
Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases

Abstract
1. Introduction
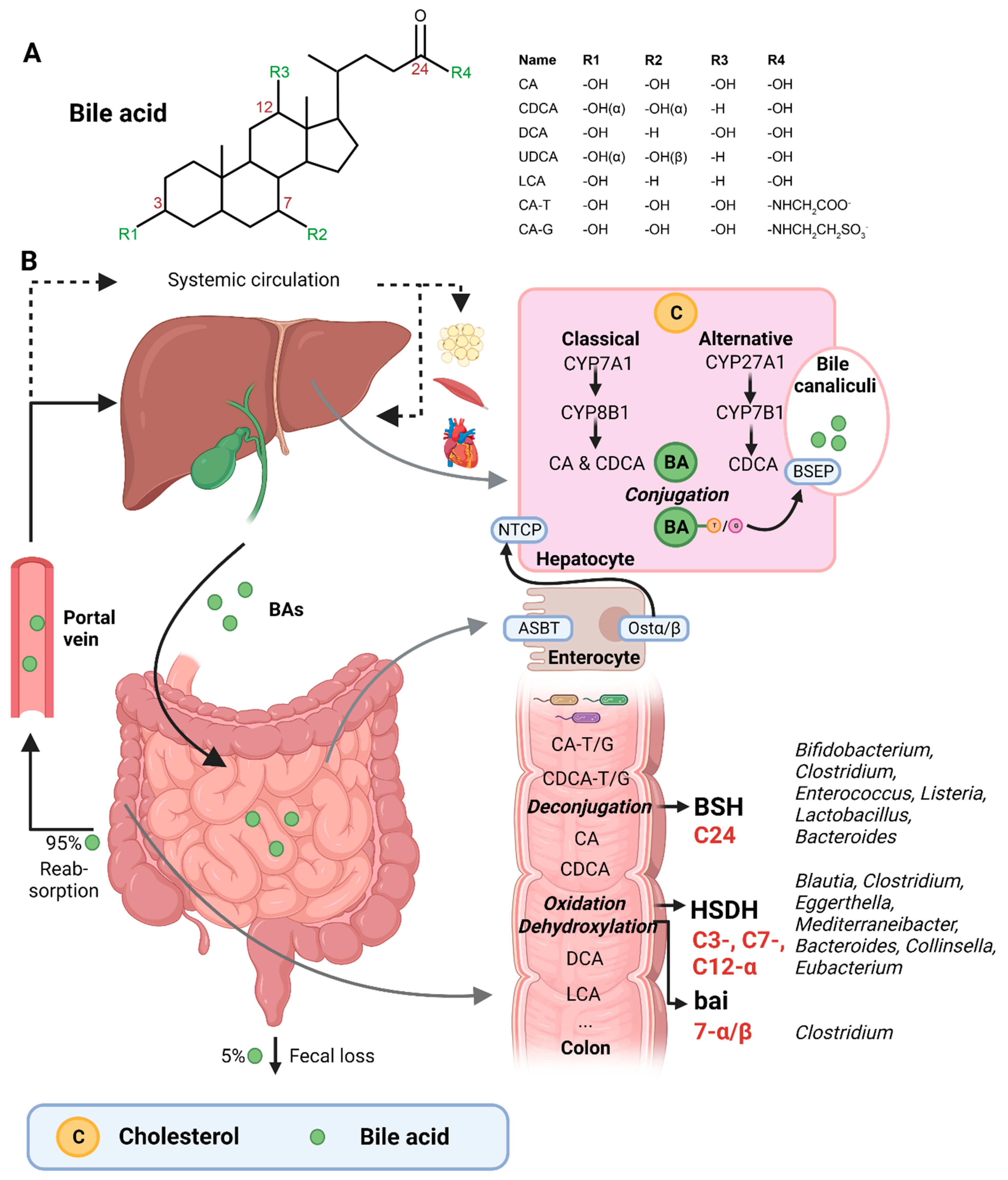
2. Bile Acids Are Synthesized by the Liver and Extensively Metabolized by the Microbiota
3. Bacteria Involved in Bile Acid Metabolism
4. Gut Microbiota Signatures in Cardiovascular Disease
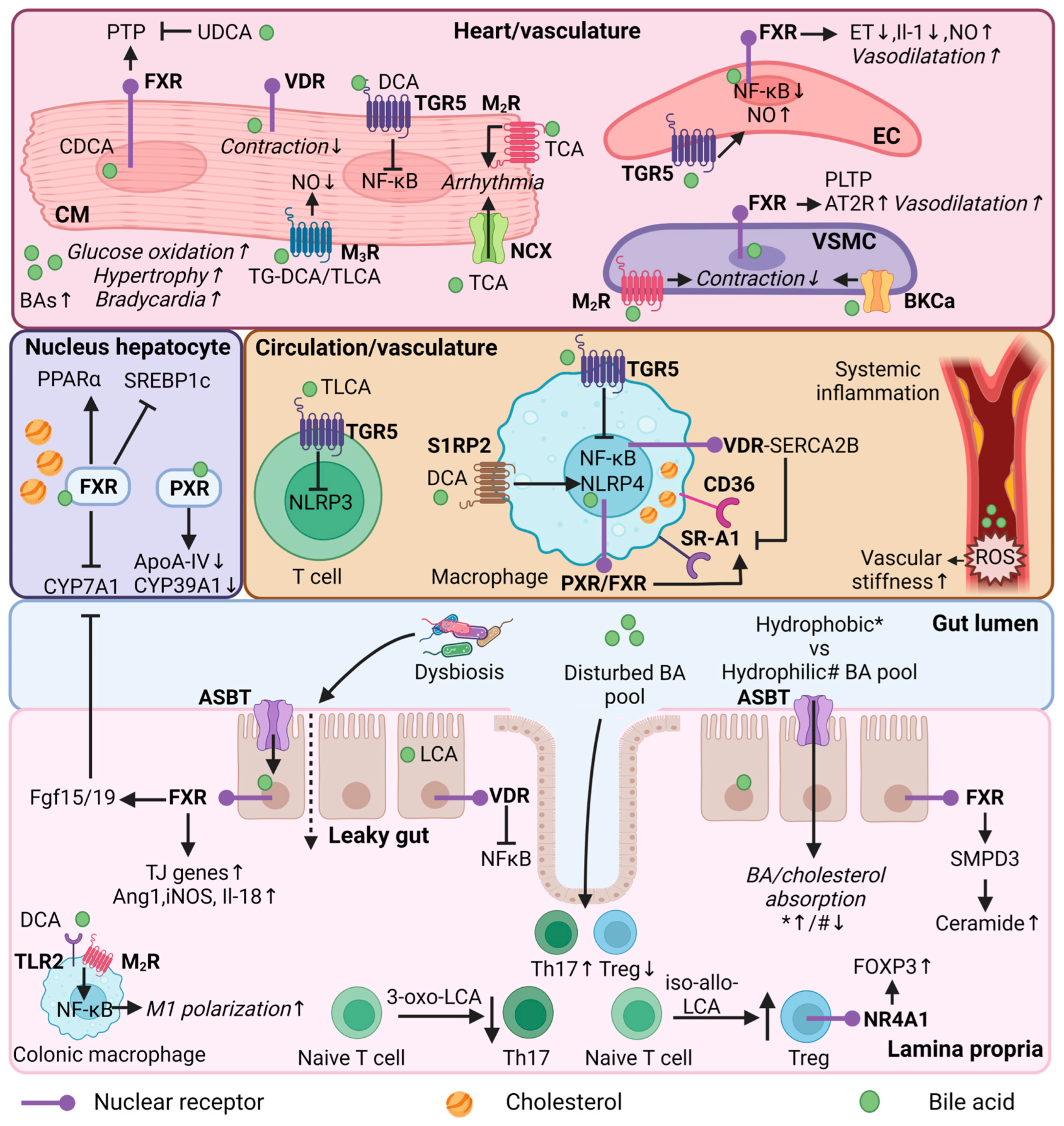
5. Altered Bile Acid Metabolism in Cardiovascular Disease
6. Bile Acids as Mediators of Cardiovascular Disease Risk
6.1. Regulation of Lipid Metabolism
6.2. Regulation of Immune Functions by Secondary Bile Acids
6.3. Regulation of Heart Function
7. Bile-Acid-Based Therapies in Cardiovascular Disease
7.1. Indirect Bile-Acid-Based Therapies
7.2. Direct Bile-Acid-Based Therapies
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cardiovascular Diseases (CVDs). 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 9 November 2022).
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.S.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, E254–E743. [Google Scholar] [CrossRef]
- Arora, S.; Stouffer, G.A.; Kucharska-Newton, A.M.; Qamar, A.; Vaduganathan, M.; Pandey, A.; Porterfield, D.S.; Blankstein, R.; Rosamond, W.D.; Bhatt, D.L.; et al. Twenty Year Trends and Sex Differences in Young Adults Hospitalized with Acute Myocardial Infarction: The ARIC Community Surveillance Study. Circulation 2019, 139, 1047–1056. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Mehta, J.L.; PSaldeen, T.G.; Rand, K. Interactive Role of Infection, Inflammation and Traditional Risk Factors in Atherosclerosis and Coronary Artery Disease. J. Am. Coll. Cardiol. 1998, 31, 1217–1225. [Google Scholar] [CrossRef]
- Dhindsa, D.S.; Sandesara, P.B.; Shapiro, M.D.; Wong, N.D. The Evolving Understanding and Approach to Residual Cardiovascular Risk Management. Front. Cardiovasc. Med. 2020, 7, 88. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Bäckhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Giugliano, G.; Gargiulo, G.; Franzone, A.; Trimarco, B.; Esposito, G.; Perrino, C. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: A systematic review and dose-response meta-analysis. Eur. Heart J. 2017, 38, 2948–2956. [Google Scholar] [CrossRef]
- Rodríguez-Morató, J.; Matthan, N.R. Nutrition and Gastrointestinal Microbiota, Microbial-Derived Secondary Bile Acids, and Cardiovascular Disease. Curr. Atheroscler. Rep. 2020, 22, 47. [Google Scholar] [CrossRef]
- Bays, H.E.; Goldberg, R.B. The Forgotten Bile Acid Sequestrants: Is Now a Good Time to Remember? Am. J. Ther. 2007, 14, 567–580. [Google Scholar] [CrossRef]
- Rifkind, B.M. Lipid research clinics coronary primary prevention trial: Results and implications. Am. J. Cardiol. 1984, 54, 30–34. [Google Scholar] [CrossRef]
- Watts, G.F.; Lewis, B.; Brunt, J.N.H.; Lewis, E.S.; Coltart, D.J.; Smith, L.D.R.; Mann, A.V. Effects on coronary artery disease of lipid-lowering diet, or diet plus cholestyramine, in the St Thomas Atherosclerosis regression study. Lancet 1992, 339, 563–569. [Google Scholar] [CrossRef]
- Watts, G.F.; Pang, J.; Chan, D.C.; Brunt, J.N.; Lewis, B. Angiographic progression of coronary atherosclerosis in patients with familial hypercholesterolaemia treated with non-statin therapy: Impact of a fat-modified diet and a resin. Atherosclerosis 2016, 252, 82–87. [Google Scholar] [CrossRef]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- Porez, G.; Prawitt, J.; Gross, B.; Staels, B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J. Lipid Res. 2012, 53, 1723–1737. [Google Scholar] [CrossRef]
- Monte, M.J.; Marin, J.J.G.; Antelo, A.; Vazquez-Tato, J. Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of Bile Acids and Bile Acid Receptors in Metabolic Regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 24, 678–693. [Google Scholar] [CrossRef]
- Li-Hawkins, J.; Gåfvels, M.; Olin, M.; Lund, E.G.; Andersson, U.; Schuster, G.; Björkhem, I.; Russell, D.W. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Investig. 2002, 110, 1191–1200. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Li, R.; Andreu-Sánchez, S.; Kuipers, F.; Fu, J. Gut microbiome and bile acids in obesity-related diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101493. [Google Scholar] [CrossRef]
- Khurana, S.; Raufman, J.P.; Pallone, T.L. Bile acids regulate cardiovascular function. Clin. Transl. Sci. 2011, 4, 210–218. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling—Mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Xu, C.; Feng, B.; Gao, X.; Liu, Z. Critical roles of bile acids in regulating intestinal mucosal immune responses. Ther. Adv. Gastroenterol. 2021, 14, 17562848211018098. [Google Scholar] [CrossRef]
- Chen, L.; van den Munckhof, I.C.L.; Schraa, K.; ter Horst, R.; Koehorst, M.; van Faassen, M.; van der Ley, C.; Doestzada, M.; Zhernakova, D.V.; Kurilshikov, A.; et al. Genetic and Microbial Associations to Plasma and Fecal Bile Acids in Obesity Relate to Plasma Lipids and Liver Fat Content. Cell Rep. 2020, 33, 108212. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Miyamoto, C.M.; Meighen, E.A.; Lee, B.H. Cloning and characterization of the bile salt hydrolase genes (bsh) from Bifidobacterium bifidum strains. Appl. Environ. Microbiol. 2004, 70, 5603–5612. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.P.; Hudson, L.L. Cloning and Characterization of a Conjugated Bile Acid Hydrolase Gene from Clostridium perfringens. Appl. Environ. Microbiol. 1995, 61, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Dussurget, O.; Cabanes, D.; Dehoux, P.; Lecuit, M.; Buchrieser, C.; Glaser, P.; Cossart, P.; European Listeria Genome Consortium. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol. Microbiol. 2002, 45, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Sleator, R.D.; Gahan, C.G.M.; Hill, C. Contribution of three bile-associated loci, bsh, pva, and btlB, to gastrointestinal persistence and bile tolerance of Listeria monocytogenes. Infect. Immun. 2005, 73, 894–904. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, X.; Mo, Y.; Smith, K.; Guo, Y.; Lin, J. Identification and characterization of a bile salt hydrolase from Lactobacillus salivarius for development of novel alternatives to antibiotic growth promoters. Appl. Environ. Microbiol. 2012, 78, 8795–8802. [Google Scholar] [CrossRef]
- Corzo, G.; Gilliland, S.E. Bile salt hydrolase activity of three strains of Lactobacillus acidophilus. J. Dairy Sci. 1999, 82, 472–480. [Google Scholar] [CrossRef]
- Stellwag, E.J.; Hylemon, P.B. Purification and characterization of bile salt hydrolase from Bacteroides fragilis subsp. fragilis. Biochim. Biophys. Acta 1976, 452, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Park, D.; Hahn, Y.; Jeon, C.O. Metagenomic analysis of the human microbiome reveals the association between the abundance of gut bile salt hydrolases and host health. Gut Microbes. 2020, 11, 1300–1313. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Melnik A v Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; Dorrestein, P.C.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Doden, H.; Sallam, L.A.; Devendran, S.; Ly, L.; Doden, G.; Daniel, S.L.; Alves, J.M.P.; Ridlon, J.M. Metabolism of oxo-bile acids and characterization of recombinant 12α- hydroxysteroid dehydrogenases from bile acid 7α-dehydroxylating human gut bacteria. Appl. Environ. Microbiol. 2018, 84, e00235-18. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef]
- Berr, F.; Kullak-Ublick, G.A.; Paumgartner, G.; Mu¨nzing, W.; Mu¨nzing, M.; Hylemon, P.B. 7a-Dehydroxylating Bacteria Enhance Deoxycholic Acid Input and Cholesterol Saturation of Bile in Patients With Gallstones. Gastroenterology 1996, 111, 1611–1620. [Google Scholar] [CrossRef]
- Brown, J.M.; Hazen, S.L. Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 2018, 16, 171–181. [Google Scholar] [CrossRef]
- Kang, D.J.; Ridlon, J.M.; Moore, D.R.; Barnes, S.; Hylemon, P.B. Clostridium scindens baiCD and baiH genes encode stereo-specific 7α/7β-hydroxy-3-oxo-Δ4-cholenoic acid oxidoreductases. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2008, 1781, 16–25. [Google Scholar] [CrossRef]
- Urdaneta, V.; Casadesús, J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front. Med. 2017, 4, 163. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Eckmann, L. How bile acids confer gut mucosal protection against bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 4333–4334. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Margolles, A.; Sánchez, B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front. Microbiol. 2013, 4, 396. [Google Scholar] [CrossRef] [PubMed]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell. Host Microbe. 2020, 28, 245–257.e6. [Google Scholar] [CrossRef]
- Le, H.H.; Lee, M.T.; Besler, K.R.; Comrie, J.M.C.; Johnson, E.L. Characterization of interactions of dietary cholesterol with the murine and human gut microbiome. Nat. Microbiol. 2022, 7, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; D’Agostino, G.D.; Park, J.; Hang, S.; Adhikari, A.A.; Zhang, Y.; Li, W.; Avila-Pacheco, J.; Bae, S.; Clish, C.B.; et al. A biosynthetic pathway for the selective sulfonation of steroidal metabolites by human gut bacteria. Nat. Microbiol. 2022, 7, 1404–1418. [Google Scholar] [CrossRef]
- Davidson, M.; Kuo, C.C.; Middaugh, J.P.; Campbell, L.A.; Wang, S.P.; Newman, W.P.; Finley, J.C.; Grayston, J.T. Confirmed Previous Infection With Chlamydia pneumoniae (TWAR) and Its Presence in Early Coronary Atherosclerosis. Circulation 1998, 98, 628–633. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4592–4598. [Google Scholar] [CrossRef]
- Chakaroun, R.M.; Olsson, L.M.; Bäckhed, F. The potential of tailoring the gut microbiome to prevent and treat cardiometabolic disease. Nat. Rev. Cardiol. 2022, 20, 217–235. [Google Scholar] [CrossRef]
- Lanter, B.B.; Sauer, K.; Davies, D.G. Bacteria present in carotid arterial plaques are found as biofilm deposits which may contribute to enhanced risk of plaque rupture. MBio 2014, 5, e01206-14. [Google Scholar] [CrossRef]
- Ott, S.J.; el Mokhtari, N.E.; Musfeldt, M.; Hellmig, S.; Freitag, S.; Rehman, A.; Kühbacher, T.; Nikolaus, S.; Namsolleck, P.; Blaut, M. Detection of diverse bacterial signatures in atherosclerotic lesions of patients with coronary heart disease. Circulation 2006, 113, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Y.; Wu, T.T.; Liu, Z.Q.; Li, A.; Guo, Q.Q.; Ma, Y.Y.; Zhang, Z.L.; Xun, Y.L.; Zhang, J.C.; Wang, W.-R.; et al. Gut Microbiome-Based Diagnostic Model to Predict Coronary Artery Disease. J. Agric. Food Chem. 2020, 68, 3548–3557. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, R.; Zhang, Y.; Pan, D.; Zhu, Y.; Zhang, X.; Yang, R.; Jiang, R.; Xu, Y.; Qin, H.; et al. Dysbiosis signatures of gut microbiota in coronary artery disease. Physiol. Genom. 2018, 50, 893–903. [Google Scholar] [CrossRef]
- Yin, J.; Liao, S.X.; He, Y.; Wang, S.; Xia, G.H.; Liu, F.T.; Zhu, J.J.; You, C.; Chen, Q.; Zhou, L.; et al. Dysbiosis of gut microbiota with reduced trimethylamine-n-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J. Am. Heart Assoc. 2015, 4, e002699. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef]
- Toya, T.; Corban, M.T.; Marrietta, E.; Horwath, I.E.; Lerman, L.O.; Murray, J.A.; Lerman, A. Coronary artery disease is associated with an altered gut microbiome composition. PLoS ONE 2020, 15, e0227147. [Google Scholar] [CrossRef]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; et al. Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 2019, 7, 68. [Google Scholar] [CrossRef]
- Feng, Q.; Liu, Z.; Zhong, S.; Li, R.; Xia, H.; Jie, Z.; Wen, B.; Chen, X.; Yan, W.; Fan, Y.; et al. Integrated metabolomics and metagenomics analysis of plasma and urine identified microbial metabolites associated with coronary heart disease. Sci. Rep. 2016, 6, 22525. [Google Scholar] [CrossRef]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef]
- Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut microbiota in hypertension and atherosclerosis: A review. Nutrients 2020, 12, 2982. [Google Scholar] [CrossRef]
- Tamanai-Shacoori, Z.; Smida, I.; Bousarghin, L.; Loreal, O.; Meuric, V.; Fong, S.B.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Roseburia spp.: A marker of health? Future Microbiol. 2017, 12, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Halder, C.V.; de Sousa Faria, A.V.; Andrade, S.S. Action and function of Faecalibacterium prausnitzii in health and disease. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, M.A.; Frank, M.O. Veillonella parvula Meningitis: Case Report and Review of Veillonella Infections. Clin. Infect. Dis. 2000, 31, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Kesavalu, L.; Lucas, A.R.; Verma, R.K.; Liu, L.; Dai, E.; Sampson, E.; Progulske-Fox, A. Increased atherogenesis during Streptococcus mutans infection in ApoE-null mice. J. Dent. Res. 2012, 91, 255–260. [Google Scholar] [CrossRef]
- Chalmers, N.I.; Palmer, R.J.; Cisar, J.O.; Kolenbrander, P.E. Characterization of a Streptococcus sp.-Veillonella sp. community micromanipulated from dental plaque. J. Bacteriol. 2008, 190, 8145–8154. [Google Scholar] [CrossRef]
- Binder, C.J.; Hörkkö, S.; Dewan, A.; Chang, M.K.; Kieu, E.P.; Goodyear, C.S.; Silverman, G.J. Pneumococcal vaccination decreases atherosclerotic lesion formation molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 2003, 9, 736–743. [Google Scholar] [CrossRef]
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef]
- Barlow, G.M.; Yu, A.; Mathur, R. Role of the gut microbiome in obesity and diabetes mellitus. Nutr. Clin. Pract. 2015, 30, 787–797. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Kaser, A. Obesity and the Microbiota. Gastroenterology 2009, 136, 1476–1483. [Google Scholar] [CrossRef]
- Huang, K.; Liu, C.; Peng, M.; Su, Q.; Liu, R.; Guo, Z.; Chen, S.; Li, Z.; Chang, G. Glycoursodeoxycholic acid ameliorates atherosclerosis and alters gut microbiota in apolipoprotein e–deficient mice. J. Am. Heart Assoc. 2021, 10, e019820. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.B.; Black, A.S.; Sobel, A.L.; Zhao, Y.; Mukherjee, P.; Molparia, B.; Moore, N.E.; Aleman Muench, G.R.; Wu, J.; Chen, W.; et al. Directed remodeling of the mouse gut microbiome inhibits the development of atherosclerosis. Nat. Biotechnol. 2020, 38, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tian, R.; Wang, H.; Feng, S.; Li, H.; Xiao, Y.; Luan, X.; Zhang, Z.; Shi, N.; Niu, H.; et al. Gut microbiota from coronary artery disease patients contributes to vascular dysfunction in mice by regulating bile acid metabolism and immune activation. J. Transl. Med. 2020, 18, 382. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.; van der Velden, S.; Ríos-Morales, M.; van Faassen, M.J.; Loreti, M.G.; de Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef]
- Chong Nguyen, C.; Duboc, D.; Rainteau, D.; Sokol, H.; Humbert, L.; Seksik, P.; Bellino, A.; Abdoul, H.; Bouazza, N.; Treluyer, J.M.; et al. Circulating bile acids concentration is predictive of coronary artery disease in human. Sci. Rep. 2021, 11, 22661. [Google Scholar] [CrossRef]
- Li, W.; Shu, S.; Cheng, L.; Hao, X.; Wang, L.; Wu, Y.; Yuan, Z.; Zhou, J. Fasting serum total bile acid level is associated with coronary artery disease, myocardial infarction and severity of coronary lesions. Atherosclerosis 2020, 292, 193–200. [Google Scholar] [CrossRef]
- Steiner, C.; Othman, A.; Saely, C.H.; Rein, P.; Drexel, H.; von Eckardstein, A.; Rentsch, K.M. Bile acid metabolites in serum: Intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS ONE 2011, 6, e25006. [Google Scholar] [CrossRef]
- Neale, G.; Lewis, B.; Weaver, V.; Panveliwalla, D. Serum bile acids in liver disease. Gut 1971, 12, 145–152. [Google Scholar] [CrossRef]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.; He, Y.; Chen, C.; Song, C.; Zhao, Y.; Bai, Y.; Wang, Y.; Pu, J.; Chen, J.; et al. Investigation of novel metabolites potentially involved in the pathogenesis of coronary heart disease using a UHPLC-QTOF/MS-based metabolomics approach. Sci. Rep. 2017, 7, 15357. [Google Scholar] [CrossRef]
- Charach, G.; Grosskopf, I.; Rabinovich, A.; Shochat, M.; Weintraub, M.; Rabinovich, P. The association of bile acid excretion and atherosclerotic coronary artery disease. Ther. Adv. Gastroenterol. 2011, 4, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Charach, G.; Karniel, E.; Novikov, I.; Galin, L.; Vons, S.; Grosskopf, I.; Charach, L. Reduced bile acid excretion is an independent risk factor for stroke and mortality: A prospective follow-up study. Atherosclerosis 2020, 293, 79–85. [Google Scholar] [CrossRef]
- Charach, G.; Argov, O.; Geiger, K.; Charach, L.; Rogowski, O.; Grosskopf, I. Diminished bile acids excretion is a risk factor for coronary artery disease: 20-year follow up and long-term outcome. Ther. Adv. Gastroenterol. 2018, 11, 1756283X17743420. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Bandsma, R.H.J.; Ouwens, M.; Van Der Sluijs, F.H.; Havinga, R.; Boer, T.; Reijngoud, D.J.; Romijn, J.A.; Kuipers, F. Hepatic VLDL Production in ob/ob Mice is Not Stimulated by Massive De Novo Lipogenesis but is Less Sensitive to the Suppressive Effects of Insulin. Diabetes 2003, 52, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Auwerx’, J. I review Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J. Lipid Res. 1996, 37, 907–925. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Preidis, G.A.; Kim, K.H.; Moore, D.D. Nutrient-sensing nuclear receptors PPAR and FXR control liver energy balance. J. Clin. Investig. 2017, 127, 1193–1201. [Google Scholar] [CrossRef]
- Srivastava, R.A.K.; Jahagirdar, R.; Azhar, S.; Sharma, S.; Bisgaier, C.L. Peroxisome proliferator-activated receptor-α selective ligand reduces adiposity, improves insulin sensitivity and inhibits atherosclerosis in LDL receptor-deficient mice. Mol. Cell. Biochem. 2006, 285, 35–50. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, B.R.; Kang, G.H.; Lee, G.J.; Park, Y.J.; Kim, H.; Jang, H.C.; Choi, S.H. The Effects of PPAR Agonists on Atherosclerosis and Nonalcoholic Fatty Liver Disease in ApoE−/−FXR−/− Mice. Endocrinol. Metab. 2021, 36, 1243–1253. [Google Scholar] [CrossRef]
- Torra, I.P.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor α gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; Mackenzie, K.I.; Latour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; King, N.; Chen, K.Y.; Breslow, J.L. Activation of PXR induces hypercholesterolemia in wild-type and accelerates atherosclerosis in apoE deficient mice. J. Lipid Res. 2009, 50, 2004–2013. [Google Scholar] [CrossRef]
- Sui, Y.; Xu, J.; Rios-Pilier, J.; Zhou, C. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J. Lipid Res. 2011, 52, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef]
- Zurkinden, L.; Sviridov, D.; Vogt, B.; Escher, G. Downregulation of Cyp7a1 by Cholic Acid and Chenodeoxycholic Acid in Cyp27a1/ApoE Double Knockout Mice: Differential Cardiovascular Outcome. Front. Endocrinol. 2020, 11, 586980. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Rockwell, C.E.; Cui, J.Y.; Klaassen, C.D. Individual bile acids have differential effects on bile acid signaling in mice. Toxicol. Appl. Pharmacol. 2015, 283, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Palmiotti, A.; de Vries, H.D.; Hovingh, M.V.; Koehorst, M.; Mulder, N.L.; Zhang, Y.; Kats, K.; Bloks, V.W.; Fu, J.; et al. Low production of 12α-hydroxylated bile acids prevents hepatic steatosis in Cyp2c70-/- mice by reducing fat absorption. J. Lipid Res. 2021, 62, 100134. [Google Scholar] [CrossRef]
- Bhat, B.G.; Rapp, S.R.; Beaudry, J.A.; Napawan, N.; Butteiger, D.N.; Hall, K.A.; Null, C.L.; Luo, Y.; Keller, B.T. Inhibition of ileal bile acid transport and reduced atherosclerosis in apoE-/- mice by SC-435. J. Lipid Res. 2003, 44, 1614–1621. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Walsh, D.T.; Warner, T.D. Expression and activation of the farnesoid X receptor in the vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 3668–3673. [Google Scholar] [CrossRef]
- Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. Antiatherosclerotic effect of farnesoid X receptor. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Li, Y.T.Y.; Swales, K.E.; Thomas, G.J.; Warner, T.D.; Bishop-Bailey, D. Farnesoid X receptor ligands inhibit vascular smooth muscle cell inflammation and migration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2606–2611. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, J.; Mu, Y.; Kuruba, R.; Ma, Z.; Wilson, A.; Alber, S.; Jiang, Y.; Stevens, T.; Watkins, S.; et al. Downregulation of endothelin-1 by farnesoid X receptor in vascular endothelial cells. Circ. Res. 2006, 98, 192–199. [Google Scholar] [CrossRef]
- Pols, T.W.H.; Nomura, M.; Harach, T.; lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell. Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef]
- Miyazaki-Anzai, S.; Masuda, M.; Kohno, S.; Levi, M.; Shiozaki, Y.; Keenan, A.L.; Miyazaki, M. Simultaneous inhibition of FXR and TGR5 exacerbates atherosclerotic formation. J. Lipid Res. 2018, 59, 1709–1713. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, K.; Xu, Y.; Xu, Y.; Li, Y.; Xu, J.; Zhu, Y.; Adorini, L.; Lee, Y.K.; Kasumov, T.; Yin, L.; et al. Reversal of metabolic disorders by pharmacological activation of bile acid receptors TGR5 and FXR. Mol. Metab. 2018, 9, 131–140. [Google Scholar] [CrossRef]
- Guo, G.L.; Santamarina-Fojo, S.; Akiyama, T.E.; Amar, M.J.A.; Paigen, B.J.; Brewer, B.; Gonzalez, F.J. Effects of FXR in foam-cell formation and atherosclerosis development. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2006, 1761, 1401–1409. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Vales, C.; Lee, F.Y.; Lee, H.; Lusis, A.J.; Edwards, P.A. FXR deficiency causes reduced atherosclerosis in Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2316–2321. [Google Scholar] [CrossRef]
- Hartman, H.B.; Gardell, S.J.; Petucci, C.J.; Wang, S.; Krueger, J.A.; Evans, M.J. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR−/− and apoE−/− mice. J. Lipid Res. 2009, 50, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, F.; Zalzala, M.; Xu, J.; Gonzalez, F.J.; Adorini, L.; Lee, Y.L.; Yin, L.; Zhang, Y. Farnesoid X Receptor Activation Increases Reverse Cholesterol Transport by Modulating Bile Acid Composition and Cholesterol Absorption in Mice. Hepatology 2016, 64, 1072–1085. [Google Scholar] [CrossRef]
- Wu, Q.; Sun, L.; Hu, X.; Wang, X.; Xu, F.; Chen, B.; Liang, X.; Xia, J.; Wang, P.; Aibara, D.; et al. Suppressing the intestinal farnesoid X receptor/sphingomyelin phosphodiesterase 3 axis decreases atherosclerosis. J. Clin. Investig. 2021, 131, e143865. [Google Scholar] [CrossRef]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef]
- Zhong, Z.; Zhang, H.; Xu, T.; Hao, J.; Chen, X.; Sun, S.; Yang, J.; Sun, J.; Lin, H.; Guo, H. Identification and verification of immune-related biomarkers and immune infiltration in diabetic heart failure. Front. Cardiovasc. Med. 2022, 9, 931066. [Google Scholar] [CrossRef]
- Hayward, S.L.; Bautista-Lopez, N.; Suzuki, K.; Atrazhev, A.; Dickie, P.; Elliott, J.F. CD4 T Cells Play Major Effector Role and CD8 T Cells Initiating Role in Spontaneous Autoimmune Myocarditis of HLA-DQ8 Transgenic IAb Knockout Nonobese Diabetic Mice 1. J. Immunol. 2006, 176, 7715–7725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Prabhu, S.D.; Bansal, S.S. CD4+ T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front. Cardiovasc. Med. 2022, 9, 992653. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xia, N.; Cheng, X. Regulatory T Cells in Chronic Heart Failure. Front. Immunol. 2021, 12, 732794. [Google Scholar] [CrossRef]
- Rurik, J.G.; Aghajanian, H.; Epstein, J.A. Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ. Res. 2021, 128, 1766–1779. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Kumar, V.; Gupta, S.; Bermeo-Blanco, O.; Stratton, M.S.; Gumina, R.J.; Bansal, S.S. Estrogen Receptor-β Agonists Modulate T-Lymphocyte Activation and Ameliorate Left Ventricular Remodeling during Chronic Heart Failure. Circ. Heart Fail. 2022, 15, E008997. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Guan, B.; Tong, J.; Hao, H.; Yang, Z.; Chen, K.; Xu, H.; Wang, A. Bile acid coordinates microbiota homeostasis and systemic immunometabolism in cardiometabolic diseases. Acta Pharm. Sin. B 2022, 12, 2129–2149. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.I.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef]
- Úbeda, M.; Lario, M.; Muñoz, L.; Borrero, M.J.; Rodríguez-Serrano, M.; Sánchez-Díaz, A.M.; Del Campo, R.; Lledó, L.; Pastor, Ó.; García-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef]
- Yao, B.; He, J.; Yin, X.; Shi, Y.; Wan, J.; Tian, Z. The protective effect of lithocholic acid on the intestinal epithelial barrier is mediated by the vitamin D receptor via a SIRT1/Nrf2 and NF-κB dependent mechanism in Caco-2 cells. Toxicol. Lett. 2019, 316, 109–118. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Jamma, T.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Tang, X.; Elloso, M.M.; Harnish, D.C. Bile acids induce adhesion molecule expression in endothelial cells through activation of reactive oxygen species, NF-B, and p38. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Guo, S.; Hu, F.; Guo, Y.; Yan, L.; Ma, W.; Wang, Y.; Wei, Y.; Zhang, Z.; Wang, Z.; et al. The associations between the polymorphisms of Vitamin D receptor and coronary artery disease. Medicine 2016, 95, e3467. [Google Scholar] [CrossRef] [PubMed]
- Szeto, F.L.; Reardon, C.A.; Yoon, D.; Wang, Y.; Wong, K.E.; Chen, Y.; Kong, J.; Liu, S.Q.; Thadhani, R.; Getz, G.S.; et al. Vitamin D receptor signaling inhibits atherosclerosis in Mice. Mol. Endocrinol. 2012, 26, 1091–1101. [Google Scholar] [CrossRef]
- Latic, N.; Erben, R.G. Vitamin D and cardiovascular disease, with emphasis on hypertension, atherosclerosis, and heart failure. Int. J. Mol. Sci. 2020, 21, 6483. [Google Scholar] [CrossRef]
- Stenman, L.K.; Holma, R.; Eggert, A.; Korpela, R. A novel mechanism for gut barrier dysfunction by dietary fat: Epithelial disruption by hydrophobic bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, 227–234. [Google Scholar] [CrossRef]
- Zhao, S.; Gong, Z.; Du, X.; Tian, C.; Wang, L.; Zhou, J.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Deoxycholic acid-mediated sphingosine-1-phosphate receptor 2 signaling exacerbates DSS-induced colitis through promoting cathepsin b release. J. Immunol. Res. 2018, 2018, 2481418. [Google Scholar] [CrossRef]
- Wang, L.; Gong, Z.; Zhang, X.; Zhu, F.; Liu, Y.; Jin, C.; Du, X.; Xu, C.; Chen, Y.; Cai, W.; et al. Gut microbial bile acid metabolite skews macrophage polarization and contributes to high-fat diet-induced colonic inflammation. Gut Microbes 2020, 12, 1819155. [Google Scholar] [CrossRef]
- Wang, J.; Xu, T.; Xu, M. Roles and Mechanisms of TGR5 in the Modulation of CD4+ T Cell Functions in Myocardial Infarction. J. Cardiovasc. Transl. Res. 2022, 15, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef]
- Yang, C.S.; Kim, J.J.; Kim, T.S.; Lee, P.Y.; Kim, S.Y.; Lee, H.M.; Shin, D.M.; Nguyen, L.T.; Lee, M.S.; Jin, H.S.; et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6115. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Riek, A.E.; Darwech, I.; Funai, K.; Shao, J.S.; Chin, K.; Sierra, O.L.; Carmeliet, G.; Ostlund, R.E.; Bernal-Mizrachi, C. Deletion of macrophage vitamin D receptor promotes insulin resistance and monocyte cholesterol transport to accelerate atherosclerosis in mice. Cell. Rep. 2015, 10, 1872–1886. [Google Scholar] [CrossRef]
- Weng, S.; Sprague, J.E.; Oh, J.; Riek, A.E.; Chin, K.; Garcia, M.; Bernal-Mizrachi, C. Vitamin D Deficiency Induces High Blood Pressure and Accelerates Atherosclerosis in Mice. PLoS ONE 2013, 8, e54625. [Google Scholar] [CrossRef]
- Dong, B.; Zhou, Y.; Wang, W.; Scott, J.; Kim, K.; Sun, Z.; Guo, Q.; Lu, Y.; Gonzales, N.M.; Wu, H.; et al. Vitamin D Receptor Activation in Liver Macrophages Ameliorates Hepatic Inflammation, Steatosis, and Insulin Resistance in Mice. Hepatology 2020, 71, 1559–1574. [Google Scholar] [CrossRef] [PubMed]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.; Yao, L.; Zhang, Y.; Bae, S.; D’Agostino, G.D.; Zhang, M.; Kim, E.; Franzosa, E.A.; Avila-Pacheco, J.; Bisanz, J.E.; et al. Human gut bacteria produce ΤH17-modulating bile acid metabolites. Nature 2022, 603, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hang, S.; Fang, Y.; Bae, S.; Zhang, Y.; Zhang, M.; Wang, G.; McCurry, M.D.; Bae, M.; Paik, D.; et al. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell. Host Microbe. 2021, 29, 1366–1377.e9. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Lahore, G.F.; Raposo, B.; Lagerguist, M.; Ohlsson, C.; Sabatier, P.; Xu, B.; Aoun, M.; James, J.; Cai, X.; Zubarev, R.A.; et al. VDR polymorphisms regulated t cells and t cell dependent inflammatory disease. Proc. Natl. Acad. Sci. USA 2020, 117, 24986–24997. [Google Scholar] [CrossRef]
- Pu, J.; Yuan, A.; Shan, P.; Gao, E.; Wang, X.; Wang, Y.; Lau, W.B.; Koch, W.; Ma, X.L.; He, B. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur. Heart J. 2013, 34, 1834–1845. [Google Scholar] [CrossRef]
- Kida, T.; Murata, T.; Hori, M.; Ozaki, H. Chronic stimulation of farnesoid X receptor impairs nitric oxide sensitivity of vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H195–H201. [Google Scholar] [CrossRef]
- Zhang, Q.; He, F.; Kuruba, R.; Gao, X.; Wilson, A.; Li, J.; Billiar, T.J.; Pitt, B.R.; Xie, W.; Li, S. FXR-mediated regulation of angiotensin type 2 receptor expression in vascular smooth muscle cells. Cardiovasc. Res. 2008, 77, 560–569. [Google Scholar] [CrossRef]
- Li, J.; Wilson, A.; Kuruba, R.; Zhang, Q.; Gao, X.; He, F.; Zhang, L.M.; Pitt, B.R.; Xie, W.; Li, S. FXR-mediated regulation of eNOS expression in vascular endothelial cells. Cardiovasc. Res. 2008, 77, 169–177. [Google Scholar] [CrossRef]
- Rajesh, K.G.; Suzuki, R.; Maeda, H.; Yamamoto, M.; Yutong, X.; Sasaguri, S. Hydrophilic bile salt ursodeoxycholic acid protects myocardium against reperfusion injury in a PI3K/Akt dependent pathway. J. Mol. Cell. Cardiol. 2005, 39, 766–776. [Google Scholar] [CrossRef]
- Thromb, A.; Biol, V. Bile Acid Receptor TGR5 Agonism Induces NO Production and Reduces Monocyte Adhesion in Vascular Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1663–1669. [Google Scholar]
- Sandoo, A.; Veldhuijzen Van Zanten, J.J.C.S.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Lin, X.; Wang, Y.; Wu, X.; Yang, F.; Gao, W.; Zhang, Y.; Sun, J.; Jiang, C.; et al. DCA-TGR5 signaling activation alleviates inflammatory response and improves cardiac function in myocardial infarction. J. Mol. Cell. Cardiol. 2021, 151, 3–14. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell. Physiol. 2007, 292, 82–97. [Google Scholar] [CrossRef]
- Desai, M.S.; Mathur, B.; Eblimit, Z.; Vasquez, H.; Taegtmeyer, H.; Karpen, S.J.; Penny, D.J.; Moore, D.D.; Anakk, S. Bile Acid Excess Induces Cardiomyopathy and Metabolic Dysfunctions in the Heart. Hepatology 2017, 65, 189–201. [Google Scholar] [CrossRef]
- Desai, M.S.; Shabier, Z.; Taylor, M.; Lam, F.; Thevananther, S.; Kosters, A.; Karpen, S.J. Hypertrophic cardiomyopathy and dysregulation of cardiac energetics in a mouse model of biliary fibrosis. Hepatology 2010, 51, 2097–2107. [Google Scholar] [CrossRef]
- Tishkoff, D.X.; Nibbelink, K.A.; Holmberg, K.H.; Dandu, L.; Simpson, R.U. Functional vitamin D receptor (VDR) in the T-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008, 149, 558–564. [Google Scholar] [CrossRef]
- Rodriguez, A.J.; Mousa, A.; Ebeling, P.R.; Scott, D.; de Courten, B. Effects of Vitamin D supplementation on inflammatory markers in heart failure: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 1169. [Google Scholar] [CrossRef]
- Ibrahim, M.; Gorelik, J.; Yacoub, M.H.; Terracciano, C.M. The structure and function of cardiac t-tubules in health and disease. Proc. R. Soc. B Biol. Sci. 2011, 278, 2714–2723. [Google Scholar] [CrossRef]
- Giovannucci, E.; Liu, Y.; Hollis, B.W.; Rimm, E.B. 25-Hydroxyvitamin D and Risk of Myocardial Infarction in Men a Prospective Study. Arch. Intern. Med. 2008, 168, 1174–1180. [Google Scholar] [CrossRef]
- Dobnig, H.; Pilz, S.; Scharnagl, H.; Renner, W.; Seelhorst, U.; Wellnitz, B.; Kinkeldei, J.; Boehm, B.O.; Weihrauch, G.; Maerz, W. Independent Association of Low Serum 25-Hydroxyvitamin D and 1,25-Dihydroxyvitamin D Levels With All-Cause and Cardiovascular Mortality. Arch. Intern. Med. 2008, 168, 1340–1349. [Google Scholar] [CrossRef]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef]
- Raufman, J.P.; Chen, Y.; Cheng, K.; Compadre, C.; Compadre, L.; Zimniak, P. Selective interaction of bile acids with muscarinic receptors: A case of molecular mimicry. Eur. J. Pharmacol. 2002, 457, 77–84. [Google Scholar] [CrossRef]
- Raufman, J.P.; Chen, Y.; Zimniak, P.; Cheng, K. Deoxycholic Acid Conjugates are Muscarinic Cholinergic Receptor Antagonists. Pharmacology 2002, 65, 215–221. [Google Scholar] [CrossRef]
- Cheng, K.; Khurana, S.; Chen, Y.; Kennedy, R.H.; Zimniak, P.; Raufman, J.P. Lithocholylcholine, a bile acid/acetylcholine hybrid, is a muscarinic receptor antagonist. J. Pharmacol. Exp. Ther. 2002, 303, 29–35. [Google Scholar] [CrossRef]
- Shah, N.; Khurana, S.; Cheng, K.; Raufman, J.P. Muscarinic receptors and ligands in cancer. Am. J. Physiol. Cell. Physiol. 2009, 296, 221–232. [Google Scholar] [CrossRef]
- Khurana, S.; Yamada, M.; Wess, J.; Kennedy, R.H.; Raufman, J.P. Deoxycholyltaurine-induced vasodilation of rodent aorta is nitric oxide- and muscarinic M3 receptor-dependent. Eur. J. Pharmacol. 2005, 517, 103–110. [Google Scholar] [CrossRef]
- Ibrahim, E.; Diakonov, I.; Arunthavarajah, D.; Swift, T.; Goodwin, M.; McIlvride, S.; Nikolova, V.; Williamson, C.; Gorelik, J. Bile acids and their respective conjugates elicit different responses in neonatal cardiomyocytes: Role of Gi protein, muscarinic receptors and TGR5. Sci. Rep. 2018, 8, 7110. [Google Scholar] [CrossRef]
- Sheikh Abdul Kadir, S.H.; Miragoli, M.; Abu-Hayyeh, S.; Moshkov A v Xie, Q.; Keitel, V.; Nikolaev, V.O.; Williamson, C.; Gorelik, J. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS ONE 2010, 5, e9689. [Google Scholar] [CrossRef]
- Dopico, A.M.; Walsh J v Singer, J.J. Natural Bile Acids and Synthetic Analogues Modulate Large Conductance Ca 2-activated K (BK Ca) Channel Activity in Smooth Muscle Cells. J. Gen. Physiol. 2002, 119, 251–273. [Google Scholar] [CrossRef]
- Bukiya, A.N.; Liu, J.; Toro, L.; Dopico, A.M. β1 (KCNMB1) subunits mediate lithocholate activation of large-conductance Ca2+-activated K+ channels and dilation in small, resistance-size arteries. Mol. Pharmacol. 2007, 72, 359–369. [Google Scholar] [CrossRef]
- Rainer, P.P.; Primessnig, U.; Harenkamp, S.; Doleschal, B.; Wallner, M.; Fauler, G.; Stojakovic, T.; Wachter, R.; Yates, A.; Groschner, K.; et al. Bile acids induce arrhythmias in human atrial myocardium-implications for altered serum bile acid composition in patients with atrial fibrillation. Heart 2013, 99, 1685–1692. [Google Scholar] [CrossRef]
- Oniszczuk, A.; Oniszczuk, T.; Gancarz, M.; Szymá Nska, J.; Hamaguchi, M. Role of Gut Microbiota, Probiotics and Prebiotics in the Cardiovascular Diseases. Molecules 2021, 26, 1172. [Google Scholar] [CrossRef]
- Bui, T.P.N.; de Vos, W.M. Next-generation therapeutic bacteria for treatment of obesity, diabetes, and other endocrine diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101504. [Google Scholar] [CrossRef]
- Lambert, J.E.; Parnell, J.A.; Tunnicliffe, J.M.; Han, J.; Sturzenegger, T.; Reimer, R.A. Consuming yellow pea fiber reduces voluntary energy intake and body fat in overweight/obese adults in a 12-week randomized controlled trial. Clin. Nutr. 2017, 36, 126–133. [Google Scholar] [CrossRef]
- Mayengbam, S.; Lambert, J.E.; Parnell, J.A.; Tunnicliffe, J.M.; Nicolucci, A.C.; Han, J.; Sturzenegger, T.; Shearer, J.; Mickiewicz, B.; Vogel, H.J.; et al. Impact of dietary fiber supplementation on modulating microbiota–host–metabolic axes in obesity. J. Nutr. Biochem. 2019, 64, 228–236. [Google Scholar] [CrossRef]
- O’Morain, V.L.; Ramji, D.P. The Potential of Probiotics in the Prevention and Treatment of Atherosclerosis. Mol. Nutr. Food Res. 2020, 64, e1900797. [Google Scholar] [CrossRef]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef]
- Hassan, A.; Din, A.U.; Zhu, Y.; Zhang, K.; Li, T.; Wang, Y.; Luo, Y.; Wang, G. Updates in understanding the hypocholesterolemia effect of probiotics on atherosclerosis. Appl. Microbiol. Biotechnol. 2019, 103, 5993–6006. [Google Scholar] [CrossRef]
- Mahdavi-Roshan, M.; Salari, A.; Kheirkhah, J.; Ghorbani, Z. The Effects of Probiotics on Inflammation, Endothelial Dysfunction, and Atherosclerosis Progression: A Mechanistic Overview. Heart Lung Circ. 2022, 31, e45–e71. [Google Scholar] [CrossRef]
- Kim, K.T.; Kim, J.W.; Kim S il Kim, S.; Nguyen, T.H.; Kang, C.H. Antioxidant and anti-inflammatory effect and probiotic properties of lactic acid bacteria isolated from canine and feline feces. Microorganisms 2021, 9, 1971. [Google Scholar] [CrossRef]
- Hibberd, A.A.; Yde, C.C.; Ziegler, M.L.; Honoré, A.H.; Saarinen, M.T.; Lahtinen, S.; Stahl, B.; Jensen, H.M.; Stenman, L.K. Probiotic or synbiotic alters the gut microbiota and metabolism in a randomised controlled trial of weight management in overweight adults. Benef. Microbes 2019, 10, 121–135. [Google Scholar] [CrossRef]
- Ghorbani, Z.; Kazemi, A.; UPBartolomaeus, T.; Martami, F.; Noormohammadi, M.; Salari, A.; Löber, U.; Balou, H.A.; Forslund, S.K.; Mahdavi-Roshan, M. The effect of probiotic and synbiotic supplementation on lipid parameters among patients with cardiometabolic risk factors: A systematic review and meta-analysis of clinical trials. Cardiovasc. Res. 2022. [Google Scholar] [CrossRef]
- Hadi, A.; Ghaedi, E.; Khalesi, S.; Pourmasoumi, M.; Arab, A. Effects of synbiotic consumption on lipid profile: A systematic review and meta-analysis of randomized controlled clinical trials. Eur. J. Nutr. 2020, 59, 2857–2874. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid x receptor agonist Obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Hodge, R.J.; Lin, J.; Vasist Johnson, L.S.; Gould, E.P.; Bowers, G.D.; Nunez, D.J. Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 Agonist, SB-756050, in Type 2 Diabetes. Clin. Pharmacol. Drug Dev. 2013, 2, 213–222. [Google Scholar] [CrossRef]
- Bhimanwar, R.S.; Mittal, A. TGR5 agonists for diabetes treatment: A patent review and clinical advancements (2012-present). Expert Opin. Ther. Pat. 2022, 32, 191–209. [Google Scholar] [CrossRef]
- de Boer, J.F.; de Vries, H.D.; Palmiotti, A.; Li, R.; Doestzada, M.; Hoogerland, J.A.; Fu, J.; La Rose, A.M.; Westerterp, M.; Mulder, N.L.; et al. Cholangiopathy and Biliary Fibrosis in Cyp2c70-Deficient Mice Are Fully Reversed by Ursodeoxycholic Acid. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1045–1069. [Google Scholar] [CrossRef]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Straniero, S.; Laskar, A.; Savva, C.; Härdfeldt, J.; Angelin, B.; Rudling, M. Of mice and men: Murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J. Lipid Res. 2020, 61, 480–491. [Google Scholar] [CrossRef]
- Yang, J.; Palmiotti, A.; Kuipers, F. Emerging roles of bile acids in control of intestinal functions. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 127–133. [Google Scholar] [CrossRef]
- Kritikou, E.; Depuydt, M.A.C.; de Vries, M.R.; Mulder, K.E.; Govaert, A.M.; Smit, M.D.; van Duijn, J.; Foks, A.C.; Wezel, A.; Smeets, H.J.; et al. Flow Cytometry-Based Characterization of mast Cells in Human Atherosclerosis. Cells 2019, 8, 334. [Google Scholar] [CrossRef]
- Erbel, C.; Okuyucu, D.; Akhavanpoor, M.; Zhao, L.; Wangler, S.; Hakimi, M.; Doesch, A.; Dengler, T.J.; Katus, H.A.; Gleissner, C.A. A human ex vivo atherosclerotic plaque model to study lesion biology. J. Vis. Exp. 2014, 87, e50542. [Google Scholar]
- Lebedeva, A.; Vorobyeva, D.; Vagida, M.; Ivanova, O.; Felker, E.; Fitzgerald, W.; Danilova, N.; Gontarenko, V.; Shpektor, A.; Vasilieva, E.; et al. Ex vivo culture of human atherosclerotic plaques: A model to study immune cells in atherogenesis. Atherosclerosis 2017, 267, 90–98. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Author | Population | Atherosclerosis Definition | Sequencing Method | Higher Abundance in Atherosclerosis | Lower Abundance in Atherosclerosis | Microbial Diversity in Atherosclerosis | Covariates in Analyses |
|---|---|---|---|---|---|---|---|
| Zheng et al., 2020 [63] | 152 patients 105 controls | Atherosclerosis (≥50% stenosis in one or more vessels) | 16S | Bulleidia, Comamonas, Enhydrobacter | Agrobacterium, Delftia, Enterobacter, Morganella | Increased | Unadjusted |
| Karlsson et al., 2012 [64] | 12 patients 13 controls | Symptomatic atherosclerosis (who had undergone CAE) | Shotgun | Collinsella (7α-HSDH BSH-T4) | Roseburia (BSH-T1), Eubacterium (7α-HSDH/BSH-T1) | NR | Smoking, diabetes, age and BMI |
| Zhu et al., 2018 [65] | 70 patients 98 controls | Atherosclerosis (confirmed by coronary angiography) | 16S | Escherichia-Shigella, Enterococcus (BSH) | Roseburia (BSH-T1), Eubacterium rectale (7α-HSDH/BSH-T1), Faecalibacterium, Enterococcus (BSH-T0) | Decreased | Unadjusted |
| Yin et al., 2015 [66] | 141 patients 94 controls | Symptomatic atherosclerosis (with TIA) | 16S | Enterobacter, Megaspaera, Desulfovibrio | Bacteroides (7α-HSDH/BSH-T5/6), Prevotella, Faecalibacterium | Increased | Unadjusted |
| Jie et al., 2017 [67] | 218 patients 187 controsl | Atherosclerosis (≥50% stenosis in one or more vessels) | Shotgun | Enterobacteriaceae, Streptococcus spp. (BSH-T2) | Roseburia (BSH-T1), Faecalibacterium Prausnitzii | No difference | Unadjusted |
| Toya et al., 2020 [68] | 53 patients 53 controls | Atherosclerosis (≥50% stenosis in one or more vessels) | 16S | Ruminococcus gnavus (3α-HSDH BSH-T1) | Lachnospiraceae NK4B4, Ruminococcus Gauvreauii (BSH-T1) | Decreased | Age, sex, race, BMI, DM, dyslipidemia |
| Liu et al., 2019 [69] | 161 patients 40 controls | Atherosclerosis (≥50% stenosis in one or more vessels) | 16S | Veillonella, Haemophilus, Klebsiella | Roseburia (BSH-T1), Faecalibacterium | No difference | Unadjusted |
| Feng et al., 2016 [70] | 59 patients 43 controls | Atherosclerosis (confirmed by coronary angiography) | Shotgun | Clostridium sp. HGF2 (BSH-T0), Streptococcus sp. M334/M143 (BSH-T2) | NR | NR | Unadjusted |
| Yoshida et al., 2018 [71] | 30 patients 30 controls with risk factors | Atherosclerosis (≥75% stenosis in one or more vessels) AND stable angina pectoris, MI | 16S | Faecalibacterium prausnitzii, Prevotella copri | Bacteroides vulgatus, Bacteroides dorei (BSH-T5/6) | No difference | Age, sex |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yntema, T.; Koonen, D.P.Y.; Kuipers, F. Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases. Nutrients 2023, 15, 1850. https://doi.org/10.3390/nu15081850
Yntema T, Koonen DPY, Kuipers F. Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases. Nutrients. 2023; 15(8):1850. https://doi.org/10.3390/nu15081850
Chicago/Turabian StyleYntema, Tess, Debby P. Y. Koonen, and Folkert Kuipers. 2023. "Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases" Nutrients 15, no. 8: 1850. https://doi.org/10.3390/nu15081850
APA StyleYntema, T., Koonen, D. P. Y., & Kuipers, F. (2023). Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases. Nutrients, 15(8), 1850. https://doi.org/10.3390/nu15081850