burdette, S.C. The zinc paradigm for metalloneurochemistry. Essays Biochem. 2017, 61, 225–235. [Google Scholar]
- Takeda, A.; Tamano, H.; Tochigi, M.; Oku, N. Zinc homeostasis in the hippocampus of zinc-deficient young adult rats. Neurochem. Int. 2005, 46, 221–225. [Google Scholar] [CrossRef]
- Choi, B.Y.; Hong, D.K.; Jeong, J.H.; Lee, B.E.; Koh, J.Y.; Suh, S.W. Zinc transporter 3 modulates cell proliferation and neuronal differentiation in the adult hippocampus. Stem Cells 2020, 38, 994–1006. [Google Scholar] [CrossRef]
- Keller, K.A.; Grider, A.; Coffield, J.A. Age-dependent influence of dietary zinc restriction on short-term memory in male rats. Physiol. Behav. 2001, 72, 339–348. [Google Scholar] [CrossRef]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxid. Med. Cell. Longev. 2018, 2018, 9156285. [Google Scholar] [CrossRef]
- Tonder, N.; Johansen, F.F.; Frederickson, C.J.; Zimmer, J.; Diemer, N.H. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci. Lett. 1990, 109, 247–252. [Google Scholar] [CrossRef]
- Li, Y.; Hough, C.J.; Suh, S.W.; Sarvey, J.M.; Frederickson, C.J. Rapid translocation of Zn2+ from presynaptic terminals into postsynaptic hippocampal neurons after physiological stimulation. J. Neurophysiol. 2001, 86, 2597–2604. [Google Scholar] [CrossRef]
- Suh, S.W.; Thompson, R.B.; Frederickson, C.J. Loss of vesicular zinc and appearance of perikaryal zinc after seizures induced by pilocarpine. Neuroreport 2001, 12, 1523–1525. [Google Scholar] [CrossRef]
- Li, M.S.; Adesina, S.E.; Ellis, C.L.; Gooch, J.L.; Hoover, R.S.; Williams, C.R. NADPH oxidase-2 mediates zinc deficiency-induced oxidative stress and kidney damage. Am. J. Physiol.-Cell Physiol. 2017, 312, C47–C55. [Google Scholar] [CrossRef]
- Choi, S.; Hong, D.K.; Choi, B.Y.; Suh, S.W. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar] [CrossRef]
- Maret, W.; Krezel, A. Cellular zinc and redox buffering capacity of metallothionein/thionein in health and disease. Mol. Med. 2007, 13, 371–375. [Google Scholar] [CrossRef]
- Quaife, C.J.; Findley, S.D.; Erickson, J.C.; Froelick, G.J.; Kelly, E.J.; Zambrowicz, B.P.; Palmiter, R.D. Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry 1994, 33, 7250–7259. [Google Scholar] [CrossRef]
- Manso, Y.; Adlard, P.A.; Carrasco, J.; Vasak, M.; Hidalgo, J. Metallothionein and brain inflammation. J. Biol. Inorg. Chem. 2011, 16, 1103–1113. [Google Scholar] [CrossRef]
- Thirumoorthy, N.; Shyam, S.A.; Manisenthil, K.K.; Senthil, K.M.; Ganesh, G.; Chatterjee, M. A review of metallothionein isoforms and their role in pathophysiology. World J. Surg. Oncol. 2011, 9, 54. [Google Scholar] [CrossRef]
- Sato, M.; Kondoh, M. Recent studies on metallothionein: Protection against toxicity of heavy metals and oxygen free radicals. Tohoku J. Exp. Med. 2002, 196, 9–22. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Ronnstrand, L.; Rosenberg, P.A. Why and how to investigate the role of protein phosphorylation in ZIP and ZnT zinc transporter activity and regulation. Cell. Mol. Life Sci. 2020, 77, 3085–3102. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Findley, S.D. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. Embo J. 1995, 14, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Robbins, C.A.; Wenzel, H.J.; Schwartzkroin, P.A.; Palmiter, R.D. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000, 39, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Olesen, R.H.; Hyde, T.M.; Kleinman, J.E.; Smidt, K.; Rungby, J.; Larsen, A. Obesity and age-related alterations in the gene expression of zinc-transporter proteins in the human brain. Transl. Psychiatr. 2016, 6, e838. [Google Scholar] [CrossRef]
- Beyer, N.; Coulson, D.T.; Heggarty, S.; Ravid, R.; Irvine, G.B.; Hellemans, J.; Johnston, J.A. ZnT3 mRNA levels are reduced in Alzheimer’s disease post-mortem brain. Mol. Neurodegener. 2009, 4, 53. [Google Scholar] [CrossRef]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef]
- Huang, L.; Tepaamorndech, S. The SLC30 family of zinc transporters—A review of current understanding of their biological and pathophysiological roles. Mol. Asp. Med. 2013, 34, 548–560. [Google Scholar] [CrossRef]
- Lichten, L.A.; Cousins, R.J. Mammalian zinc transporters: Nutritional and physiologic regulation. Annu. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef]
- Jeong, J.; Eide, D.J. The SLC39 family of zinc transporters. Mol. Asp. Med. 2013, 34, 612–619. [Google Scholar] [CrossRef]
- Wang, F.; Dufner-Beattie, J.; Kim, B.E.; Petris, M.J.; Andrews, G.; Eide, D.J. Zinc-stimulated endocytosis controls activity of the mouse ZIP1 and ZIP3 zinc uptake transporters. J. Biol. Chem. 2004, 279, 24631–24639. [Google Scholar] [CrossRef]
- Boycott, K.M.; Beaulieu, C.L.; Kernohan, K.D.; Gebril, O.H.; Mhanni, A.; Chudley, A.E.; Redl, D.; Qin, W.; Hampson, S.; Kury, S.; et al. Autosomal-Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8. Am. J. Hum. Genet. 2015, 97, 886–893. [Google Scholar] [CrossRef]
- Baum, L.; Chan, I.H.; Cheung, S.K.; Goggins, W.B.; Mok, V.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; Woo, J.; et al. Serum zinc is decreased in Alzheimer’s disease and serum arsenic correlates positively with cognitive ability. Biometals 2010, 23, 173–179. [Google Scholar] [CrossRef]
- Wang, Z.X.; Tan, L.; Wang, H.F.; Ma, J.; Liu, J.; Tan, M.S.; Sun, J.H.; Zhu, X.C.; Jiang, T.; Yu, J.T. Serum Iron, Zinc, and Copper Levels in Patients with Alzheimer’s Disease: A Replication Study and Meta-Analyses. J. Alzheimers Dis. 2015, 47, 565–581. [Google Scholar] [CrossRef]
- Corona, C.; Masciopinto, F.; Silvestri, E.; Viscovo, A.D.; Lattanzio, R.; Sorda, R.L.; Ciavardelli, D.; Goglia, F.; Piantelli, M.; Canzoniero, L.M.; et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 2010, 1, e91. [Google Scholar] [CrossRef]
- Loef, M.; von Stillfried, N.; Walach, H. Zinc diet and Alzheimer’s disease: A systematic review. Nutr. Neurosci. 2012, 15, 2–12. [Google Scholar] [CrossRef]
- Maes, M.; D’Haese, P.C.; Scharpe, S.; D’Hondt, P.; Cosyns, P.; De Broe, M.E. Hypozincemia in depression. J. Affect. Disord. 1994, 31, 135–140. [Google Scholar] [CrossRef]
- Lai, J.; Moxey, A.; Nowak, G.; Vashum, K.; Bailey, K.; McEvoy, M. The efficacy of zinc supplementation in depression: Systematic review of randomised controlled trials. J. Affect. Disord. 2012, 136, e31–e39. [Google Scholar] [CrossRef]
- Yosaee, S.; Clark, C.; Keshtkaran, Z.; Ashourpour, M.; Keshani, P.; Soltani, S. Zinc in depression: From development to treatment: A comparative/dose response meta-analysis of observational studies and randomized controlled trials. Gen. Hosp. Psychiatry 2022, 74, 110–117. [Google Scholar] [CrossRef]
- Aoki, C.; Imai, K.; Owaki, T.; Kobayashi-Nakano, T.; Ushida, T.; Iitani, Y.; Nakamura, N.; Kajiyama, H.; Kotani, T. The Possible Effects of Zinc Supplementation on Postpartum Depression and Anemia. Med. Lith. 2022, 58, 731. [Google Scholar] [CrossRef]
- Du, K.; Liu, M.Y.; Zhong, X.; Wei, M.J. Decreased circulating Zinc levels in Parkinson’s disease: A meta-analysis study. Sci. Rep. 2017, 7, 3902. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liu, X.; Ge, H.; Wang, T.; Wang, Y.; Li, W. Association Between Serum Zinc Levels and the Risk of Parkinson’s Disease: A Meta-Analysis. Biol. Trace Elem. Res. 2017, 179, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Adani, G.; Filippini, T.; Michalke, B.; Vinceti, M. Selenium and Other Trace Elements in the Etiology of Parkinson’s Disease: A Systematic Review and Meta-Analysis of Case-Control Studies. Neuroepidemiology 2020, 54, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Schaffner, W. Zinc supplement greatly improves the condition of parkin mutant Drosophila. Biol. Chem. 2010, 391, 513–518. [Google Scholar] [CrossRef]
- Bredholt, M.; Frederiksen, J.L. Zinc in Multiple Sclerosis: A Systematic Review and Meta-Analysis. Asn Neuro 2016, 8, 1759091416651511. [Google Scholar] [CrossRef]
- Nirooei, E.; Kashani, S.; Owrangi, S.; Malekpour, F.; Niknam, M.; Moazzen, F.; Nowrouzi-Sohrabi, P.; Farzinmehr, S.; Akbari, H. Blood Trace Element Status in Multiple Sclerosis: A Systematic Review and Meta-analysis. Biol. Trace Elem. Res. 2022, 200, 13–26. [Google Scholar] [CrossRef]
- Stoye, D.; Schubert, C.; Goihl, A.; Guttek, K.; Reinhold, A.; Brocke, S.; Grungreiff, K.; Reinhold, D. Zinc aspartate suppresses T cell activation in vitro and relapsing experimental autoimmune encephalomyelitis in SJL/J mice. Biometals 2012, 25, 529–539. [Google Scholar] [CrossRef]
- Schubert, C.; Guttek, K.; Grungreiff, K.; Thielitz, A.; Buhling, F.; Reinhold, A.; Brocke, S.; Reinhold, D. Oral zinc aspartate treats experimental autoimmune encephalomyelitis. Biometals 2014, 27, 1249–1262. [Google Scholar] [CrossRef]
- Joe, P.; Petrilli, M.; Malaspina, D.; Weissman, J. Zinc in schizophrenia: A meta-analysis. Gen. Hosp. Psych. 2018, 53, 19–24. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Mahmoudi, M.; Shahrokhi, S.; Mojarrad, M.; Dastmardi, M.; Mirbeyk, M.; Rezaei, N. Trace elements in schizophrenia: A systematic review and meta-analysis of 39 studies (N = 5151 participants). Nutr. Rev. 2020, 78, 278–303. [Google Scholar] [CrossRef]
- Mortazavi, M.; Farzin, D.; Zarhghami, M.; Hosseini, S.H.; Mansoori, P.; Nateghi, G. Efficacy of Zinc Sulfate as an Add-on Therapy to Risperidone Versus Risperidone Alone in Patients with Schizophrenia: A Double-Blind Randomized Placebo-Controlled Trial. Iran. J. Psychiatry Behav. Sci. 2015, 9, e853. [Google Scholar] [CrossRef]
- Onaolapo, O.J.; Ademakinwa, O.Q.; Olalekan, T.O.; Onaolapo, A.Y. Ketamine-induced behavioural and brain oxidative changes in mice: An assessment of possible beneficial effects of zinc as mono- or adjunct therapy. Psychopharmacology 2017, 234, 2707–2725. [Google Scholar] [CrossRef]
- Alizadeh, F.; Davoodian, N.; Kazemi, H.; Ghasemi-Kasman, M.; Shaerzadeh, F. Prenatal zinc supplementation attenuates lipopolysaccharide-induced behavioral impairments in maternal immune activation model. Behav. Brain Res. 2020, 377, 112247. [Google Scholar] [CrossRef]
- Wojciak, R.W.; Mojs, E.; Stanislawska-Kubiak, M.; Samborski, W. The serum zinc, copper, iron, and chromium concentrations in epileptic children. Epilepsy Res. 2013, 104, 40–44. [Google Scholar] [CrossRef]
- Seven, M.; Basaran, S.Y.; Cengiz, M.; Unal, S.; Yuksel, A. Deficiency of selenium and zinc as a causative factor for idiopathic intractable epilepsy. Epilepsy Res. 2013, 104, 35–39. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Mahmoudi, M.; Meysamie, A.; Gharedaghi, M.; Zamponi, G.W.; Rezaei, N. Possible role of trace elements in epilepsy and febrile seizures: A meta-analysis. Nutr. Rev. 2015, 73, 760–779. [Google Scholar] [CrossRef]
- Baraka, A.M.; Hassab El Nabi, W.; El Ghotni, S. Investigating the role of zinc in a rat model of epilepsy. CNS Neurosci. Ther. 2012, 18, 327–333. [Google Scholar] [CrossRef]
- Saad, K.; El-Houfey, A.A.; Abd El-Hamed, M.A.; El-Asheer, O.M.; Al-Atram, A.A.; Tawfeek, M.S.K. A randomized, double-blind, placebo-controlled clinical trial of the efficacy of treatment with zinc in children with intractable epilepsy. Funct. Neurol. 2015, 30, 181–185. [Google Scholar] [CrossRef]
- Kirazlar, M.; Erdogan, M.A.; Erbas, O. Anti-seizure effect of zinc on PTZ-induced epilepsy in rat model. Bratisl. Lek. Listy 2022, 123, 648–652. [Google Scholar] [CrossRef]
- McClain, C.J.; Twyman, D.L.; Ott, L.G.; Rapp, R.P.; Tibbs, P.A.; Norton, J.A.; Kasarskis, E.J.; Dempsey, R.J.; Young, B. Serum and urine zinc response in head-injured patients. J. Neurosurg. 1986, 64, 224–230. [Google Scholar] [CrossRef]
- Cope, E.C.; Morris, D.R.; Scrimgeour, A.G.; Levenson, C.W. Use of zinc as a treatment for traumatic brain injury in the rat: Effects on cognitive and behavioral outcomes. Neurorehabilit. Neural Repair 2012, 26, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Young, B.; Ott, L.; Kasarskis, E.; Rapp, R.; Moles, K.; Dempsey, R.J.; Tibbs, P.A.; Kryscio, R.; McClain, C. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J. Neurotrauma 1996, 13, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. A possible role of zinc in the pathology of dementia. Lancet 1981, 1, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. Embo Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Grilli, M.; Goffi, F.; Memo, M.; Spano, P. Interleukin-1beta and glutamate activate the NF-kappaB/Rel binding site from the regulatory region of the amyloid precursor protein gene in primary neuronal cultures. J. Biol. Chem. 1996, 271, 15002–15007. [Google Scholar] [CrossRef]
- Cuesta, A.; Zambrano, A.; Royo, M.; Pascual, A. The tumour suppressor p53 regulates the expression of amyloid precursor protein (APP). Biochem. J. 2009, 418, 643–650. [Google Scholar] [CrossRef]
- Xie, Z.; Wu, H.; Zhao, J. Multifunctional roles of zinc in Alzheimer’s disease. Neurotoxicology 2020, 80, 112–123. [Google Scholar] [CrossRef]
- Dahms, S.O.; Konnig, I.; Roeser, D.; Guhrs, K.H.; Mayer, M.C.; Kaden, D.; Multhaup, G.; Than, M.E. Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J. Mol. Biol. 2012, 416, 438–452. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado, S.A.; Vissoci, R.E. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Cristovao, J.S.; Santos, R.; Gomes, C.M. Metals and Neuronal Metal Binding Proteins Implicated in Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2016, 2016, 9812178. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Religa, D.; Strozyk, D.; Cherny, R.A.; Volitakis, I.; Haroutunian, V.; Winblad, B.; Naslund, J.; Bush, A.I. Elevated cortical zinc in Alzheimer disease. Neurology 2006, 67, 69–75. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef]
- Kawahara, M.; Mizuno, D.; Koyama, H.; Konoha, K.; Ohkawara, S.; Sadakane, Y. Disruption of zinc homeostasis and the pathogenesis of senile dementia. Metallomics 2014, 6, 209–219. [Google Scholar] [CrossRef]
- Arispe, N.; Diaz, J.C.; Simakova, O. Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim. Biophys Acta 2007, 1768, 1952–1965. [Google Scholar] [CrossRef]
- Yuan, Y.; Niu, F.; Liu, Y.; Lu, N. Zinc and its effects on oxidative stress in Alzheimer’s disease. Neurol. Sci. 2014, 35, 923–928. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Goldstein, L.E.; Nunomura, A.; Smith, M.A.; Lim, J.T.; Atwood, C.S.; Huang, X.; Farrag, Y.W.; Perry, G.; Bush, A.I. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J. Biol. Chem. 2000, 275, 19439–19442. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.J.; Paradis, M.D.; Tanzi, R.E. Modulation of A beta adhesiveness and secretase site cleavage by zinc. J. Biol. Chem. 1994, 269, 12152–12158. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.Y.; Wang, C.Y.; Wang, T.; Li, Y.C.; Wang, Z.Y. Glial S100A6 Degrades beta-amyloid Aggregation through Targeting Competition with Zinc Ions. Aging Dis. 2019, 10, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Segawa, S.; Matsuo, T.; Nakamura, S.; Kikkawa, Y.; Nishida, K.; Nagasawa, K. Microglial zinc uptake via zinc transporters induces ATP release and the activation of microglia. Glia 2011, 59, 1933–1945. [Google Scholar] [CrossRef]
- Hwang, J.J.; Park, M.H.; Choi, S.Y.; Koh, J.Y. Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J. Biol. Chem. 2005, 280, 11995–12001. [Google Scholar] [CrossRef]
- Buchman, A.S.; Yu, L.; Boyle, P.A.; Schneider, J.A.; De Jager, P.L.; Bennett, D.A. Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology 2016, 86, 735–741. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Riffault, B.; Medina, I.; Dumon, C.; Thalman, C.; Ferrand, N.; Friedel, P.; Gaiarsa, J.L.; Porcher, C. Pro-brain-derived neurotrophic factor inhibits GABAergic neurotransmission by activating endocytosis and repression of GABAA receptors. J. Neurosci. 2014, 34, 13516–13534. [Google Scholar] [CrossRef]
- Busche, M.A.; Konnerth, A. Neuronal hyperactivity—A key defect in Alzheimer’s disease? Bioessays 2015, 37, 624–632. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 2021, 296, 100105. [Google Scholar] [CrossRef]
- Datki, Z.; Galik-Olah, Z.; Janosi-Mozes, E.; Szegedi, V.; Kalman, J.; Hunya, A.G.; Fulop, L.; Tamano, H.; Takeda, A.; Adlard, P.A.; et al. Alzheimer risk factors age and female sex induce cortical Abeta aggregation by raising extracellular zinc. Mol. Psychiatr. 2020, 25, 2728–2741. [Google Scholar] [CrossRef]
- Smith, J.L.; Xiong, S.; Lovell, M.A. 4-Hydroxynonenal disrupts zinc export in primary rat cortical cells. Neurotoxicology 2006, 27, 1–5. [Google Scholar] [CrossRef]
- Xiong, Y.; Jing, X.P.; Zhou, X.W.; Wang, X.L.; Yang, Y.; Sun, X.Y.; Qiu, M.; Cao, F.Y.; Lu, Y.M.; Liu, R.; et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol. Aging 2013, 34, 745–756. [Google Scholar] [CrossRef]
- Moreira, G.G.; Cristovao, J.S.; Torres, V.M.; Carapeto, A.P.; Rodrigues, M.S.; Landrieu, I.; Cordeiro, C.; Gomes, C.M. Zinc Binding to Tau Influences Aggregation Kinetics and Oligomer Distribution. Int. J. Mol. Sci. 2019, 20, 5979. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, Z.; Cao, Y.; Lang, M.; Lu, B.; Zhou, B. Zinc binding directly regulates tau toxicity independent of tau hyperphosphorylation. Cell Rep. 2014, 8, 831–842. [Google Scholar] [CrossRef]
- Stoltenberg, M.; Bush, A.I.; Bach, G.; Smidt, K.; Larsen, A.; Rungby, J.; Lund, S.; Doering, P.; Danscher, G. Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 2007, 150, 357–369. [Google Scholar] [CrossRef]
- Linkous, D.H.; Adlard, P.A.; Wanschura, P.B.; Conko, K.M.; Flinn, J.M. The effects of enhanced zinc on spatial memory and plaque formation in transgenic mice. J. Alzheimers Dis. 2009, 18, 565–579. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, T.; Zheng, W.; Zhao, B.L.; Danscher, G.; Chen, Y.H.; Wang, Z.Y. Zinc overload enhances APP cleavage and Abeta deposition in the Alzheimer mouse brain. PLoS ONE 2010, 5, e15349. [Google Scholar] [CrossRef]
- Craven, K.M.; Kochen, W.R.; Hernandez, C.M.; Flinn, J.M. Zinc Exacerbates Tau Pathology in a Tau Mouse Model. J. Alzheimers Dis. 2018, 64, 617–630. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Du, T.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Xilinas, M.E.; Hoke, D.E.; Holsinger, R.M.; Evin, G.; Cherny, R.A.; et al. Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J. Biol. Chem. 2006, 281, 17670–17680. [Google Scholar] [CrossRef] [PubMed]
- Tassabehji, N.M.; Corniola, R.S.; Alshingiti, A.; Levenson, C.W. Zinc deficiency induces depression-like symptoms in adult rats. Physiol. Behav. 2008, 95, 365–369. [Google Scholar] [CrossRef]
- Siwek, M.; Dudek, D.; Schlegel-Zawadzka, M.; Morawska, A.; Piekoszewski, W.; Opoka, W.; Zieba, A.; Pilc, A.; Popik, P.; Nowak, G. Serum zinc level in depressed patients during zinc supplementation of imipramine treatment. J. Affect. Disord. 2010, 126, 447–452. [Google Scholar] [CrossRef]
- Carroll, B.J.; Feinberg, M.; Greden, J.F.; Tarika, J.; Albala, A.A.; Haskett, R.F.; James, N.M.; Kronfol, Z.; Lohr, N.; Steiner, M.; et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch. Gen. Psychiatry 1981, 38, 15–22. [Google Scholar] [CrossRef]
- Holsboer, F. The dexamethasone suppression test in depressed patients: Clinical and biochemical aspects. J. Steroid Biochem. Mol. Biol. 1983, 19, 251–257. [Google Scholar] [CrossRef]
- Arana, G.W.; Baldessarini, R.J.; Ornsteen, M. The dexamethasone suppression test for diagnosis and prognosis in psychiatry. Commentary and review. Arch. Gen. Psychiatry 1985, 42, 1193–1204. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H.; Kan, F.; Itoh, H.; Oku, N. Anxiety-like behavior of young rats after 2-week zinc deprivation. Behav. Brain Res. 2007, 177, 1–6. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H.; Kan, F.; Hanajima, T.; Yamada, K.; Oku, N. Enhancement of social isolation-induced aggressive behavior of young mice by zinc deficiency. Life Sci. 2008, 82, 909–914. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H.; Ogawa, T.; Takada, S.; Ando, M.; Oku, N.; Watanabe, M. Significance of serum glucocorticoid and chelatable zinc in depression and cognition in zinc deficiency. Behav. Brain Res. 2012, 226, 259–264. [Google Scholar] [CrossRef]
- Gregus, A.; Wintink, A.J.; Davis, A.C.; Kalynchuk, L.E. Effect of repeated corticosterone injections and restraint stress on anxiety and depression-like behavior in male rats. Behav. Brain Res. 2005, 156, 105–114. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, R.; Shen, J.; Su, H.; Xing, D.; Du, L. A mouse model of depression induced by repeated corticosterone injections. Eur. J. Pharmacol. 2008, 581, 113–120. [Google Scholar] [CrossRef]
- Watanabe, M.; Tamano, H.; Kikuchi, T.; Takeda, A. Susceptibility to stress in young rats after 2-week zinc deprivation. Neurochem. Int. 2010, 56, 410–416. [Google Scholar] [CrossRef]
- Mlyniec, K. Zinc in the Glutamatergic Theory of Depression. Curr. Neuropharmacol. 2015, 13, 505–513. [Google Scholar] [CrossRef]
- Nowak, G.; Schlegel-Zawadzka, M. Alterations in serum and brain trace element levels after antidepressant treatment: Part I. Zinc. Biol. Trace Elem. Res. 1999, 67, 85–92. [Google Scholar] [CrossRef]
- Doboszewska, U.; Szewczyk, B.; Sowa-Kucma, M.; Mlyniec, K.; Rafalo, A.; Ostachowicz, B.; Lankosz, M.; Nowak, G. Antidepressant activity of fluoxetine in the zinc deficiency model in rats involves the NMDA receptor complex. Behav. Brain Res. 2015, 287, 323–330. [Google Scholar] [CrossRef]
- Szewczyk, B.; Poleszak, E.; Wlaz, P.; Wrobel, A.; Blicharska, E.; Cichy, A.; Dybala, M.; Siwek, A.; Pomierny-Chamiolo, L.; Piotrowska, A.; et al. The involvement of serotonergic system in the antidepressant effect of zinc in the forced swim test. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 323–329. [Google Scholar] [CrossRef]
- Cichy, A.; Sowa-Kucma, M.; Legutko, B.; Pomierny-Chamiolo, L.; Siwek, A.; Piotrowska, A.; Szewczyk, B.; Poleszak, E.; Pilc, A.; Nowak, G. Zinc-induced adaptive changes in NMDA/glutamatergic and serotonergic receptors. Pharmacol. Rep. 2009, 61, 1184–1191. [Google Scholar] [CrossRef]
- Satala, G.; Duszynska, B.; Stachowicz, K.; Rafalo, A.; Pochwat, B.; Luckhart, C.; Albert, P.R.; Daigle, M.; Tanaka, K.F.; Hen, R.; et al. Concentration-Dependent Dual Mode of Zn2+ Action at Serotonin 5-HT1A Receptors: In Vitro and In Vivo Studies. Mol. Neurobiol. 2016, 53, 6869–6881. [Google Scholar] [CrossRef]
- Barrondo, S.; Salles, J. Allosteric modulation of 5-HT(1A) receptors by zinc: Binding studies. Neuropharmacology 2009, 56, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Satala, G.; Duszynska, B.; Lenda, T.; Nowak, G.; Bojarski, A.J. Allosteric Inhibition of Serotonin 5-HT7 Receptors by Zinc Ions. Mol. Neurobiol. 2018, 55, 2897–2910. [Google Scholar] [CrossRef] [PubMed]
- Tena-Campos, M.; Ramon, E.; Borroto-Escuela, D.O.; Fuxe, K.; Garriga, P. The zinc binding receptor GPR39 interacts with 5-HT1A and GalR1 to form dynamic heteroreceptor complexes with signaling diversity. Biochim. Biophys. Acta 2015, 1852, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Tena-Campos, M.; Ramon, E.; Lupala, C.S.; Perez, J.J.; Koch, K.W.; Garriga, P. Zinc Is Involved in Depression by Modulating G Protein-Coupled Receptor Heterodimerization. Mol. Neurobiol. 2016, 53, 2003–2015. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Narvaez, M.; Marcellino, D.; Parrado, C.; Narvaez, J.A.; Tarakanov, A.O.; Agnati, L.F.; Diaz-Cabiale, Z.; Fuxe, K. Galanin receptor-1 modulates 5-hydroxtryptamine-1A signaling via heterodimerization. Biochem. Biophys. Res. Commun. 2010, 393, 767–772. [Google Scholar] [CrossRef]
- Petrilli, M.A.; Kranz, T.M.; Kleinhaus, K.; Joe, P.; Getz, M.; Johnson, P.; Chao, M.V.; Malaspina, D. The Emerging Role for Zinc in Depression and Psychosis. Front. Pharmacol. 2017, 8, 414. [Google Scholar] [CrossRef]
- Szewczyk, B.; Poleszak, E.; Sowa-Kucma, M.; Wrobel, A.; Slotwinski, S.; Listos, J.; Wlaz, P.; Cichy, A.; Siwek, A.; Dybala, M.; et al. The involvement of NMDA and AMPA receptors in the mechanism of antidepressant-like action of zinc in the forced swim test. Amino Acids 2010, 39, 205–217. [Google Scholar] [CrossRef]
- Poleszak, E.; Szewczyk, B.; Wlaz, A.; Fidecka, S.; Wlaz, P.; Pilc, A.; Nowak, G. D-serine, a selective glycine/N-methyl-D-aspartate receptor agonist, antagonizes the antidepressant-like effects of magnesium and zinc in mice. Pharmacol. Rep. 2008, 60, 996–1000. [Google Scholar]
- Doboszewska, U.; Sowa-Kucma, M.; Mlyniec, K.; Pochwat, B.; Holuj, M.; Ostachowicz, B.; Pilc, A.; Nowak, G.; Szewczyk, B. Zinc deficiency in rats is associated with up-regulation of hippocampal NMDA receptor. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 56, 254–263. [Google Scholar] [CrossRef]
- Belozertseva, I.V.; Kos, T.; Popik, P.; Danysz, W.; Bespalov, A.Y. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur. Neuropsychopharmacol. 2007, 17, 172–179. [Google Scholar] [CrossRef]
- Chaki, S.; Yoshikawa, R.; Hirota, S.; Shimazaki, T.; Maeda, M.; Kawashima, N.; Yoshimizu, T.; Yasuhara, A.; Sakagami, K.; Okuyama, S.; et al. MGS0039: A potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology 2004, 46, 457–467. [Google Scholar] [CrossRef]
- Szewczyk, B.; Poleszak, E.; Sowa-Kucma, M.; Siwek, M.; Dudek, D.; Ryszewska-Pokrasniewicz, B.; Radziwon-Zaleska, M.; Opoka, W.; Czekaj, J.; Pilc, A.; et al. Antidepressant activity of zinc and magnesium in view of the current hypotheses of antidepressant action. Pharmacol. Rep. 2008, 60, 588–589. [Google Scholar]
- Holst, B.; Egerod, K.L.; Schild, E.; Vickers, S.P.; Cheetham, S.; Gerlach, L.O.; Storjohann, L.; Stidsen, C.E.; Jones, R.; Beck-Sickinger, A.G.; et al. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology 2007, 148, 13–20. [Google Scholar] [CrossRef]
- Mlyniec, K.; Budziszewska, B.; Holst, B.; Ostachowicz, B.; Nowak, G. GPR39 (zinc receptor) knockout mice exhibit depression-like behavior and CREB/BDNF down-regulation in the hippocampus. Int. J. Neuropsychopharmacol. 2014, 18, pyu002. [Google Scholar] [CrossRef]
- Mlyniec, K.; Doboszewska, U.; Szewczyk, B.; Sowa-Kucma, M.; Misztak, P.; Piekoszewski, W.; Trela, F.; Ostachowicz, B.; Nowak, G. The involvement of the GPR39-Zn(2+)-sensing receptor in the pathophysiology of depression. Studies in rodent models and suicide victims. Neuropharmacology 2014, 79, 290–297. [Google Scholar] [CrossRef]
- Omar, N.N.; Tash, R.F. Fluoxetine coupled with zinc in a chronic mild stress model of depression: Providing a reservoir for optimum zinc signaling and neuronal remodeling. Pharmacol. Biochem. Behav. 2017, 160, 30–38. [Google Scholar] [CrossRef]
- Saadi, R.A.; He, K.; Hartnett, K.A.; Kandler, K.; Hershfinkel, M.; Aizenman, E. SNARE-dependent upregulation of potassium chloride co-transporter 2 activity after metabotropic zinc receptor activation in rat cortical neurons in vitro. Neuroscience 2012, 210, 38–46. [Google Scholar] [CrossRef]
- Siodlak, D.; Nowak, G.; Mlyniec, K. Interaction between zinc, the GPR39 zinc receptor and the serotonergic system in depression. Brain Res. Bull. 2021, 170, 146–154. [Google Scholar] [CrossRef]
- Perez-Rosello, T.; Anderson, C.T.; Schopfer, F.J.; Zhao, Y.; Gilad, D.; Salvatore, S.R.; Freeman, B.A.; Hershfinkel, M.; Aizenman, E.; Tzounopoulos, T. Synaptic Zn2+ inhibits neurotransmitter release by promoting endocannabinoid synthesis. J. Neurosci. 2013, 33, 9259–9272. [Google Scholar] [CrossRef]
- Mlyniec, K.; Gawel, M.; Nowak, G. Study of antidepressant drugs in GPR39 (zinc receptor(−)/(−)) knockout mice, showing no effect of conventional antidepressants, but effectiveness of NMDA antagonists. Behav. Brain Res. 2015, 287, 135–138. [Google Scholar] [CrossRef]
- Sen, S.; Duman, R.; Sanacora, G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: Meta-analyses and implications. Biol. Psychiatry 2008, 64, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.D.; Duman, R.S. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology 2010, 35, 2378–2391. [Google Scholar] [CrossRef] [PubMed]
- Mlyniec, K. Interaction between Zinc, GPR39, BDNF and Neuropeptides in Depression. Curr. Neuropharmacol. 2021, 19, 2012–2019. [Google Scholar] [CrossRef] [PubMed]
- Sowa-Kucma, M.; Legutko, B.; Szewczyk, B.; Novak, K.; Znojek, P.; Poleszak, E.; Papp, M.; Pilc, A.; Nowak, G. Antidepressant-like activity of zinc: Further behavioral and molecular evidence. J. Neural Transm. 2008, 115, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.L.; Posser, T.; Brocardo, P.S.; Trevisan, R.; Uliano-Silva, M.; Gabilan, N.H.; Santos, A.R.; Leal, R.B.; Rodrigues, A.L.; Farina, M.; et al. Involvement of glutathione, ERK1/2 phosphorylation and BDNF expression in the antidepressant-like effect of zinc in rats. Behav. Brain Res. 2008, 188, 316–323. [Google Scholar] [CrossRef]
- Huang, Y.Z.; Pan, E.; Xiong, Z.Q.; McNamara, J.O. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 2008, 57, 546–558. [Google Scholar] [CrossRef]
- Nagappan, G.; Woo, N.H.; Lu, B. Ama “zinc” link between TrkB transactivation and synaptic plasticity. Neuron 2008, 57, 477–479. [Google Scholar] [CrossRef]
- Mlyniec, K.; Starowicz, G.; Gawel, M.; Frackiewicz, E.; Nowak, G. Potential antidepressant-like properties of the TC G-1008, a GPR39 (zinc receptor) agonist. J. Affect. Disord. 2016, 201, 179–184. [Google Scholar] [CrossRef]
- Corniola, R.S.; Tassabehji, N.M.; Hare, J.; Sharma, G.; Levenson, C.W. Zinc deficiency impairs neuronal precursor cell proliferation and induces apoptosis via p53-mediated mechanisms. Brain Res. 2008, 1237, 52–61. [Google Scholar] [CrossRef]
- Gao, H.L.; Zheng, W.; Xin, N.; Chi, Z.H.; Wang, Z.Y.; Chen, J.; Wang, Z.Y. Zinc deficiency reduces neurogenesis accompanied by neuronal apoptosis through caspase-dependent and -independent signaling pathways. Neurotox. Res. 2009, 16, 416–425. [Google Scholar] [CrossRef]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef] [PubMed]
- Drzyzga, L.R.; Marcinowska, A.; Obuchowicz, E. Antiapoptotic and neurotrophic effects of antidepressants: A review of clinical and experimental studies. Brain Res. Bull. 2009, 79, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Gower-Winter, S.D.; Corniola, R.S.; Morgan, T.J.; Levenson, C.W. Zinc deficiency regulates hippocampal gene expression and impairs neuronal differentiation. Nutr. Neurosci. 2013, 16, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Herken, H.; Gurel, A.; Selek, S.; Armutcu, F.; Ozen, M.E.; Bulut, M.; Kap, O.; Yumru, M.; Savas, H.A.; Akyol, O. Adenosine deaminase, nitric oxide, superoxide dismutase, and xanthine oxidase in patients with major depression: Impact of antidepressant treatment. Arch. Med. Res. 2007, 38, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.O.; Lin, J.; Calixto, J.B.; Santos, A.R.; Rodrigues, A.L. Involvement of NMDA receptors and L-arginine-nitric oxide pathway in the antidepressant-like effects of zinc in mice. Behav. Brain Res. 2003, 144, 87–93. [Google Scholar] [CrossRef]
- Brocardo, P.S.; Assini, F.; Franco, J.L.; Pandolfo, P.; Muller, Y.M.; Takahashi, R.N.; Dafre, A.L.; Rodrigues, A.L. Zinc attenuates malathion-induced depressant-like behavior and confers neuroprotection in the rat brain. Toxicol. Sci. 2007, 97, 140–148. [Google Scholar] [CrossRef]
- Dorri, S.A.; Hosseinzadeh, H.; Abnous, K.; Hasani, F.V.; Robati, R.Y.; Razavi, B.M. Involvement of brain-derived neurotrophic factor (BDNF) on malathion induced depressive-like behavior in subacute exposure and protective effects of crocin. Iran. J. Basic Med. Sci. 2015, 18, 958–966. [Google Scholar]
- Bhalla, P.; Chadha, V.D.; Dhar, R.; Dhawan, D.K. Neuroprotective effects of zinc on antioxidant defense system in lithium treated rat brain. Indian J. Exp. Biol. 2007, 45, 954–958. [Google Scholar]
- Swardfager, W.; Herrmann, N.; McIntyre, R.S.; Mazereeuw, G.; Goldberger, K.; Cha, D.S.; Schwartz, Y.; Lanctot, K.L. Potential roles of zinc in the pathophysiology and treatment of major depressive disorder. Neurosci. Biobehav. Rev. 2013, 37, 911–929. [Google Scholar] [CrossRef]
- Kelly, J.P.; Wrynn, A.S.; Leonard, B.E. The olfactory bulbectomized rat as a model of depression: An update. Pharmacol. Ther. 1997, 74, 299–316. [Google Scholar] [CrossRef]
- You, Z.; Luo, C.; Zhang, W.; Chen, Y.; He, J.; Zhao, Q.; Zuo, R.; Wu, Y. Pro- and anti-inflammatory cytokines expression in rat’s brain and spleen exposed to chronic mild stress: Involvement in depression. Behav. Brain Res. 2011, 225, 135–141. [Google Scholar] [CrossRef]
- Lindqvist, D.; Dhabhar, F.S.; James, S.J.; Hough, C.M.; Jain, F.A.; Bersani, F.S.; Reus, V.I.; Verhoeven, J.E.; Epel, E.S.; Mahan, L.; et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2017, 76, 197–205. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef]
- Cousins, R.J.; Leinart, A.S. Tissue-specific regulation of zinc metabolism and metallothionein genes by interleukin 1. Faseb J. 1988, 2, 2884–2890. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, K.J.; Kim, D.H.; Jeong, T.C.; Lee, E.S.; Choi, Y.M.; Jeong, H.G. Cytokine-mediated induction of metallothionein in Hepa-1c1c7 cells by oleanolic acid. Biochem. Biophys. Res. Commun. 2004, 325, 792–797. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Lichten, L.A.; Rivera, S.; Blanchard, R.K.; Aydemir, T.B.; Knutson, M.D.; Ganz, T.; Cousins, R.J. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc. Natl. Acad. Sci. USA 2005, 102, 6843–6848. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; De Jongh, R.; Kenis, G.; Vandoolaeghe, E.; Neels, H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 1997, 9, 853–858. [Google Scholar] [CrossRef]
- Van Hunsel, F.; Wauters, A.; Vandoolaeghe, E.; Neels, H.; Demedts, P.; Maes, M. Lower total serum protein, albumin, and beta- and gamma-globulin in major and treatment-resistant depression: Effects of antidepressant treatments. Psychiatry Res. 1996, 65, 159–169. [Google Scholar] [CrossRef]
- Maes, M.; De Vos, N.; Demedts, P.; Wauters, A.; Neels, H. Lower serum zinc in major depression in relation to changes in serum acute phase proteins. J. Affect. Disord. 1999, 56, 189–194. [Google Scholar] [CrossRef]
- Prasad, A.S.; Meftah, S.; Abdallah, J.; Kaplan, J.; Brewer, G.J.; Bach, J.F.; Dardenne, M. Serum thymulin in human zinc deficiency. J. Clin. Investig. 1988, 82, 1202–1210. [Google Scholar] [CrossRef]
- Maes, M.; Vandoolaeghe, E.; Neels, H.; Demedts, P.; Wauters, A.; Meltzer, H.Y.; Altamura, C.; Desnyder, R. Lower serum zinc in major depression is a sensitive marker of treatment resistance and of the immune/inflammatory response in that illness. Biol. Psychiatry. 1997, 42, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Beck, F.W.; Grabowski, S.M.; Kaplan, J.; Mathog, R.H. Zinc deficiency: Changes in cytokine production and T-cell subpopulations in patients with head and neck cancer and in noncancer subjects. Proc. Assoc. Am. Phys. 1997, 109, 68–77. [Google Scholar] [PubMed]
- Beck, F.W.; Prasad, A.S.; Kaplan, J.; Fitzgerald, J.T.; Brewer, G.J. Changes in cytokine production and T cell subpopulations in experimentally induced zinc-deficient humans. Am. J. Physiol. 1997, 272, E1002–E1007. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, T.; Chen, P.; Ouyang, J.; Xu, G.; Zeng, Z.; Sun, Y. Emerging tendency towards autoimmune process in major depressive patients: A novel insight from Th17 cells. Psychiatry Res. 2011, 188, 224–230. [Google Scholar] [CrossRef]
- Kitabayashi, C.; Fukada, T.; Kanamoto, M.; Ohashi, W.; Hojyo, S.; Atsumi, T.; Ueda, N.; Azuma, I.; Hirota, H.; Murakami, M.; et al. Zinc suppresses Th17 development via inhibition of STAT3 activation. Int. Immunol. 2010, 22, 375–386. [Google Scholar] [CrossRef]
- Robertson, M.J.; Schacterle, R.S.; Mackin, G.A.; Wilson, S.N.; Bloomingdale, K.L.; Ritz, J.; Komaroff, A.L. Lymphocyte subset differences in patients with chronic fatigue syndrome, multiple sclerosis and major depression. Clin. Exp. Immunol. 2005, 141, 326–332. [Google Scholar] [CrossRef]
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and Parkinson’s Disease: Mechanisms and Biochemical Processes. Curr. Med. Chem. 2018, 25, 2198–2214. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Brewer, G.J.; Kanzer, S.H.; Zimmerman, E.A.; Molho, E.S.; Celmins, D.F.; Heckman, S.M.; Dick, R. Subclinical zinc deficiency in Alzheimer’s disease and Parkinson’s disease. Am. J. Alzheimers Dis. Other Dement. 2010, 25, 572–575. [Google Scholar] [CrossRef]
- Zhao, H.W.; Lin, J.; Wang, X.B.; Cheng, X.; Wang, J.Y.; Hu, B.L.; Zhang, Y.; Zhang, X.; Zhu, J.H. Assessing plasma levels of selenium, copper, iron and zinc in patients of Parkinson’s disease. PLoS ONE 2013, 8, e83060. [Google Scholar] [CrossRef]
- Alimonti, A.; Ristori, G.; Giubilei, F.; Stazi, M.A.; Pino, A.; Visconti, A.; Brescianini, S.; Sepe, M.M.; Forte, G.; Stanzione, P.; et al. Serum chemical elements and oxidative status in Alzheimer’s disease, Parkinson disease and multiple sclerosis. Neurotoxicology 2007, 28, 450–456. [Google Scholar] [CrossRef]
- Hegde, M.L.; Shanmugavelu, P.; Vengamma, B.; Rao, T.S.; Menon, R.B.; Rao, R.V.; Rao, K.S. Serum trace element levels and the complexity of inter-element relations in patients with Parkinson’s disease. J. Trace Elem. Med. Biol. 2004, 18, 163–171. [Google Scholar] [CrossRef]
- Forte, G.; Bocca, B.; Senofonte, O.; Petrucci, F.; Brusa, L.; Stanzione, P.; Zannino, S.; Violante, N.; Alimonti, A.; Sancesario, G. Trace and major elements in whole blood, serum, cerebrospinal fluid and urine of patients with Parkinson’s disease. J. Neural Transm. 2004, 111, 1031–1040. [Google Scholar] [CrossRef]
- Quiroga, M.J.; Carroll, D.W.; Brown, T.M. Ascorbate- and zinc-responsive parkinsonism. Ann. Pharmacother. 2014, 48, 1515–1520. [Google Scholar] [CrossRef]
- Sikora, J.; Ouagazzal, A.M. Synaptic Zinc: An Emerging Player in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4724. [Google Scholar] [CrossRef]
- Gaki, G.S.; Papavassiliou, A.G. Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson’s disease. Neuromol. Med. 2014, 16, 217–230. [Google Scholar] [CrossRef]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Olanow, C.W. Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef]
- Mbiydzenyuy, N.E.; Ninsiima, H.I.; Valladares, M.B.; Pieme, C.A. Zinc and linoleic acid pre-treatment attenuates biochemical and histological changes in the midbrain of rats with rotenone-induced Parkinsonism. BMC Neurosci. 2018, 19, 29. [Google Scholar] [CrossRef]
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Son, H.J.; Choi, J.H.; Cho, E.; Kim, J.; Chung, S.J.; Hwang, O.; Koh, J.Y. Cytosolic labile zinc accumulation in degenerating dopaminergic neurons of mouse brain after MPTP treatment. Brain Res. 2009, 1286, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Tarohda, T.; Ishida, Y.; Kawai, K.; Yamamoto, M.; Amano, R. Regional distributions of manganese, iron, copper, and zinc in the brains of 6-hydroxydopamine-induced parkinsonian rats. Anal. Bioanal. Chem. 2005, 383, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Sheline, C.T.; Zhu, J.; Zhang, W.; Shi, C.; Cai, A.L. Mitochondrial inhibitor models of Huntington’s disease and Parkinson’s disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo. Neurodegener. Dis. 2013, 11, 49–58. [Google Scholar] [CrossRef]
- Xu, J.; Kao, S.Y.; Lee, F.J.; Song, W.; Jin, L.W.; Yankner, B.A. Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 2002, 8, 600–606. [Google Scholar] [CrossRef]
- Ajjimaporn, A.; Phansuwan-Pujito, P.; Ebadi, M.; Govitrapong, P. Zinc protects SK-N-SH cells from methamphetamine-induced alpha-synuclein expression. Neurosci. Lett. 2007, 419, 59–63. [Google Scholar] [CrossRef]
- Tsunemi, T.; Krainc, D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 2791–2801. [Google Scholar] [CrossRef]
- Choi, B.Y.; Jung, J.W.; Suh, S.W. The Emerging Role of Zinc in the Pathogenesis of Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 2070. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Simao, A.; Oliveira, S.R.; Flauzino, T.; Alfieri, D.F.; de Carvalho, J.P.W.; Kallaur, A.P.; Lozovoy, M.; Kaimen-Maciel, D.R.; Maes, M.; et al. Antioxidant and Anti-inflammatory Diagnostic Biomarkers in Multiple Sclerosis: A Machine Learning Study. Mol. Neurobiol. 2020, 57, 2167–2178. [Google Scholar] [CrossRef]
- Pawlitzki, M.; Uebelhor, J.; Sweeney-Reed, C.M.; Stephanik, H.; Hoffmann, J.; Lux, A.; Reinhold, D. Lower Serum Zinc Levels in Patients with Multiple Sclerosis Compared to Healthy Controls. Nutrients 2018, 10, 967. [Google Scholar] [CrossRef]
- Armon-Omer, A.; Waldman, C.; Simaan, N.; Neuman, H.; Tamir, S.; Shahien, R. New Insights on the Nutrition Status and Antioxidant Capacity in Multiple Sclerosis Patients. Nutrients 2019, 11, 427. [Google Scholar] [CrossRef]
- Matar, A.; Jennani, S.; Abdallah, H.; Mohsen, N.; Borjac, J. Serum Iron and Zinc Levels in Lebanese Multiple Sclerosis Patients. Acta Neurol. Taiwan 2020, 29, 5–11. [Google Scholar]
- Castro, A.; Albuquerque, L.; Melo, M.; D’Almeida, J.; Braga, R.; Assis, R.C.; Marreiro, D.; Matos, W.O.; Maia, C. Relationship between zinc-related nutritional status and the progression of multiple sclerosis. Mult. Scler. Relat. Disord. 2022, 66, 104063. [Google Scholar] [CrossRef]
- Ho, S.Y.; Catalanotto, F.A.; Lisak, R.P.; Dore-Duffy, P. Zinc in multiple sclerosis. II: Correlation with disease activity and elevated plasma membrane-bound zinc in erythrocytes from patients with multiple sclerosis. Ann. Neurol. 1986, 20, 712–715. [Google Scholar] [CrossRef]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Seo, J.N.; Wu, G.; Sohn, M.; Chung, T.N.; Suh, S.W. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol. Dis. 2013, 54, 382–391. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Kho, A.R.; Lee, S.H.; Lee, B.E.; Sohn, M.; Koh, J.Y.; Suh, S.W. Zinc transporter 3 (ZnT3) gene deletion reduces spinal cord white matter damage and motor deficits in a murine MOG-induced multiple sclerosis model. Neurobiol. Dis. 2016, 94, 205–212. [Google Scholar] [CrossRef]
- Choi, B.Y.; Jeong, J.H.; Eom, J.W.; Koh, J.Y.; Kim, Y.H.; Suh, S.W. A Novel Zinc Chelator, 1H10, Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating Zinc Toxicity and AMPK Activation. Int. J. Mol. Sci. 2020, 21, 3375. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, J.H.; Kho, A.R.; Kim, I.Y.; Lee, S.H.; Lee, B.E.; Choi, E.; Sohn, M.; Stevenson, M.; Chung, T.N.; et al. Inhibition of NADPH oxidase activation reduces EAE-induced white matter damage in mice. J. Neuroinflamm. 2015, 12, 104. [Google Scholar] [CrossRef]
- Domercq, M.; Mato, S.; Soria, F.N.; Sanchez-gomez, M.V.; Alberdi, E.; Matute, C. Zn2+ -induced ERK activation mediates PARP-1-dependent ischemic-reoxygenation damage to oligodendrocytes. Glia 2013, 61, 383–393. [Google Scholar] [CrossRef]
- Krone, A.; Fu, Y.; Schreiber, S.; Kotrba, J.; Borde, L.; Notzold, A.; Thurm, C.; Negele, J.; Franz, T.; Stegemann-Koniszewski, S.; et al. Ionic mitigation of CD4(+) T cell metabolic fitness, Th1 central nervous system autoimmunity and Th2 asthmatic airway inflammation by therapeutic zinc. Sci. Rep. 2022, 12, 1943. [Google Scholar] [CrossRef] [PubMed]
- Elitt, C.M.; Fahrni, C.J.; Rosenberg, P.A. Zinc homeostasis and zinc signaling in white matter development and injury. Neurosci. Lett. 2019, 707, 134247. [Google Scholar] [CrossRef] [PubMed]
- Lang, U.E.; Puls, I.; Muller, D.J.; Strutz-Seebohm, N.; Gallinat, J. Molecular mechanisms of schizophrenia. Cell Physiol. Biochem. 2007, 20, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Mousaviyan, R.; Davoodian, N.; Alizadeh, F.; Ghasemi-Kasman, M.; Mousavi, S.A.; Shaerzadeh, F.; Kazemi, H. Zinc Supplementation During Pregnancy Alleviates Lipopolysaccharide-Induced Glial Activation and Inflammatory Markers Expression in a Rat Model of Maternal Immune Activation. Biol. Trace Elem. Res. 2021, 199, 4193–4204. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Zhang, T.; Yao, Y.; Shen, C.; Xue, Y. Association of Serum Trace Elements with Schizophrenia and Effects of Antipsychotic Treatment. Biol. Trace Elem. Res. 2018, 181, 22–30. [Google Scholar] [CrossRef]
- Santa, C.E.; Madrid, K.C.; Arruda, M.; Sussulini, A. Association between trace elements in serum from bipolar disorder and schizophrenia patients considering treatment effects. J. Trace Elem. Med. Biol. 2020, 59, 126467. [Google Scholar] [CrossRef]
- Cai, L.; Chen, T.; Yang, J.; Zhou, K.; Yan, X.; Chen, W.; Sun, L.; Li, L.; Qin, S.; Wang, P.; et al. Serum trace element differences between Schizophrenia patients and controls in the Han Chinese population. Sci. Rep. 2015, 5, 15013. [Google Scholar] [CrossRef]
- Carrera, N.; Arrojo, M.; Sanjuan, J.; Ramos-Rios, R.; Paz, E.; Suarez-Rama, J.J.; Paramo, M.; Agra, S.; Brenlla, J.; Martinez, S.; et al. Association study of nonsynonymous single nucleotide polymorphisms in schizophrenia. Biol. Psychiatry 2012, 71, 169–177. [Google Scholar] [CrossRef]
- Li, S.; Ma, C.; Li, Y.; Chen, R.; Liu, Y.; Wan, L.P.; Xiong, Q.; Wang, C.; Huo, Y.; Dang, X.; et al. The schizophrenia-associated missense variant rs13107325 regulates dendritic spine density. Transl. Psychiatr. 2022, 12, 361. [Google Scholar] [CrossRef]
- Tseng, W.C.; Reinhart, V.; Lanz, T.A.; Weber, M.L.; Pang, J.; Le, K.X.V.; Bell, R.D.; O’Donnell, P.; Buhl, D.L. Schizophrenia-associated SLC39A8 polymorphism is a loss-of-function allele altering glutamate receptor and innate immune signaling. Transl. Psychiatr. 2021, 11, 136. [Google Scholar] [CrossRef]
- Scarr, E.; Udawela, M.; Greenough, M.A.; Neo, J.; Suk, S.M.; Money, T.T.; Upadhyay, A.; Bush, A.I.; Everall, I.P.; Thomas, E.A.; et al. Increased cortical expression of the zinc transporter SLC39A12 suggests a breakdown in zinc cellular homeostasis as part of the pathophysiology of schizophrenia. Npj Schizophr. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Perez-Becerril, C.; Morris, A.G.; Mortimer, A.; McKenna, P.J.; de Belleroche, J. Allelic variants in the zinc transporter-3 gene, SLC30A3, a candidate gene identified from gene expression studies, show gender-specific association with schizophrenia. Eur. Psychiat. 2014, 29, 172–178. [Google Scholar] [CrossRef]
- Marger, L.; Schubert, C.R.; Bertrand, D. Zinc: An underappreciated modulatory factor of brain function. Biochem. Pharmacol. 2014, 91, 426–435. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, D.; Liang, J.; Bao, Y.P.; Meng, S.Q.; Lu, L.; Shi, J. Association between variants of zinc finger genes and psychiatric disorders: Systematic review and meta-analysis. Schizophr. Res. 2015, 162, 124–137. [Google Scholar] [CrossRef]
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef]
- Dalic, L.; Cook, M.J. Managing drug-resistant epilepsy: Challenges and solutions. Neuropsychiatr. Dis. Treat. 2016, 12, 2605–2616. [Google Scholar] [CrossRef]
- Galanopoulou, A.S.; Buckmaster, P.S.; Staley, K.J.; Moshé, S.L.; Perucca, E.; Engel, J.J.; Löscher, W.; Noebels, J.L.; Pitkänen, A.; Stables, J.; et al. Identification of new epilepsy treatments: Issues in preclinical methodology. Epilepsia 2012, 53, 571–582. [Google Scholar] [CrossRef]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef]
- Saad, K.; Hammad, E.; Hassan, A.F.; Badry, R. Trace element, oxidant, and antioxidant enzyme values in blood of children with refractory epilepsy. Int. J. Neurosci. 2014, 124, 181–186. [Google Scholar] [CrossRef]
- Jia, W.; Song, Y.; Yang, L.; Kong, J.; Boczek, T.; He, Z.; Wang, Y.; Zhang, X.; Hu, H.; Shao, D.; et al. The changes of serum zinc, copper, and selenium levels in epileptic patients: A systematic review and meta-analysis. Expert Rev. Clin. Pharmacol. 2020, 13, 1047–1058. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nature reviews. Neurology 2011, 7, 31–40. [Google Scholar] [PubMed]
- Vezzani, A.; Baram, T.Z. New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy Curr. 2007, 7, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.J.; Lee, S.; Kim, T.; Cho, J.; Koh, J. Zinc and 4-hydroxy-2-nonenal mediate lysosomal membrane permeabilization induced by H2O2 in cultured hippocampal neurons. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 3114–3122. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D.C. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Trans. 2007, 35, 421–423. [Google Scholar] [CrossRef]
- Qian, J.; Xu, K.; Yoo, J.; Chen, T.T.; Andrews, G.; Noebels, J.L. Knockout of Zn2+ transporters Zip-1 and Zip-3 attenuates seizure-induced CA1 neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 97–104. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef]
- Morris, D.R.; Levenson, C.W. Zinc in traumatic brain injury: From neuroprotection to neurotoxicity. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 708–711. [Google Scholar] [CrossRef]
- Gower-Winter, S.D.; Levenson, C.W. Zinc in the central nervous system: From molecules to behavior. BioFactors 2012, 38, 186–193. [Google Scholar] [CrossRef]
- Hellmich, H.L.; Frederickson, C.J.; DeWitt, D.S.; Saban, R.; Parsley, M.O.; Stephenson, R.; Velasco, M.; Uchida, T.; Shimamura, M.; Prough, D.S. Protective effects of zinc chelation in traumatic brain injury correlate with upregulation of neuroprotective genes in rat brain. Neurosci. Lett. 2004, 355, 221–225. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, Q.; Ma, S.; Zhang, Y.; Liang, P. TPEN Attenuates Neural Autophagy Induced by Synaptically-released Zinc Translocation and Improves Histological Outcomes after Traumatic Brain Injury in Rats. Ann. Clin. Lab. Sci. 2018, 48, 446–452. [Google Scholar]
- Levenson, C.W. Zinc and Traumatic Brain Injury: From Chelation to Supplementation. Med. Sci. 2020, 8, 36. [Google Scholar] [CrossRef]
- Yeiser, E.C.; Vanlandingham, J.W.; Levenson, C.W. Moderate zinc deficiency increases cell death after brain injury in the rat. Nutr. Neurosci. 2002, 5, 345–352. [Google Scholar] [CrossRef]
- Kim, K.H.; Ro, Y.S.; Yoon, H.; Lee, S.G.W.; Jung, E.; Moon, S.B.; Park, G.J.; Shin, S.D. Serum Zinc and Long-Term Prognosis after Acute Traumatic Brain Injury with Intracranial Injury: A Multicenter Prospective Study. J. Clin. Med. 2022, 11, 6496. [Google Scholar] [CrossRef]
- Hellmich, H.L.; Eidson, K.; Cowart, J.; Crookshanks, J.; Boone, D.K.; Shah, S.; Uchida, T.; DeWitt, D.S.; Prough, D.S. Chelation of neurotoxic zinc levels does not improve neurobehavioral outcome after traumatic brain injury. Neurosci. Lett. 2008, 440, 155–159. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J. Trace Elem. Med. Biol. 2014, 28, 474–481. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disorder | Serum/Plasma Zn2+ Status | Zn2+ Supplementation |
|---|---|---|
| Alzheimer’s disease (AD) | ↓ [54,55] | Delay memory deficits and reduce both β-amyloid (Aβ) and tau in 3xTg-AD mice [56] |
| No conclusive evidence [57] | ||
| Depression | ↓ [58] | Lower depressive symptom scores as an adjunct [59] |
| Improve depression status as a monotherapy [60] | ||
| Postpartum Zn2+ supplementation reduced the risk of postpartum depression [61] | ||
| Parkinson’s disease (PD) | ↓ [62,63,64] | Improve both lifespan and motor abilities in a drosophila model with a mutant Parkin [65] |
| Multiple sclerosis (MS) | ↓ [66,67] | Reduce clinical signs in animal models [68,69] |
| Schizophrenia (SCZ) | ↓ [70,71] | Effective as an adjuvant agent [72] |
| Effective as a therapeutic agent [73] | ||
| Prenatal Zn2+ supplementation attenuated the behavioral impairments [74] | ||
| Epilepsy | ↓ [75,76] ↑ [77] | A medium dose of Zn2+ reduced the severity of pilocarpine-induced limbic seizures either as a monotherapy or in combination with valproic acid in a rat model of epilepsy [78] |
| Reduce seizure frequency in children with refractory epilepsy [79] | ||
| Mitigate seizures by alleviating inflammation and oxidative stress [78,80] | ||
| Traumatic brain injury (TBI) | ↓ [81] | Effective in improving cognitive impairment and depression in animal models [82] |
| Reduce mortality rate in a double-blinded controlled trial [83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Fang, T.; Chen, H. Zinc and Central Nervous System Disorders. Nutrients 2023, 15, 2140. https://doi.org/10.3390/nu15092140
Wang B, Fang T, Chen H. Zinc and Central Nervous System Disorders. Nutrients. 2023; 15(9):2140. https://doi.org/10.3390/nu15092140
Chicago/Turabian StyleWang, Bangqi, Tianshu Fang, and Hongping Chen. 2023. "Zinc and Central Nervous System Disorders" Nutrients 15, no. 9: 2140. https://doi.org/10.3390/nu15092140
APA StyleWang, B., Fang, T., & Chen, H. (2023). Zinc and Central Nervous System Disorders. Nutrients, 15(9), 2140. https://doi.org/10.3390/nu15092140