
The Genetic Basis of Childhood Obesity: A Systematic Review



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Eligibility Criteria
2.3. Literature Search
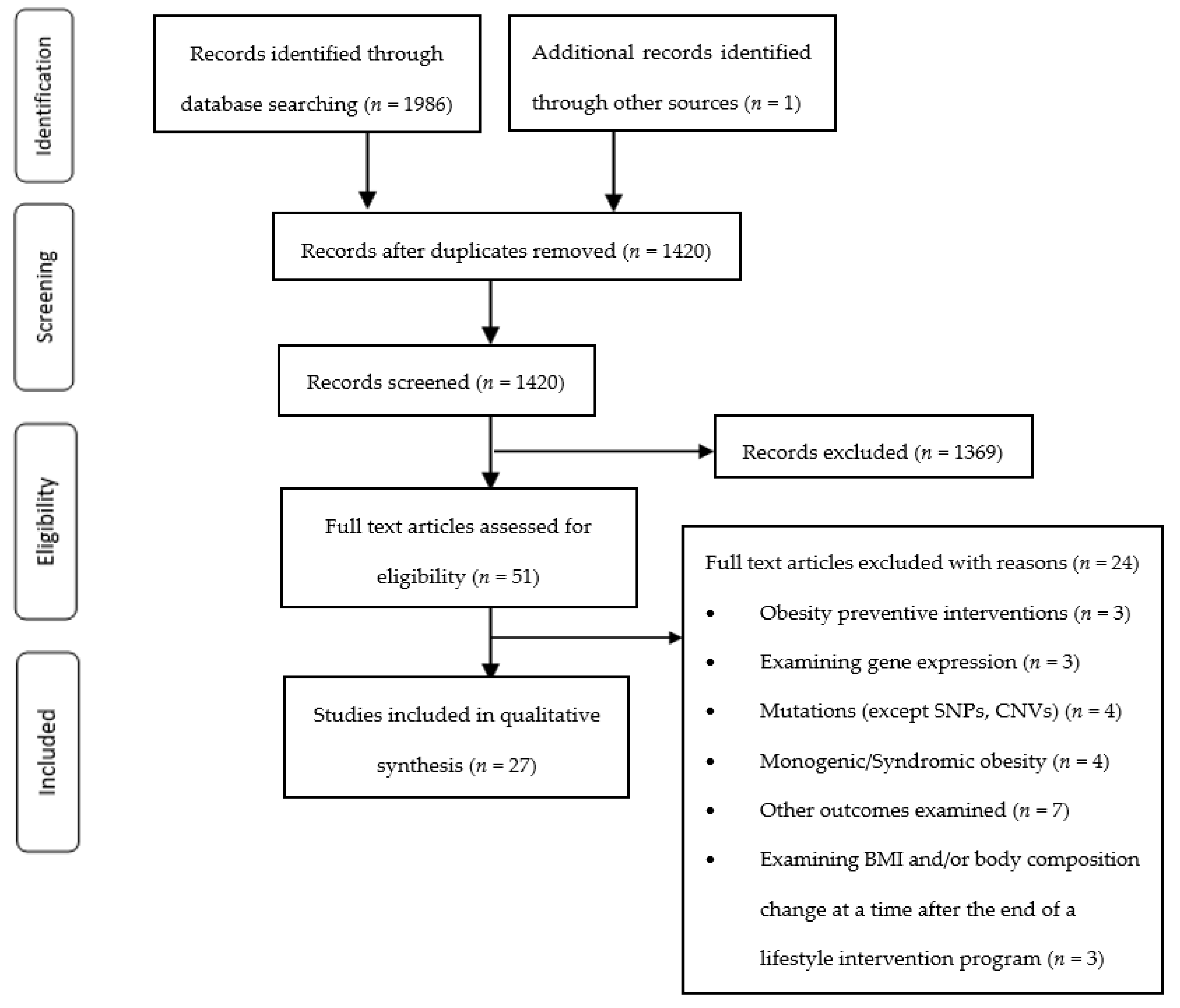
2.4. Study Selection
2.5. Data Extraction, Outcomes and Data Synthesis
2.6. Validity Assessment
3. Results
4. Discussion
4.1. Central Nervous System and Obesity
4.2. Adipose Tissue and Obesity
4.3. Adipose Tissue Metabolism
4.4. Adipose Tissue Inflammation and Obesity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 9 June 2021).
- Simmonds, M.; Llewellyn, A.; Owen, C.G.; Woolacott, N. Predicting adult obesity from childhood obesity: A systematic review and meta-analysis. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2016, 17, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kelly, A.S. Review of Childhood Obesity: From Epidemiology, Etiology, and Comorbidities to Clinical Assessment and Treatment. Mayo Clin. Proc. 2017, 92, 251–265. [Google Scholar] [CrossRef] [PubMed]
- WHO-Europe. Data and Statistics. Available online: http://www.euro.who.int/en/health-topics/noncommunicable-diseases/obesity/data-and-statistics (accessed on 25 May 2022).
- WHO. International Classification of Diseases for Mortality and Morbidity Statistics (11th Revision). Available online: https://icd.who.int/browse11/l-m/en (accessed on 29 May 2022).
- Styne, D.M.; Arslanian, S.A.; Connor, E.L.; Farooqi, I.S.; Murad, M.H.; Silverstein, J.H.; Yanovski, J.A. Pediatric Obesity-Assessment, Treatment, and Prevention: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2017, 102, 709–757. [Google Scholar] [CrossRef]
- Weihrauch-Blüher, S.; Schwarz, P.; Klusmann, J.H. Childhood obesity: Increased risk for cardiometabolic disease and cancer in adulthood. Metab. Clin. Exp. 2019, 92, 147–152. [Google Scholar] [CrossRef]
- Singh, M. Anthropometric Measures during Infancy and Childhood and the Risk of Developing Cardiovascular Disease or Diabetes Mellitus Type 2 in Later Life: A Systematic Review; ICMR Advanced Centre For Evidence Based Child Health, PGIMER, Eds.; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Zhang, T.; Zhang, H.; Li, Y.; Li, S.; Fernandez, C.; Bazzano, L.; He, J.; Xue, F.; Chen, W. Long-term Impact of Temporal Sequence from Childhood Obesity to Hyperinsulinemia on Adult Metabolic Syndrome and Diabetes: The Bogalusa Heart Study. Sci. Rep. 2017, 7, 43422. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Yaniv, G.; Levine, H.; Leiba, A.; Goldberger, N.; Derazne, E.; Ben-Ami Shor, D.; Tzur, D.; Afek, A.; Shamiss, A.; et al. Body-Mass Index in 2.3 Million Adolescents and Cardiovascular Death in Adulthood. N. Engl. J. Med. 2016, 374, 2430–2440. [Google Scholar] [CrossRef]
- Reilly, J.J.; Kelly, J. Long-term impact of overweight and obesity in childhood and adolescence on morbidity and premature mortality in adulthood: Systematic review. Int. J. Obes. 2011, 35, 891–898. [Google Scholar] [CrossRef]
- Park, M.H.; Falconer, C.; Viner, R.M.; Kinra, S. The impact of childhood obesity on morbidity and mortality in adulthood: A systematic review. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2012, 13, 985–1000. [Google Scholar] [CrossRef]
- Ramachandrappa, S.; Farooqi, I.S. Genetic approaches to understanding human obesity. J. Clin. Investig. 2011, 121, 2080–2086. [Google Scholar] [CrossRef]
- Tounian, P. Programming towards Childhood Obesity. Ann. Nutr. Metab. 2011, 58 (Suppl. S2), 30–41. [Google Scholar] [CrossRef]
- van der Klaauw, A.A.; Farooqi, I.S. The Hunger Genes: Pathways to Obesity. Cell 2015, 161, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Reddon, H.; Guéant, J.L.; Meyre, D. The importance of gene-environment interactions in human obesity. Clin. Sci. 2016, 130, 1571–1597. [Google Scholar] [CrossRef]
- Swinburn, B.; Egger, G.; Raza, F. Dissecting obesogenic environments: The development and application of a framework for identifying and prioritizing environmental interventions for obesity. Prev. Med. 1999, 29, 563–570. [Google Scholar] [CrossRef]
- Bouchard, C. Defining the genetic architecture of the predisposition to obesity: A challenging but not insurmountable task. Am. J. Clin. Nutr. 2009, 91, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Silventoinen, K.; Rokholm, B.; Kaprio, J.; Sørensen, T.I. The genetic and environmental influences on childhood obesity: A systematic review of twin and adoption studies. Int. J. Obes. 2010, 34, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Elks, C.E.; den Hoed, M.; Zhao, J.H.; Sharp, S.J.; Wareham, N.J.; Loos, R.J.F.; Ong, K.K. Variability in the heritability of body mass index: A systematic review and meta-regression. Front. Endocrinol. 2012, 3, 29. [Google Scholar] [CrossRef]
- Schleinitz, D.; Böttcher, Y.; Blüher, M.; Kovacs, P. The genetics of fat distribution. Diabetologia 2014, 57, 1276–1286. [Google Scholar] [CrossRef]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015, 518, 187–196. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef]
- Frazer, K.A.; Murray, S.S.; Schork, N.J.; Topol, E.J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 2009, 10, 241–251. [Google Scholar] [CrossRef]
- Vogelezang, S.; Bradfield, J.P.; Ahluwalia, T.S.; Curtin, J.A.; Lakka, T.A.; Grarup, N.; Scholz, M.; van der Most, P.J.; Monnereau, C.; Stergiakouli, E.; et al. Novel loci for childhood body mass index and shared heritability with adult cardiometabolic traits. PLoS Genet. 2020, 16, e1008718. [Google Scholar] [CrossRef]
- Bradfield, J.P.; Vogelezang, S.; Felix, J.F.; Chesi, A.; Helgeland, Ø.; Horikoshi, M.; Karhunen, V.; Lowry, E.; Cousminer, D.L.; Ahluwalia, T.S.; et al. A trans-ancestral meta-analysis of genome-wide association studies reveals loci associated with childhood obesity. Hum. Mol. Genet. 2019, 28, 3327–3338. [Google Scholar] [CrossRef]
- Felix, J.F.; Bradfield, J.P.; Monnereau, C.; van der Valk, R.J.; Stergiakouli, E.; Chesi, A.; Gaillard, R.; Feenstra, B.; Thiering, E.; Kreiner-Møller, E.; et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum. Mol. Genet. 2016, 25, 389–403. [Google Scholar] [CrossRef]
- Phillips, M.; Babu, J.R.; Wang, X.; Geetha, T. DNA copy number and structural variation (CNV) contributions to adult and childhood obesity. Biochem. Soc. Trans. 2020, 48, 1819–1828. [Google Scholar] [CrossRef]
- Yengo, L.; Sidorenko, J.; Kemper, K.E.; Zheng, Z.; Wood, A.R.; Weedon, M.N.; Frayling, T.M.; Hirschhorn, J.; Yang, J.; Visscher, P.M. Meta-analysis of genome-wide association studies for height and body mass index in ∼700,000 individuals of European ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef] [PubMed]
- Ghanemi, A.; Yoshioka, M.; St-Amand, J. Broken Energy Homeostasis and Obesity Pathogenesis: The Surrounding Concepts. J. Clin. Med. 2018, 7, 453. [Google Scholar] [CrossRef]
- van Galen, K.A.; Ter Horst, K.W.; Serlie, M.J. Serotonin, food intake, and obesity. Obes. Rev. 2021, 22, e13210. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D.; Scherer, P.E. Adipocyte metabolism and obesity. J. Lipid Res. 2009, 50, S395–S399. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Giralt, M.; Villarroya, F. White, brown, beige/brite: Different adipose cells for different functions? Endocrinology 2013, 154, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Matsushita, M.; Yoneshiro, T.; Okamatsu-Ogura, Y. Brown Adipose Tissue, Diet-Induced Thermogenesis, and Thermogenic Food Ingredients: From Mice to Men. Front. Endocrinol. 2020, 11, 222. [Google Scholar] [CrossRef]
- Mikhailova, S.V.; Ivanoshchuk, D.E. Innate-Immunity Genes in Obesity. J. Pers. Med. 2021, 11, 1201. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Nikolajczyk, B.S. Tissue Immune Cells Fuel Obesity-Associated Inflammation in Adipose Tissue and Beyond. Front. Immunol. 2019, 10, 01587. [Google Scholar] [CrossRef]
- Tian, Y.; Jennings, J.; Gong, Y.; Sang, Y. Viral Infections and Interferons in the Development of Obesity. Biomolecules 2019, 9, 726. [Google Scholar] [CrossRef]
- Tan, P.Y.; Mitra, S.R.; Amini, F. Lifestyle Interventions for Weight Control Modified by Genetic Variation: A Review of the Evidence. Public Health Genom. 2018, 21, 169–185. [Google Scholar] [CrossRef]
- Xiang, L.; Wu, H.; Pan, A.; Patel, B.; Xiang, G.; Qi, L.; Kaplan, R.C.; Hu, F.; Wylie-Rosett, J.; Qi, Q. FTO genotype and weight loss in diet and lifestyle interventions: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2016, 103, 1162–1170. [Google Scholar] [CrossRef]
- Kilpeläinen, T.O.; Qi, L.; Brage, S.; Sharp, S.J.; Sonestedt, E.; Demerath, E.; Ahmad, T.; Mora, S.; Kaakinen, M.; Sandholt, C.H.; et al. Physical activity attenuates the influence of FTO variants on obesity risk: A meta-analysis of 218,166 adults and 19,268 children. PLoS Med. 2011, 8, e1001116. [Google Scholar] [CrossRef]
- Rendo, T.; Moleres, A.; Marti Del Moral, A. Effects of the FTO gene on lifestyle intervention studies in children. Obes. Facts 2009, 2, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Koochakpoor, G.; Hosseini-Esfahani, F.; Daneshpour, M.S.; Hosseini, S.A.; Mirmiran, P. Effect of interactions of polymorphisms in the Melanocortin-4 receptor gene with dietary factors on the risk of obesity and Type 2 diabetes: A systematic review. Diabet. Med. A J. Br. Diabet. Assoc. 2016, 33, 1026–1034. [Google Scholar] [CrossRef]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef]
- Weber, D.R.; Leonard, M.B.; Zemel, B.S. Body composition analysis in the pediatric population. Pediatr. Endocrinol. Rev. PER 2012, 10, 130–139. [Google Scholar]
- Barbian, C.D.; Reuter, C.P.; Renner, J.D.P.; da Silva, P.T.; Klinger, E.I.; Hobkirk, J.P.; de Moura Valim, A.R.; Burgos, M.S. Anthropometric and Metabolic Responses in FTO rs9939609 Gene Polymorphism after a Multidisciplinary Lifestyle Intervention in Overweight and Obese Adolescents. J. Pediatr. Genet. 2020, 9, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Corgosinho, F.C.; Almeida, S.S.; Tock, L.; Pesquero, J.B.; Araújo, R.C.; Clemente, A.P.G.; Dal’Molin Netto, B.; da Silveira Campos, R.M.; Masquio, D.C.L.; de Carvalho Ferreira, J.P.; et al. LEPR polymorphism may affect energy balance during weight loss among Brazilians obese adolescents. Neuropeptides 2017, 66, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Deram, S.; Nicolau, C.Y.; Perez-Martinez, P.; Guazzelli, I.; Halpern, A.; Wajchenberg, B.L.; Ordovas, J.M.; Villares, S.M. Effects of perilipin (PLIN) gene variation on metabolic syndrome risk and weight loss in obese children and adolescents. J. Clin. Endocrinol. Metab. 2008, 93, 4933–4940. [Google Scholar] [CrossRef]
- do Nascimento, G.A.; Teixeira, M.D.; Furtado-Alle, L.; Leite, N.; de Souza, R.L.R.; Saliba, L.F.; Milano, G.E.; da Silva, L.R.; Pizzi, J.; Lopes, W.A.; et al. FTO rs9939609 A allele influences anthropometric outcome in response to dietary intervention, but not in response to physical exercise program. Eur. J. Nutr. 2019, 58, 325–334. [Google Scholar] [CrossRef]
- Gajewska, J.; Kuryłowicz, A.; Mierzejewska, E.; Ambroszkiewicz, J.; Chełchowska, M.; Weker, H.; Puzianowska-Kuźnicka, M. Complementary Effects of Genetic Variations in LEPR on Body Composition and Soluble Leptin Receptor Concentration after 3-Month Lifestyle Intervention in Prepubertal Obese Children. Nutrients 2016, 8, 328. [Google Scholar] [CrossRef]
- Gao, R.R.; Wang, M.; Hu, Y.; Xu, C.Y.; Li, Y.C.; Zhang, Z.Y.; Chen, S.Y.; Mao, X.Y. Impact of LPL gene rs283 polymorphism on exercise-induced changes in metabolism of obese adolescents and the regulatory mechanisms behind it. Exp. Physiol. 2015, 100, 698–707. [Google Scholar] [CrossRef]
- Hagman, E.; Hecht, L.; Marko, L.; Azmanov, H.; Groop, L.; Santoro, N.; Caprio, S.; Weiss, R. Predictors of responses to clinic-based childhood obesity care. Pediatr. Diabetes 2018, 19, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Heitkamp, M.; Siegrist, M.; Molnos, S.; Brandmaier, S.; Wahl, S.; Langhof, H.; Grallert, H.; Halle, M. Obesity Genes and Weight Loss During Lifestyle Intervention in Children With Obesity. JAMA Pediatr. 2021, 175, e205142. [Google Scholar] [CrossRef]
- Hollensted, M.; Fogh, M.; Schnurr, T.M.; Kloppenborg, J.T.; Have, C.T.; Ruest Haarmark Nielsen, T.; Rask, J.; Asp Vonsild Lund, M.; Frithioff-Bøjsøe, C.; Østergaard Johansen, M.; et al. Genetic Susceptibility for Childhood BMI has no Impact on Weight Loss Following Lifestyle Intervention in Danish Children. Obesity 2018, 26, 1915–1922. [Google Scholar] [CrossRef]
- Holzapfel, C.; Siegrist, M.; Rank, M.; Langhof, H.; Grallert, H.; Baumert, J.; Irimie, C.; Klopp, N.; Wolfarth, B.; Illig, T.; et al. Association of a MTNR1B gene variant with fasting glucose and HOMA-B in children and adolescents with high BMI-SDS. Eur. J. Endocrinol. 2011, 164, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Knoll, N.; Volckmar, A.L.; Pütter, C.; Scherag, A.; Kleber, M.; Hebebrand, J.; Hinney, A.; Reinehr, T. The fatty acid amide hydrolase (FAAH) gene variant rs324420 AA/AC is not associated with weight loss in a 1-year lifestyle intervention for obese children and adolescents. Horm. Metab. Res. 2012, 44, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Chen, W.; Helm, K. Effects of Visfatin Gene Polymorphism RS4730153 on Exercise-induced Weight Loss of Obese Children and Adolescents of Han Chinese. Int. J. Biol. Sci. 2013, 9, 16–21. [Google Scholar] [CrossRef]
- Leite, N.; Silva, L.; Jesus, Í.; Consentino, C.; Radominski, R.; Souza, R.; Tureck, L.; Furtado-Alle, L.; Lopes, W.; Cavaglieri, C. ADRB2 Gln27Glu polymorphism influenced changes in leptin but not body composition or metabolic and other inflammatory parameters after twelve weeks of combined training in overweight adolescents. Mot. Rev. De Educ. Física 2018, 13, 23. [Google Scholar] [CrossRef]
- Moleres, A.; Rendo-Urteaga, T.; Zulet, M.A.; Marcos, A.; Campoy, C.; Garagorri, J.M.; Martínez, J.A.; Azcona-Sanjulián, M.C.; Marti, A. Obesity susceptibility loci on body mass index and weight loss in Spanish adolescents after a lifestyle intervention. J. Pediatr. 2012, 161, 466–470.e462. [Google Scholar] [CrossRef]
- Moleres, A.; Milagro, F.I.; Marcos, A.; González Zorzano, E.; Campoy, C.; Garagorri, J.M.; Azcona-Sanjulian, M.C.; Martínez, J.A.; Marti, A. Common variants in genes related to lipid and energy metabolism are associated with weight loss after an intervention in overweight/obese adolescents. Nutr. Hosp. 2014, 30, 75–83. [Google Scholar] [CrossRef]
- Moraes, G.G.; Reuter, C.P.; Renner, J.D.; Klinger, E.I.; Ferreira, M.B.; Mello, E.D.; Valim, A.M.; Burgos, M.S. Genotypic carriers of the obesity-associated FTO polymorphism exhibit different cardiometabolic profiles after an intervention. An. Acad. Bras. Cienc. 2016, 88, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.D.; Hinney, A.; Scherag, A.; Nguyen, T.T.; Schreiner, F.; Schäfer, H.; Hebebrand, J.; Roth, C.L.; Reinehr, T. ‘Fat mass and obesity associated’ gene (FTO): No significant association of variant rs9939609 with weight loss in a lifestyle intervention and lipid metabolism markers in German obese children and adolescents. BMC Med. Genet. 2008, 9, 85. [Google Scholar] [CrossRef]
- Reinehr, T.; Hinney, A.; Nguyen, T.T.; Hebebrand, J. Evidence of an Influence of a Polymorphism Near the INSIG2 on Weight Loss During a Lifestyle Intervention in Obese Children and Adolescents. Diabetes 2008, 57, 623–626. [Google Scholar] [CrossRef]
- Reinehr, T.; Hinney, A.; Toschke, A.M.; Hebebrand, J. Aggravating effect of INSIG2 and FTO on overweight reduction in a one-year lifestyle intervention. Arch. Dis. Child. 2009, 94, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Hinney, A.; Schur, E.A.; Elfers, C.T.; Reinehr, T. Association analyses for dopamine receptor gene polymorphisms and weight status in a longitudinal analysis in obese children before and after lifestyle intervention. BMC Pediatr. 2013, 13, 197. [Google Scholar] [CrossRef]
- Santoro, N.; Perrone, L.; Cirillo, G.; Raimondo, P.; Amato, A.; Brienza, C.; Del Giudice, E.M. Effect of the melanocortin-3 receptor C17A and G241A variants on weight loss in childhood obesity. Am. J. Clin. Nutr. 2007, 85, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Scherag, A.; Kleber, M.; Boes, T.; Kolbe, A.L.; Ruth, A.; Grallert, H.; Illig, T.; Heid, I.M.; Toschke, A.M.; Grau, K.; et al. SDCCAG8 obesity alleles and reduced weight loss after a lifestyle intervention in overweight children and adolescents. Obesity 2012, 20, 466–470. [Google Scholar] [CrossRef]
- Schum, J.; Blumenstock, G.; Weber, K.; Schweizer, R.; Pfaff, C.; Schurr, N.; Ranke, M.B.; Binder, G.; Ehehalt, S. Variants of the FTO gene in obese children and their impact on body composition and metabolism before and after lifestyle intervention. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2012, 120, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.I.; Boes, T.; Reinehr, T.; Roth, C.L.; Scherag, S.; Scherag, A.; Hebebrand, J.; Hinney, A. Common variants near MC4R: Exploring gender effects in overweight and obese children and adolescents participating in a lifestyle intervention. Obes. Facts 2011, 4, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Volckmar, A.L.; Pütter, C.; Song, J.Y.; Graniger, J.; Knoll, N.; Wolters, B.; Hebebrand, J.; Scherag, A.; Reinehr, T.; Hinney, A. Analyses of non-synonymous obesity risk alleles in SH2B1 (rs7498665) and APOB48R (rs180743) in obese children and adolescents undergoing a 1-year lifestyle intervention. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2013, 121, 334–337. [Google Scholar] [CrossRef]
- Zlatohlavek, L.; Vrablik, M.; Motykova, E.; Ceska, R.; Vasickova, L.; Dlouha, D.; Hubacek, J.A. FTO and MC4R gene variants determine BMI changes in children after intensive lifestyle intervention. Clin. Biochem. 2013, 46, 313–316. [Google Scholar] [CrossRef]
- Zlatohlavek, L.; Maratka, V.; Tumova, E.; Ceska, R.; Lanska, V.; Vrablik, M.; Hubacek, J.A. Body Adiposity Changes After Lifestyle Interventions in Children/Adolescents and the NYD-SP18 and TMEM18 Variants. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 7493–7498. [Google Scholar] [CrossRef]
- Wells, G.; Shea, B.; O’Connell, D.; Peterson, j.; Welch, V.; Losos, M.; Tugwell, P. The Newcastle–Ottawa Scale (NOS) for Assessing the Quality of Non-Randomized Studies in Meta-Analysis; Ottawa Hospital Research Institute: Ottawa, ON, Canada, 2000. [Google Scholar]
- Bondyra-Wiśniewska, B.; Myszkowska-Ryciak, J.; Harton, A. Impact of Lifestyle Intervention Programs for Children and Adolescents with Overweight or Obesity on Body Weight and Selected Cardiometabolic Factors-A Systematic Review. Int J Env. Res Public Health 2021, 18, 2061. [Google Scholar] [CrossRef]
- Biederer, T. Bioinformatic characterization of the SynCAM family of immunoglobulin-like domain-containing adhesion molecules. Genomics 2006, 87, 139–150. [Google Scholar] [CrossRef]
- Thomas, L.A.; Akins, M.R.; Biederer, T. Expression and adhesion profiles of SynCAM molecules indicate distinct neuronal functions. J. Comp. Neurol. 2008, 510, 47–67. [Google Scholar] [CrossRef]
- Yan, X.; Wang, Z.; Schmidt, V.; Gauert, A.; Willnow, T.E.; Heinig, M.; Poy, M.N. Cadm2 regulates body weight and energy homeostasis in mice. Mol. Metab. 2018, 8, 180–188. [Google Scholar] [CrossRef]
- Fogel, A.I.; Akins, M.R.; Krupp, A.J.; Stagi, M.; Stein, V.; Biederer, T. SynCAMs Organize Synapses through Heterophilic Adhesion. J. Neurosci. 2007, 27, 12516–12530. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Allen, H.L.; Lindgren, C.M.; Luan, J.a.; Mägi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Rathjen, T.; Yan, X.; Kononenko, N.L.; Ku, M.C.; Song, K.; Ferrarese, L.; Tarallo, V.; Puchkov, D.; Kochlamazashvili, G.; Brachs, S.; et al. Regulation of body weight and energy homeostasis by neuronal cell adhesion molecule 1. Nat. Neurosci. 2017, 20, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Luquet, S.; Small, D.M. DRD2: Bridging the Genome and Ingestive Behavior. Trends Cogn. Sci. 2017, 21, 372–384. [Google Scholar] [CrossRef]
- Cameron, J.D.; Riou, M.; Tesson, F.; Goldfield, G.S.; Rabasa-Lhoret, R.; Brochu, M.; Doucet, É. The TaqIA RFLP is associated with attenuated intervention-induced body weight loss and increased carbohydrate intake in post-menopausal obese women. Appetite 2013, 60, 111–116. [Google Scholar] [CrossRef]
- Winkler, J.K.; Woehning, A.; Schultz, J.H.; Brune, M.; Beaton, N.; Challa, T.D.; Minkova, S.; Roeder, E.; Nawroth, P.P.; Friederich, H.C.; et al. TaqIA polymorphism in dopamine D2 receptor gene complicates weight maintenance in younger obese patients. Nutrition 2012, 28, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.D.; Chaput, J.P.; Sjödin, A.M.; Goldfield, G.S. Brain on Fire: Incentive Salience, Hedonic Hot Spots, Dopamine, Obesity, and Other Hunger Games. Annu. Rev. Nutr. 2017, 37, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Szczypka, M.S.; Kwok, K.; Brot, M.D.; Marck, B.T.; Matsumoto, A.M.; Donahue, B.A.; Palmiter, R.D. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron 2001, 30, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Sotak, B.N.; Hnasko, T.S.; Robinson, S.; Kremer, E.J.; Palmiter, R.D. Dysregulation of dopamine signaling in the dorsal striatum inhibits feeding. Brain Res. 2005, 1061, 88–96. [Google Scholar] [CrossRef]
- Klein, T.A.; Neumann, J.; Reuter, M.; Hennig, J.; von Cramon, D.Y.; Ullsperger, M. Genetically determined differences in learning from errors. Science 2007, 318, 1642–1645. [Google Scholar] [CrossRef]
- DiFeliceantonio, A.G.; Small, D.M. Dopamine and diet-induced obesity. Nat. Neurosci. 2019, 22, 1–2. [Google Scholar] [CrossRef]
- Stice, E.; Spoor, S.; Bohon, C.; Small, D.M. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science 2008, 322, 449–452. [Google Scholar] [CrossRef]
- Stice, E.; Yokum, S.; Blum, K.; Bohon, C. Weight gain is associated with reduced striatal response to palatable food. J. Neurosci. 2010, 30, 13105–13109. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Booth, F.W. Running from Disease: Molecular Mechanisms Associating Dopamine and Leptin Signaling in the Brain with Physical Inactivity, Obesity, and Type 2 Diabetes. Front. Endocrinol. 2017, 8, 109. [Google Scholar] [CrossRef]
- OMIM. LMX1B Gene. Available online: https://omim.org/entry/602575?search=LMX1B&highlight=lmx1b#10 (accessed on 19 May 2022).
- Zhu, Q.; Xue, K.; Guo, H.W.; Yang, Y.H. Lmx1b rs10733682 polymorphism interacts with macronutrients, dietary patterns on the risk of obesity in han chinese girls. Nutrients 2020, 12, 1227. [Google Scholar] [CrossRef]
- Monnereau, C.; Jansen, P.W.; Tiemeier, H.; Jaddoe, V.W.; Felix, J.F. Influence of genetic variants associated with body mass index on eating behavior in childhood. Obesity 2017, 25, 765–772. [Google Scholar] [CrossRef]
- Donovan, L.J.; Spencer, W.C.; Kitt, M.M.; Eastman, B.A.; Lobur, K.J.; Jiao, K.; Silver, J.; Deneris, E.S. Lmx1b is required at multiple stages to build expansive serotonergic axon architectures. eLife 2019, 8, e48788. [Google Scholar] [CrossRef] [PubMed]
- Doucet-Beaupré, H.; Ang, S.L.; Lévesque, M. Cell fate determination, neuronal maintenance and disease state: The emerging role of transcription factors Lmx1a and Lmx1b. FEBS Lett. 2015, 589, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Yabut, J.M.; Crane, J.D.; Green, A.E.; Keating, D.J.; Khan, W.I.; Steinberg, G.R. Emerging Roles for Serotonin in Regulating Metabolism: New Implications for an Ancient Molecule. Endocr. Rev. 2019, 40, 1092–1107. [Google Scholar] [CrossRef]
- Koopman, K.E.; Booij, J.; Fliers, E.; Serlie, M.J.; la Fleur, S.E. Diet-induced changes in the Lean Brain: Hypercaloric high-fat-high-sugar snacking decreases serotonin transporters in the human hypothalamic region. Mol. Metab. 2013, 2, 417–422. [Google Scholar] [CrossRef]
- Banas, S.M.; Rouch, C.; Kassis, N.; Markaki, E.M.; Gerozissis, K. A dietary fat excess alters metabolic and neuroendocrine responses before the onset of metabolic diseases. Cell. Mol. Neurobiol. 2009, 29, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, R.I.; Schrantee, A.; Adriaanse, S.M.; Unmehopa, U.A.; Booij, J.; Reneman, L.; Fliers, E.; la Fleur, S.E.; Serlie, M.J. Timing of caloric intake during weight loss differentially affects striatal dopamine transporter and thalamic serotonin transporter binding. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.W.; Myers, M.G., Jr. Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 2018, 19, 95–105. [Google Scholar] [CrossRef]
- Mammès, O.; Aubert, R.; Betoulle, D.; Péan, F.; Herbeth, B.; Visvikis, S.; Siest, G.; Fumeron, F. LEPR gene polymorphisms: Associations with overweight, fat mass and response to diet in women. Eur. J. Clin. Investig. 2001, 31, 398–404. [Google Scholar] [CrossRef] [PubMed]
- de Luis Roman, D.; de la Fuente, R.A.; Sagrado, M.G.; Izaola, O.; Vicente, R.C. Leptin receptor Lys656Asn polymorphism is associated with decreased leptin response and weight loss secondary to a lifestyle modification in obese patients. Arch. Med. Res. 2006, 37, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Münzberg, H.; Leinninger, G.M.; Leshan, R.L. The geometry of leptin action in the brain: More complicated than a simple ARC. Cell Metab. 2009, 9, 117–123. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and obesity: Role and clinical implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Fatima, M.T.; Ahmed, I.; Fakhro, K.A.; Akil, A.S.A. Melanocortin-4 receptor complexity in energy homeostasis, obesity and drug development strategies. Diabetes Obes. Metab. 2022, 24, 583–598. [Google Scholar] [CrossRef]
- Jääskeläinen, A.; Schwab, U.; Kolehmainen, M.; Kaakinen, M.; Savolainen, M.J.; Froguel, P.; Cauchi, S.; Järvelin, M.R.; Laitinen, J. Meal frequencies modify the effect of common genetic variants on body mass index in adolescents of the northern Finland birth cohort 1986. PLoS ONE 2013, 8, e73802. [Google Scholar] [CrossRef]
- Koletzko, B.; Toschke, A.M. Meal patterns and frequencies: Do they affect body weight in children and adolescents? Crit. Rev. Food Sci. Nutr. 2010, 50, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Zhang, D.D.; Wang, H.; Zhang, Y.; Liang, L.; Fu, J.F.; Xiong, F.; Liu, G.L.; Gong, C.X.; Luo, F.H.; et al. Genetic variations in SEC16B, MC4R, MAP2K5 and KCTD15 were associated with childhood obesity and interacted with dietary behaviors in Chinese school-age population. Gene 2015, 560, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.H.; Cho, G.B.; Sohn, J.W. Understanding melanocortin-4 receptor control of neuronal circuits: Toward novel therapeutics for obesity syndrome. Pharm. Res 2018, 129, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Kühnen, P.; Krude, H.; Biebermann, H. Melanocortin-4 Receptor Signalling: Importance for Weight Regulation and Obesity Treatment. Trends Mol. Med. 2019, 25, 136–148. [Google Scholar] [CrossRef]
- Yu, K.; Li, L.; Zhang, L.; Guo, L.; Wang, C. Association between MC4R rs17782313 genotype and obesity: A meta-analysis. Gene 2020, 733, 144372. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; Chandak, G.R.; Shen, Y.; Wang, Q.; Zhou, D. Association between common polymorphism near the MC4R gene and obesity risk: A systematic review and meta-analysis. PLoS ONE 2012, 7, e45731. [Google Scholar] [CrossRef] [PubMed]
- Dastgheib, S.A.; Bahrami, R.; Setayesh, S.; Salari, S.; Mirjalili, S.R.; Noorishadkam, M.; Sadeghizadeh-Yazdi, J.; Akbarian, E.; Neamatzadeh, H. Evidence from a meta-analysis for association of MC4R rs17782313 and FTO rs9939609 polymorphisms with susceptibility to obesity in children. Diabetes Metab. Syndr. 2021, 15, 102234. [Google Scholar] [CrossRef]
- Resende, C.M.M.; Silva, H.A.M.d.; Campello, C.P.; Ferraz, L.A.A.; de Lima, E.L.S.; Beserra, M.A.; Muniz, M.T.C.; da Silva, L.M.P. Polymorphisms on rs9939609 FTO and rs17782313 MC4R genes in children and adolescent obesity: A systematic review. Nutrition 2021, 91–92, 111474. [Google Scholar] [CrossRef] [PubMed]
- Lotta, L.A.; Mokrosiński, J.; Mendes de Oliveira, E.; Li, C.; Sharp, S.J.; Luan, J.; Brouwers, B.; Ayinampudi, V.; Bowker, N.; Kerrison, N.; et al. Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 2019, 177, 597–607.e599. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Girardet, C.; Mavrikaki, M.; Trevaskis, J.L.; Macarthur, H.; Marks, D.L.; Farr, S.A. A Life without Hunger: The Ups (and Downs) to Modulating Melanocortin-3 Receptor Signaling. Front. Neurosci. 2017, 11, 128. [Google Scholar] [CrossRef]
- Demidowich, A.P.; Jun, J.Y.; Yanovski, J.A. Polymorphisms and mutations in the melanocortin-3 receptor and their relation to human obesity. Biochim. Et Biophys. Acta (BBA) -Mol. Basis Dis. 2017, 1863, 2468–2476. [Google Scholar] [CrossRef]
- Santos, J.L.; De la Cruz, R.; Holst, C.; Grau, K.; Naranjo, C.; Maiz, A.; Astrup, A.; Saris, W.H.; MacDonald, I.; Oppert, J.M.; et al. Allelic variants of melanocortin 3 receptor gene (MC3R) and weight loss in obesity: A randomised trial of hypo-energetic high- versus low-fat diets. PLoS ONE 2011, 6, e19934. [Google Scholar] [CrossRef] [PubMed]
- Savastano, D.M.; Tanofsky-Kraff, M.; Han, J.C.; Ning, C.; Sorg, R.A.; Roza, C.A.; Wolkoff, L.E.; Anandalingam, K.; Jefferson-George, K.S.; Figueroa, R.E.; et al. Energy intake and energy expenditure among children with polymorphisms of the melanocortin-3 receptor. Am. J. Clin. Nutr. 2009, 90, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Young, S.F.; Aguilera, G.; Puricelli, E.; Adler-Wailes, D.C.; Sebring, N.G.; Yanovski, J.A. Co-occurrence of Two Partially Inactivating Polymorphisms of MC3R Is Associated With Pediatric-Onset Obesity. Diabetes 2005, 54, 2663–2667. [Google Scholar] [CrossRef]
- Aris, I.M.; Tint, M.T.; Teh, A.L.; Holbrook, J.D.; Quah, P.L.; Chong, M.F.-F.; Lin, X.; Soh, S.E.; Saw, S.-M.; Kwek, K.; et al. MC3R gene polymorphisms are associated with early childhood adiposity gain and infant appetite in an Asian population. Pediatr. Obes. 2016, 11, 450–458. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Lee, P.L.; Tang, Y.; Li, H.; Guertin, D.A. Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab. 2016, 5, 422–432. [Google Scholar] [CrossRef]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Rüegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Magdalon, J.; Festuccia, W.T. Regulation of adiposity by mTORC1. Einstein 2017, 15, 507–511. [Google Scholar] [CrossRef] [PubMed]
- OMIM. FTO ALPHA-KETOGLUTARATE-DEPENDENT DIOXYGENASE; FTO * 610966. Available online: https://www.omim.org/entry/610966 (accessed on 21 June 2022).
- Cards, G. FTO Gene—FTO Alpha-Ketoglutarate Dependent Dioxygenase. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=FTO (accessed on 15 June 2022).
- Claussnitzer, M.; Hui, C.C.; Kellis, M. FTO Obesity Variant and Adipocyte Browning in Humans. N. Engl. J. Med. 2016, 374, 192–193. [Google Scholar] [CrossRef]
- OMIM. ETS PROTOONCOGENE 2, TRANSCRIPTION FACTOR.; ETS2. Available online: https://www.omim.org/entry/164740?search=ETS2&highlight=ets2 (accessed on 20 June 2022).
- Birsoy, K.; Berry, R.; Wang, T.; Ceyhan, O.; Tavazoie, S.; Friedman, J.M.; Rodeheffer, M.S. Analysis of gene networks in white adipose tissue development reveals a role for ETS2 in adipogenesis. Development 2011, 138, 4709–4719. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jiang, Z.; Li, N.; Jiang, W.; Gao, P.; Yang, M.; Yu, X.; Wang, G.; Zhang, Y. Ets2 suppresses inflammatory cytokines through MAPK/NF-κB signaling and directly binds to the IL-6 promoter in macrophages. Aging 2019, 11, 10610–10625. [Google Scholar] [CrossRef]
- Gallant, S.; Gilkeson, G. ETS transcription factors and regulation of immunity. Arch. Immunol. Et Ther. Exp. 2006, 54, 149–163. [Google Scholar] [CrossRef]
- Quinn, S.R.; Mangan, N.E.; Caffrey, B.E.; Gantier, M.P.; Williams, B.R.G.; Hertzog, P.J.; McCoy, C.E.; O’Neill, L.A.J. The Role of Ets2 Transcription Factor in the Induction of MicroRNA-155 (miR-155) by Lipopolysaccharide and Its Targeting by Interleukin-10. J. Biol. Chem. 2014, 289, 4316–4325. [Google Scholar] [CrossRef]
- Wei, G.; Guo, J.; Doseff, A.I.; Kusewitt, D.F.; Man, A.K.; Oshima, R.G.; Ostrowski, M.C. Activated Ets2 is required for persistent inflammatory responses in the motheaten viable model. J. Immunol. 2004, 173, 1374–1379. [Google Scholar] [CrossRef]
- KAT8 O. LYSINE ACETYLTRANSFERASE 8; KAT8. Available online: https://www.omim.org/entry/609912?search=kat8&highlight=kat8 (accessed on 27 June 2022).
- Mellert, H.S.; McMahon, S.B. hMOF, a KAT(8) with Many Lives. Mol. Cell 2009, 36, 174–175. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Thomas, T.; Dixon, M.P.; Kueh, A.J.; Voss, A.K. Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol. Cell. Biol. 2008, 28, 5093–5105. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, Y.; Liu, Y.; Chen, J.; Zong, C.; Yu, C.; Cui, S.; Gao, W.; Qin, D.; Sun, W.; et al. Signal transducer and activator of transcription 5B (STAT5B) modulates adipocyte differentiation via MOF. Cell. Signal. 2015, 27, 2434–2443. [Google Scholar] [CrossRef]
- Lin, H.P.; Cheng, Z.L.; He, R.Y.; Song, L.; Tian, M.X.; Zhou, L.S.; Groh, B.S.; Liu, W.R.; Ji, M.B.; Ding, C.; et al. Destabilization of Fatty Acid Synthase by Acetylation Inhibits De Novo Lipogenesis and Tumor Cell Growth. Cancer Res. 2016, 76, 6924–6936. [Google Scholar] [CrossRef]
- Burrell, J.A.; Stephens, J.M. KAT8, lysine acetyltransferase 8, is required for adipocyte differentiation in vitro. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2021, 1867, 166103. [Google Scholar] [CrossRef]
- Pessoa Rodrigues, C.; Chatterjee, A.; Wiese, M.; Stehle, T.; Szymanski, W.; Shvedunova, M.; Akhtar, A. Histone H4 lysine 16 acetylation controls central carbon metabolism and diet-induced obesity in mice. Nat. Commun. 2021, 12, 6212. [Google Scholar] [CrossRef]
- Brenachot, X.; Rigault, C.; Nédélec, E.; Laderrière, A.; Khanam, T.; Gouazé, A.; Chaudy, S.; Lemoine, A.; Datiche, F.; Gascuel, J.; et al. The histone acetyltransferase MOF activates hypothalamic polysialylation to prevent diet-induced obesity in mice. Mol. Metab. 2014, 3, 619–629. [Google Scholar] [CrossRef] [PubMed]
- OMIM. TRANSMEMBRANE PROTEIN 18; TMEM18 * 613220. Available online: https://www.omim.org/entry/613220#2 (accessed on 28 June 2022).
- Jurvansuu, J.; Zhao, Y.; Leung, D.S.; Boulaire, J.; Yu, Y.H.; Ahmed, S.; Wang, S. Transmembrane protein 18 enhances the tropism of neural stem cells for glioma cells. Cancer Res. 2008, 68, 4614–4622. [Google Scholar] [CrossRef] [PubMed]
- Almén, M.S.; Jacobsson, J.A.; Shaik, J.H.; Olszewski, P.K.; Cedernaes, J.; Alsiö, J.; Sreedharan, S.; Levine, A.S.; Fredriksson, R.; Marcus, C.; et al. The obesity gene, TMEM18, is of ancient origin, found in majority of neuronal cells in all major brain regions and associated with obesity in severely obese children. BMC Med. Genet. 2010, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Koj, N.; Grochowalski, Ł.; Jarczak, J.; Wójtowicz, W.; Sobalska-Kwapis, M.; Słomka, M.; Marciniak, B.; Strapagiel, D. The association between polymorphisms near TMEM18 and the risk of obesity: A meta-analysis. BMC Med. Genom. 2021, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Speliotes, E.K.; Loos, R.J.; Li, S.; Lindgren, C.M.; Heid, I.M.; Berndt, S.I.; Elliott, A.L.; Jackson, A.U.; Lamina, C.; et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009, 41, 25–34. [Google Scholar] [CrossRef]
- Zhao, J.; Bradfield, J.P.; Li, M.; Wang, K.; Zhang, H.; Kim, C.E.; Annaiah, K.; Glessner, J.T.; Thomas, K.; Garris, M.; et al. The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity 2009, 17, 2254–2257. [Google Scholar] [CrossRef] [PubMed]
- Thorleifsson, G.; Walters, G.B.; Gudbjartsson, D.F.; Steinthorsdottir, V.; Sulem, P.; Helgadottir, A.; Styrkarsdottir, U.; Gretarsdottir, S.; Thorlacius, S.; Jonsdottir, I.; et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009, 41, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, K.; Klöting, N.; Gericke, M.; Maixner, N.; Guiu-Jurado, E.; Scholz, M.; Witte, A.V.; Beyer, F.; Schwartze, J.T.; Lacher, M. The obesity-susceptibility gene TMEM18 promotes adipogenesis through activation of PPARG. Cell Rep. 2020, 33, 108295. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Jacobsson, J.A.; Moschonis, G.; Chavan, R.A.; Sikder, M.A.N.; Allzén, E.; Alsiö, J.; Chrousos, G.P.; Manios, Y.; Fredriksson, R.; et al. Association of TMEM18 variants with BMI and waist circumference in children and correlation of mRNA expression in the PFC with body weight in rats. Eur. J. Hum. Genet. 2012, 20, 192–197. [Google Scholar] [CrossRef]
- Larder, R.; Sim, M.F.M.; Gulati, P.; Antrobus, R.; Tung, Y.C.L.; Rimmington, D.; Ayuso, E.; Polex-Wolf, J.; Lam, B.Y.H.; Dias, C.; et al. Obesity-associated gene TMEM18 has a role in the central control of appetite and body weight regulation. Proc. Natl. Acad. Sci. USA 2017, 114, 9421–9426. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, F.; Landgraf, K.; Klöting, N.; Berthold, A.; Büttner, P.; Friebe, D.; Kiess, W.; Kovacs, P.; Blüher, M.; Körner, A. Functional relevance of genes implicated by obesity genome-wide association study signals for human adipocyte biology. Diabetologia 2013, 56, 311–322. [Google Scholar] [CrossRef]
- OMIM. PPARG Gene—Peroxisome Proliferator Activated Receptor Gamma * 601487. Available online: https://www.omim.org/entry/601487 (accessed on 6 June 2022).
- Tugwood, J.D.; Montague, C.T. Biology and toxicology of PPARg ligands. Hum. Exp. Toxicol. 2002, 21, 429–437. [Google Scholar] [CrossRef]
- Cataldi, S.; Costa, V.; Ciccodicola, A.; Aprile, M. PPARγ and Diabetes: Beyond the Genome and Towards Personalized Medicine. Curr. Diabetes Rep. 2021, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Faghfouri, A.H.; Khajebishak, Y.; Payahoo, L.; Faghfuri, E.; Alivand, M. PPAR-gamma agonists: Potential modulators of autophagy in obesity. Eur. J. Pharmacol. 2021, 912, 174562. [Google Scholar] [CrossRef]
- Shearer, B.G.; Hoekstra, W.J. Recent advances in peroxisome proliferator-activated receptor science. Curr. Med. Chem. 2003, 10, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Cecil, J.; Dalton, M.; Finlayson, G.; Blundell, J.; Hetherington, M.; Palmer, C. Obesity and eating behaviour in children and adolescents: Contribution of common gene polymorphisms. Int. Rev. Psychiatry 2012, 24, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, A.; Amini, M.; Kolahdooz, F.; Seyedrezazadeh, E. Obesity and Pro12Ala Polymorphism of Peroxisome Proliferator-Activated Receptor-Gamma Gene in Healthy Adults: A Systematic Review and Meta-Analysis. Ann. Nutr. Metab. 2015, 67, 104–118. [Google Scholar] [CrossRef]
- Lee, J.-E.; Ge, K. Transcriptional and epigenetic regulation of PPARγ expression during adipogenesis. Cell Biosci. 2014, 4, 29. [Google Scholar] [CrossRef]
- Motawi, T.K.; Shaker, O.G.; Ismail, M.F.; Sayed, N.H. Peroxisome Proliferator-Activated Receptor Gamma in Obesity and Colorectal Cancer: The Role of Epigenetics. Sci. Rep. 2017, 7, 10714. [Google Scholar] [CrossRef] [PubMed]
- Masugi, J.; Tamori, Y.; Mori, H.; Koike, T.; Kasuga, M. Inhibitory effect of a proline-to-alanine substitution at codon 12 of peroxisome proliferator-activated receptor-gamma 2 on thiazolidinedione-induced adipogenesis. Biochem. Biophys. Res. Commun. 2000, 268, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Gouda, H.N.; Sagoo, G.S.; Harding, A.H.; Yates, J.; Sandhu, M.S.; Higgins, J.P. The association between the peroxisome proliferator-activated receptor-gamma2 (PPARG2) Pro12Ala gene variant and type 2 diabetes mellitus: A HuGE review and meta-analysis. Am. J. Epidemiol. 2010, 171, 645–655. [Google Scholar] [CrossRef]
- Luan, J.; Browne, P.O.; Harding, A.H.; Halsall, D.J.; O’Rahilly, S.; Chatterjee, V.K.; Wareham, N.J. Evidence for gene-nutrient interaction at the PPARgamma locus. Diabetes 2001, 50, 686–689. [Google Scholar] [CrossRef]
- OMIM * 613524. SHH SIGNALING AND CILIOGENESIS REGULATOR SDCCAG8; SDCCAG8. Available online: https://omim.org/entry/613524#4 (accessed on 21 June 2022).
- Cards, G. SDCCAG8 Gene—SHH Signaling and Ciliogenesis Regulator SDCCAG8. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=SDCCAG8 (accessed on 16 June 2022).
- Scherag, A.; Dina, C.; Hinney, A.; Vatin, V.; Scherag, S.; Vogel, C.I.; Müller, T.D.; Grallert, H.; Wichmann, H.E.; Balkau, B.; et al. Two new Loci for body-weight regulation identified in a joint analysis of genome-wide association studies for early-onset extreme obesity in French and german study groups. PLoS Genet. 2010, 6, e1000916. [Google Scholar] [CrossRef]
- Mariman, E.C.; Vink, R.G.; Roumans, N.J.; Bouwman, F.G.; Stumpel, C.T.; Aller, E.E.; van Baak, M.A.; Wang, P. The cilium: A cellular antenna with an influence on obesity risk. Br. J. Nutr. 2016, 116, 576–592. [Google Scholar] [CrossRef]
- GeneCards. CPNE8 Gene—Copine 8. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CPNE8 (accessed on 2 July 2022).
- Arruda, A.P.; Hotamisligil, G.S. Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes. Cell Metab. 2015, 22, 381–397. [Google Scholar] [CrossRef]
- Su, X.; Peng, D. The exchangeable apolipoproteins in lipid metabolism and obesity. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 503, 128–135. [Google Scholar] [CrossRef]
- Ruan, X.; Li, Z.; Zhang, Y.; Yang, L.; Pan, Y.; Wang, Z.; Feng, G.S.; Chen, Y. Apolipoprotein A-I possesses an anti-obesity effect associated with increase of energy expenditure and up-regulation of UCP1 in brown fat. J. Cell. Mol. Med. 2011, 15, 763–772. [Google Scholar] [CrossRef]
- Wei, H.; Averill, M.M.; McMillen, T.S.; Dastvan, F.; Mitra, P.; Subramanian, S.; Tang, C.; Chait, A.; Leboeuf, R.C. Modulation of adipose tissue lipolysis and body weight by high-density lipoproteins in mice. Nutr. Diabetes 2014, 4, e108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Badenhop, R.B.; Sim, A.S.; Wilcken, D.E. The effect on transcription efficiency of the apolipoprotein AI gene of DNA variants at the 5’ untranslated region. Int. J. Clin. Lab. Res. 1998, 28, 235–241. [Google Scholar] [CrossRef] [PubMed]
- de Luis, D.A.; Izaola, O.; Primo, D.; Aller, R. Role of rs670 variant of APOA1 gene on lipid profile, insulin resistance and adipokine levels in obese subjects after weight loss with a dietary intervention. Diabetes Res. Clin. Pract. 2018, 142, 139–145. [Google Scholar] [CrossRef] [PubMed]
- de Luis, D.; Izaola, O.; Primo, D.; Aller, R. Role of rs670 variant of APOA1 gene on metabolic response after a high fat vs. a low fat hypocaloric diets in obese human subjects. J. Diabetes Its Complicat. 2019, 33, 249–254. [Google Scholar] [CrossRef]
- Phillips, C.M.; Goumidi, L.; Bertrais, S.; Field, M.R.; McManus, R.; Hercberg, S.; Lairon, D.; Planells, R.; Roche, H.M. Gene-nutrient interactions and gender may modulate the association between ApoA1 and ApoB gene polymorphisms and metabolic syndrome risk. Atherosclerosis 2011, 214, 408–414. [Google Scholar] [CrossRef]
- Barter, P.J.; Brewer, H.B., Jr.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein: A novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef]
- Armitage, J.; Holmes, M.V.; Preiss, D. Cholesteryl Ester Transfer Protein Inhibition for Preventing Cardiovascular Events: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 477–487. [Google Scholar] [CrossRef]
- Thompson, A.; Di Angelantonio, E.; Sarwar, N.; Erqou, S.; Saleheen, D.; Dullaart, R.P.; Keavney, B.; Ye, Z.; Danesh, J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA 2008, 299, 2777–2788. [Google Scholar] [CrossRef]
- Wuni, R.; Kuhnle, G.G.C.; Wynn-Jones, A.A.; Vimaleswaran, K.S. A Nutrigenetic Update on CETP Gene-Diet Interactions on Lipid-Related Outcomes. Curr. Atheroscler. Rep. 2022, 24, 119–132. [Google Scholar] [CrossRef]
- Aller, R.; Izaola, O.; Primo, D.; de Luis, D. Cholesteryl Ester Transfer Protein Variant (RS1800777) with Liver Histology in Non-Alcoholic Fatty Liver Disease Patients. Ann. Nutr. Metab. 2018, 73, 265–270. [Google Scholar] [CrossRef]
- de Luis, D.; Izaola, O.; Primo, D.; Gomez, E.; Lopez, J.J.; Ortola, A.; Aller, R. Association of a cholesteryl ester transfer protein variant (rs1800777) with fat mass, HDL cholesterol levels, and metabolic syndrome. Endocrinol. Diabetes Y Nutr. 2018, 65, 387–393. [Google Scholar] [CrossRef]
- Agerholm-Larsen, B.; Tybjaerg-Hansen, A.; Schnohr, P.; Steffensen, R.; Nordestgaard, B.G. Common cholesteryl ester transfer protein mutations, decreased HDL cholesterol, and possible decreased risk of ischemic heart disease: The Copenhagen City Heart Study. Circulation 2000, 102, 2197–2203. [Google Scholar] [CrossRef]
- Tsai, M.Y.; Johnson, C.; Kao, W.H.; Sharrett, A.R.; Arends, V.L.; Kronmal, R.; Jenny, N.S.; Jacobs, D.R., Jr.; Arnett, D.; O’Leary, D.; et al. Cholesteryl ester transfer protein genetic polymorphisms, HDL cholesterol, and subclinical cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2008, 200, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Li, N.; Sharrett, A.R.; Shea, S.; Jacobs, D.R., Jr.; Tracy, R.; Arnett, D.; Arends, V.; Post, W. Associations of genetic variants in ATP-binding cassette A1 and cholesteryl ester transfer protein and differences in lipoprotein subclasses in the multi-ethnic study of atherosclerosis. Clin. Chem. 2009, 55, 481–488. [Google Scholar] [CrossRef]
- Kakko, S.; Tamminen, M.; Kesäniemi, Y.A.; Savolainen, M.J. R451Q mutation in the cholesteryl ester transfer protein (CETP) gene is associated with high plasma CETP activity. Atherosclerosis 1998, 136, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Kakko, S.; Tamminen, M.; Päivänsalo, M.; Kauma, H.; Rantala, A.O.; Lilja, M.; Reunanen, A.; Kesäniemi, Y.A.; Savolainen, M.J. Cholesteryl ester transfer protein gene polymorphisms are associated with carotid atherosclerosis in men. Eur. J. Clin. Investig. 2000, 30, 18–25. [Google Scholar] [CrossRef]
- OMIM. LIPOPROTEIN LIPASE; LPL * 609708. Available online: https://www.omim.org/entry/609708 (accessed on 21 June 2022).
- Wang, H.; Eckel, R.H. Lipoprotein lipase: From gene to obesity. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E271–E288. [Google Scholar] [CrossRef] [PubMed]
- OMIM. INSULIN-INDUCED GENE 2; INSIG2 * 608660. Available online: https://www.omim.org/entry/608660 (accessed on 5 June 2022).
- GeneCards. INSIG2 Gene—Insulin Induced Gene 2. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=INSIG2 (accessed on 9 June 2022).
- Burgdörfer, E.; Korenkov, M.; Jonas, D.; Weise, D.; Haaf, T.; Zechner, U.; Bartsch, O. FTO and INSIG2 Genotyping Combined with Metabolic and Anthropometric Phenotyping of Morbidly Obese Patients. Mol. Syndromol. 2013, 4, 273–279. [Google Scholar] [CrossRef] [PubMed]
- SNPedia. rs7566605. Available online: https://www.snpedia.com/index.php/Rs7566605 (accessed on 13 June 2022).
- Heid, I.M.; Huth, C.; Loos, R.J.; Kronenberg, F.; Adamkova, V.; Anand, S.S.; Ardlie, K.; Biebermann, H.; Bjerregaard, P.; Boeing, H.; et al. Meta-analysis of the INSIG2 association with obesity including 74,345 individuals: Does heterogeneity of estimates relate to study design? PLoS Genet. 2009, 5, e1000694. [Google Scholar] [CrossRef]
- Andreasen, C.H.; Mogensen, M.S.; Borch-Johnsen, K.; Sandbaek, A.; Lauritzen, T.; Sørensen, T.I.; Hansen, L.; Almind, K.; Jørgensen, T.; Pedersen, O.; et al. Non-replication of genome-wide based associations between common variants in INSIG2 and PFKP and obesity in studies of 18,014 Danes. PLoS ONE 2008, 3, e2872. [Google Scholar] [CrossRef]
- Zavattari, P.; Loche, A.; Civolani, P.; Pilia, S.; Moi, L.; Casini, M.R.; Minerba, L.; Loche, S. An INSIG2 polymorphism affects glucose homeostasis in Sardinian obese children and adolescents. Ann. Hum. Genet. 2010, 74, 381–386. [Google Scholar] [CrossRef]
- Elbein, S.C.; Das, S.K.; Hallman, D.M.; Hanis, C.L.; Hasstedt, S.J. Genome-wide linkage and admixture mapping of type 2 diabetes in African American families from the American Diabetes Association GENNID (Genetics of NIDDM) Study Cohort. Diabetes 2009, 58, 268–274. [Google Scholar] [CrossRef]
- Talbert, M.E.; Langefeld, C.D.; Ziegler, J.T.; Haffner, S.M.; Norris, J.M.; Bowden, D.W. INSIG2 SNPs associated with obesity and glucose homeostasis traits in Hispanics: The IRAS Family Study. Obesity 2009, 17, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.E.; Ordovás, J.M. Update on perilipin polymorphisms and obesity. Nutr. Rev. 2012, 70, 611–621. [Google Scholar] [CrossRef]
- Kimmel, A.R.; Sztalryd, C. The Perilipins: Major Cytosolic Lipid Droplet–Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr. 2016, 36, 471–509. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Shen, H.; Larson, I.; Schaefer, E.J.; Greenberg, A.S.; Tregouet, D.A.; Corella, D.; Ordovas, J.M. Gender-specific association of a perilipin gene haplotype with obesity risk in a white population. Obes. Res. 2004, 12, 1758–1765. [Google Scholar] [CrossRef]
- Qi, L.; Tai, E.S.; Tan, C.E.; Shen, H.; Chew, S.K.; Greenberg, A.S.; Corella, D.; Ordovas, J.M. Intragenic linkage disequilibrium structure of the human perilipin gene (PLIN) and haplotype association with increased obesity risk in a multiethnic Asian population. J. Mol. Med. 2005, 83, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Tokgöz, Y.; Işık, I.A.; Akbari, S.; Kume, T.; Sayın, O.; Erdal, E.; Arslan, N. Perilipin polymorphisms are risk factors for the development of obesity in adolescents? A case-control study. Lipids Health Dis. 2017, 16, 52. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, O.Y.; Lee, J.H.; Koh, S.J.; Chae, J.S.; Kim, J.Y.; Park, S.; Cho, H.; Lee, J.E.; Ordovas, J.M. Genetic variation at the perilipin locus is associated with changes in serum free fatty acids and abdominal fat following mild weight loss. Int. J. Obes. 2006, 30, 1601–1608. [Google Scholar] [CrossRef]
- Tai, E.S.; Ordovas, J.M. The role of perilipin in human obesity and insulin resistance. Curr. Opin. Lipidol. 2007, 18, 152–156. [Google Scholar] [CrossRef]
- Corella, D.; Qi, L.; Tai, E.S.; Deurenberg-Yap, M.; Tan, C.E.; Chew, S.K.; Ordovas, J.M. Perilipin gene variation determines higher susceptibility to insulin resistance in Asian women when consuming a high-saturated fat, low-carbohydrate diet. Diabetes Care 2006, 29, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Soenen, S.; Mariman, E.C.M.; Vogels, N.; Bouwman, F.G.; den Hoed, M.; Brown, L.; Westerterp-Plantenga, M.S. Relationship between perilipin gene polymorphisms and body weight and body composition during weight loss and weight maintenance. Physiol. Behav. 2009, 96, 723–728. [Google Scholar] [CrossRef]
- Corella, D.; Qi, L.; Sorlí, J.V.; Godoy, D.; Portolés, O.; Coltell, O.; Greenberg, A.S.; Ordovas, J.M. Obese Subjects Carrying the 11482G > A Polymorphism at the Perilipin Locus Are Resistant to Weight Loss after Dietary Energy Restriction. J. Clin. Endocrinol. Metab. 2005, 90, 5121–5126. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Corella, D.; Sorlí, J.V.; Portolés, O.; Shen, H.; Coltell, O.; Godoy, D.; Greenberg, A.S.; Ordovas, J.M. Genetic variation at the perilipin (PLIN) locus is associated with obesity-related phenotypes in White women. Clin. Genet. 2004, 66, 299–310. [Google Scholar] [CrossRef]
- Ruiz, J.R.; Larrarte, E.; Margareto, J.; Ares, R.; Alkorta, P.; Labayen, I. Preliminary findings on the role of PLIN1 polymorphisms on body composition and energy metabolism response to energy restriction in obese women. Br. J. Nutr. 2011, 106, 486–490. [Google Scholar] [CrossRef]
- Qi, L.; Zhang, C.; Greenberg, A.; Hu, F.B. Common variations in perilipin gene, central obesity, and risk of type 2 diabetes in US women. Obesity 2008, 16, 1061–1065. [Google Scholar] [CrossRef]
- Mottagui-Tabar, S.; Rydén, M.; Löfgren, P.; Faulds, G.; Hoffstedt, J.; Brookes, A.J.; Andersson, I.; Arner, P. Evidence for an important role of perilipin in the regulation of human adipocyte lipolysis. Diabetologia 2003, 46, 789–797. [Google Scholar] [CrossRef] [PubMed]
- OMIM. ADIPOCYTE-, C1q-, AND COLLAGEN DOMAIN-CONTAINING; ADIPOQ * 605441. Available online: https://www.omim.org/entry/605441 (accessed on 21 June 2022).
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Hoefle, G.; Muendlein, A.; Saely, C.H.; Risch, L.; Rein, P.; Koch, L.; Schmid, F.; Aczel, S.; Marte, T.; Langer, P.; et al. The -11377 C>G promoter variant of the adiponectin gene, prevalence of coronary atherosclerosis, and incidence of vascular events in men. Thromb. Haemost. 2007, 97, 451–457. [Google Scholar] [CrossRef]
- Paltoglou, G.; Schoina, M.; Valsamakis, G.; Salakos, N.; Avloniti, A.; Chatzinikolaou, A.; Margeli, A.; Skevaki, C.; Papagianni, M.; Kanaka-Gantenbein, C.; et al. Interrelations among the adipocytokines leptin and adiponectin, oxidative stress and aseptic inflammation markers in pre- and early-pubertal normal-weight and obese boys. Endocrine 2017, 55, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Cards, G. IFNGR1 Gene—Interferon Gamma Receptor 1. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IFNGR1&keywords=IFNGR1 (accessed on 21 June 2022).
- Ivashkiv, L. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, F.C.; Chiquoine, E.H.; Hinkle, C.C.; Kim, R.J.; Shah, R.; Roche, H.M.; Smyth, E.M.; Reilly, M.P. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J. Biol. Chem. 2009, 284, 31936–31944. [Google Scholar] [CrossRef]
- OMIM. SLC39A8 Gene—Solute Carrier Family 39 Member 8. Available online: https://www.omim.org/entry/608732 (accessed on 23 June 2022).
- Nebert, D.W.; Liu, Z. SLC39A8 gene encoding a metal ion transporter: Discovery and bench to bedside. Hum. Genom. 2019, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.S.; Xu, Y.M.; Lau, A.T.Y. Molecular and pathophysiological aspects of metal ion uptake by the zinc transporter ZIP8 (SLC39A8). Toxicol. Res. 2016, 5, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Waterworth, D.M.; Ricketts, S.L.; Song, K.; Chen, L.; Zhao, J.H.; Ripatti, S.; Aulchenko, Y.S.; Zhang, W.; Yuan, X.; Lim, N.; et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2264–2276. [Google Scholar] [CrossRef]
- Fukunaka, A.; Fujitani, Y. Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity. Int. J. Mol. Sci. 2018, 19, 476. [Google Scholar] [CrossRef]
- Voruganti, V.S.; Cai, G.; Klohe, D.M.; Jordan, K.C.; Lane, M.A.; Freeland-Graves, J.H. Short-term weight loss in overweight/obese low-income women improves plasma zinc and metabolic syndrome risk factors. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2010, 24, 271–276. [Google Scholar] [CrossRef]
- Abdollahi, S.; Toupchian, O.; Jayedi, A.; Meyre, D.; Tam, V.; Soltani, S. Zinc Supplementation and Body Weight: A Systematic Review and Dose-Response Meta-analysis of Randomized Controlled Trials. Adv. Nutr. 2020, 11, 398–411. [Google Scholar] [CrossRef]
- OMIM. INTERLEUKIN 6; IL6. Available online: https://www.omim.org/entry/147620 (accessed on 3 June 2022).
- Timper, K.; Denson, J.L.; Steculorum, S.M.; Heilinger, C.; Engström-Ruud, L.; Wunderlich, C.M.; Rose-John, S.; Wunderlich, F.T.; Brüning, J.C. IL-6 Improves Energy and Glucose Homeostasis in Obesity via Enhanced Central IL-6 trans-Signaling. Cell Rep. 2017, 19, 267–280. [Google Scholar] [CrossRef]
- Ghanemi, A.; St-Amand, J. Interleukin-6 as a “metabolic hormone”. Cytokine 2018, 112, 132–136. [Google Scholar] [CrossRef]
- Todendi, P.F.; Klinger, E.I.; Ferreira, M.B.; Reuter, C.P.; Burgos, M.S.; Possuelo, L.G.; Valim, A.R. Association of IL-6 and CRP gene polymorphisms with obesity and metabolic disorders in children and adolescents. An. Da Acad. Bras. De Cienc. 2015, 87, 915–924. [Google Scholar] [CrossRef]
- Qi, L.; Zhang, C.; van Dam, R.M.; Hu, F.B. Interleukin-6 genetic variability and adiposity: Associations in two prospective cohorts and systematic review in 26,944 individuals. J. Clin. Endocrinol. Metab. 2007, 92, 3618–3625. [Google Scholar] [CrossRef]
- Eder, K.; Baffy, N.; Falus, A.; Fulop, A.K. The major inflammatory mediator interleukin-6 and obesity. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2009, 58, 727–736. [Google Scholar] [CrossRef]
- Hu, M.; Yu, Z.; Luo, D.; Zhang, H.; Li, J.; Liang, F.; Chen, R. Association between -174G>C polymorphism in the IL-6 promoter region and the risk of obesity: A meta-analysis. Medicine 2018, 97, e11773. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Sharifi, F.; Shahriari, S.; Khoshnevisan, K.; Larijani, B.; Amoli, M.M. Association of interleukin-6 polymorphisms with obesity: A systematic review and meta-analysis. Cytokine 2019, 123, 154769. [Google Scholar] [CrossRef] [PubMed]
- Corpeleijn, E.; Petersen, L.; Holst, C.; Saris, W.H.; Astrup, A.; Langin, D.; MacDonald, I.; Martinez, J.A.; Oppert, J.M.; Polak, J.; et al. Obesity-related polymorphisms and their associations with the ability to regulate fat oxidation in obese Europeans: The NUGENOB study. Obesity 2010, 18, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Goyenechea, E.; Dolores Parra, M.; Alfredo Martínez, J. Weight regain after slimming induced by an energy-restricted diet depends on interleukin-6 and peroxisome-proliferator-activated-receptor-gamma2 gene polymorphisms. Br. J. Nutr. 2006, 96, 965–972. [Google Scholar] [CrossRef] [PubMed]
- GeneCards. IL6 Gene—Interleukin 6. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL6 (accessed on 19 June 2022).
- Ropelle, E.R.; Flores, M.B.; Cintra, D.E.; Rocha, G.Z.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Moraes, J.C.; Prada, P.O.; Guadagnini, D.; et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol. 2010, 8, e1000465. [Google Scholar] [CrossRef] [PubMed]
- Wing, R.R.; Phelan, S. Long-term weight loss maintenance. Am. J. Clin. Nutr. 2005, 82, 222S–225S. [Google Scholar] [CrossRef]
- Singh, N.; Stewart, R.A.H.; Benatar, J.R. Intensity and duration of lifestyle interventions for long-term weight loss and association with mortality: A meta-analysis of randomised trials. BMJ Open 2019, 9, e029966. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Population | Overweight and Obese Children and Adolescents |
|---|---|
| Intervention | Obesity lifestyle management interventions |
| Comparison | Carriers of at-risk genotypes (SNPs or CNVs) for obesity versus non-carriers of at-risk genotypes for obesity |
| Outcome | Change of BMI or other measures of body composition |
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| Age ≤ 20 years | Age > 20 years |
| Any language | In vitro or animal studies |
| Any geographic location | Reviews, editorials, books and book chapters, notes, letters, conference papers, surveys |
| Any publication dates | Preventive intervention programs for obesity development |
| Species: Humans | Monogenic and/or syndromic obesity |
| Obesity lifestyle interventional studies | Investigation for other mutations except SNPs or CNVs |
| BMI: overweight/obesity | Studies examining ΒΜΙ and/or body composition change at a time after the end of a lifestyle intervention program |
| Common/polygenic obesity | Pharmacological or bariatric surgery obesity management interventions |
| Outcomes examined: change in ΒΜΙ and/or body composition in relation to the genotype | Other outcomes examined and not ΒΜΙ and/or body composition change (e.g., gene expression) |
| Examining the effect of SNPs and/or CNVs |
| References | Study Type | Sample Size, N | Country | Age (Years), Mean Age ± SD or Range | Female (%) | Pubertal Status | Obesity Definition |
|---|---|---|---|---|---|---|---|
| Barbian et al., J. Pediatr. Genet., 2019 [48] | Quasi-Experimental | 34 | Brazil | 10–15 | 73.5% | Prepubertal: 41.1% Continuous Maturation: 50% Matured: 8.9% | WHO |
| Corgosinho et al., Neuropeptides, 2017 [49] | Cohort | 76 | Brazil | 15–19 | N/A | 100% Post-Pubertal | CDC |
| Deram et al., J. Clin. Endocrinol. Metab., 2008 [50] | Cohort | 335 | Brazil | 10.7 ± 1.3 | 66.6% | 49% Pubescent | CDC |
| do Nascimento et al., Eur. J. Nutr., 2017 [51] | Longitudinal | Children: 136 OW/OB, 172 NW Women: 126 | Brazil | Children: 8–17, 13.55 ± 2 | 33.7% | N/A | WHO |
| Gajewska et al., Nutrients, 2016 [52] | Cohort | 100 | Poland | 5–10 | 53% | 100% Prepubertal | Polish Ref. Chart |
| Gao et al., Exp. Physiol., 2015 [53] | Cohort | 55 | China | 16.55 ± 2.87 | 49% | N/A | Chinese Ref. Chart |
| Hagman et al., Pediatr. Diabetes, 2018 [54] | Cohort | 434 (Overall) 214 (FTO genotyping) | USA | 4–20, 12.4 ± 2.7 | 64.5% | 31.3% Prepubertal | CDC |
| Heitkamp et al., JAMA Pediatr., 2020 [55] | Cohort | 1198 | Germany | 14.0 ± 2.2 | 56% | N/A | IOTF |
| Hollensted et al., Obesity, 2018 [56] | Baseline: Case Control Follow-Up: Cohort | Baseline: Cases: 920 OW/OB Controls: 698 NW Follow-Up: 754 | Denmark | Cases: 11.63 (9.59–13.87) Controls: 12.50 (10.09–15.10) | 57.7% | N/A | Danish Ref. Chart |
| Holzapfel et al., Eur. J. Endocrinol., 2011 [57] | Cohort | 310 | Germany | 8–19, 14 ± 2 | 60.3% | N/A | German Ref. Chart |
| Knoll et al., Horm. Metab. Res., 2012 [58] | Cohort | 453 | Germany | 10.8 ± 2.6 | 55% | N/A | German Ref. Chart |
| Lai et al., Int. J. Biol. Sci, 2013 [59] | Cohort | 88 | China | 14.11 ± 3.63 | 50% | N/A | N/A |
| Leite et al., Mortiz, 2017 [60] | Randomized Control Trial | 47 | Brazil | 12–16, 15.05 ± 1.07 | 44.6% | Tanner Stage: 4 or 5 | WHO |
| Moleres et al., J. Pediatr., 2012 [61] | Cohort | 168 | Spain | 12–16, 14.6 ± 0.09 | 62% | N/A | IOTF |
| Moleres et al., Nutr. Hosp., 2014 [62] | Cohort | 199 | Spain | 12–16, 14.5 ± 0.08 | 61% | N/A | IOTF |
| Moraes et al., An. Acad. Bras. Cienc., 2016 [63] | Cohort | 36 Control Group: 17 Intervention Group: 19 | Brazil | 8–16 Control Group: 11.3 ± 1.6 Ιnterventiοn Group: 10.2 ± 2.2 | 58.3% | N/A | CDC |
| Müller et al., BMC Med. Genet., 2008 [64] | Baseline: Case Control Follow-Up: Cohort | Baseline: Cases: 519 Children Controls: 178 Adults Follow up: 207 | Germany | Follow-Up: 10.79 ± 2.52 | Follow up: 54.5% | N/A | IOTF |
| Reinehr et al., Diabetes, 2008 [65] | Cohort | 293 | Germany | 6–16, 10.8 ± 2.7 | 55% | 51% Prepubertal, 30% Pubertal, 19% Post-Pubertal | IOTF |
| Reinehr et al., Arch. Dis. Child., 2009 [66] | Cohort | 280 | Germany | 10.8 (4.5–16.5) | 45% | N/A | German Ref. Chart |
| Roth et al., BMC Pediatr., 2013 [67] | Βaseline: Case Control Follow-Up: Longitudinal | 451 (28 OW, 423 OB) 583 Lean Adults | Germany | Children: 12.0 (10.0–13.7) Adults: 25.3 (22.5–26.8) | 54.9% | N/A | IOTF |
| Santoro et al., Am. J. Clin. Nutr., 2007 [68] | Baseline: Case Control Follow-Up: Cohort | 184 OB 100 Non-OΒ Controls | Italy | 9.2 ± 2 | 41.8% | 82% Prepubertal | Italian Ref. Chart |
| Scherag et al., Obesity, 2011 [69] | Longitudinal Cohort | 401 Children 626 Adults | Germany | 10.74 ± 2.55 | 54.6% | 53.9% Prepubertal | IOTF |
| Schum et al., Exp. Clin. Endocrinol. Diabetes, 2012 [70] | Longitudinal | 75 | Germany | 12.6 ± 2.6 | 46.6% | Pubertal: Heterozygous: 55.8% Homozygous: 76.2% | N/A |
| Vogel et al., Obes. Facts, 2011 [71] | Baseline: Case Control Follow-Up: Cohort | Baseline Cases: 889 Controls: 442 Follow-Up: 367 | Germany | Baseline: Cases: 10.69 ± 2.98 Controls: 18.31 ± 1.10 Follow-Up: 10.77 ± 2.66 | Baseline: Cases: 53.2% Controls: 61.3% Follow-Up: 55.9% | N/A | IOTF |
| Volckmar et al., Exp. Clin. Endocrinol. Diabetes, 2013 [72] | Baseline: Case Control Follow-Up: Cohort | Baseline: Cases: 454 Controls: 435 Follow-Up: 454 | Germany | 6–16, 10.8 ± 2.6 | 55% | N/A | German Ref. Chart |
| Zlatohlavek et al., Clin. Biochem., 2013 [73] | Cohort | 357 | Czech Republic | 8–15, 13.7 ± 4.9 | 61% | N/A | N/A |
| Zlatohlavek et al., Med. Sci. Monit., 2018 [74] | Cohort | 684 | Czech Republic | 12.7 ± 2.1 | 59% | N/A | N/A |
| References | Genes | Main Findings |
|---|---|---|
| Central nervous system and obesity | ||
| Heitkamp et al., JAMA Pediatr., 2020 [55] | CADM2 | The G allele in rs13078960 SNP is associated with decreased BMI-SDS reduction |
| Roth et al., BMC Pediatr., 2013 [67] | DRD2 | Homozygosity for the Τ allele in rs18000497 SNP is associated with decreased BMI-SDS reduction |
| Heitkamp et al., JAMA Pediatr., 2020 [55] | LMX1B | Homozygosity for the A allele in rs10733682 SNP is associated with greater weight loss |
| Hollensted et al., Obesity, 2018 [56] | The T allele in rs3829849 SNP is correlated with decreased BMI-SDS reduction | |
| Gajewska et al., Nutrients, 2016 [52] | LEPR | Carrying at least one minor G allele in Q223R together with the wild-type K665N is associated with the greatest weight loss and fat mass reduction |
| Corgosinho et al., Neuropeptides, 2017 [49] | Homozygosity for the T allele in rs2767485 SNP is associated with greater BMI reduction | |
| Zlatohlavek et al., Clin. Biochem., 2013 [73] | MC4R | Homozygosity for the C allele in s17782313 SNP is associated with greater weight loss outcomes |
| Moleres et al., J. Pediatr., 2012 [61] | The C allele in s17782313 SNP is associated with greater BMI and body composition reduction as part of a GRS | |
| Vogel et al., Obes. Facts, 2011 [71] | The C allele in rs17782313 SNP or the A allele in rs12970134 SNP in females are associated with more efficient BMI-SDS reduction than in males | |
| Santoro et al., Am. J. Clin. Nutr., 2007 [68] | MC3R | The 6Lys allele of rs3746619 and the 81Ile allele of rs3827103 are associated with reduced BMI change |
| Adipose tissue and obesity | ||
| Heitkamp et al., JAMA Pediatr., 2020 [55] | RPTOR | Homozygosity for the G allele in the rs12940622 SNP is associated with reduced weight loss |
| Barbian et al. [48], do Nascimento et al. [51], Müller et al. [64], Moraes et al. [63], Hollensted et al. [56], Scherag et al. [69], Schum et al. [70] | FTO | Τhe rs9939609, rs1421085, rs1558902, rs1421085, rs17817449, rs9939609 SNPs are not associated with BMI or body composition change |
| Moleres et al., J. Pediatr., 2012 [61] | The A allele in rs9939609 SNP is associated with greater BMI reduction both individually and as a part of a GRS | |
| Reinehr et al., Arch. Dis. Child., 2009 [66] | Homozygosity for the A allele in rs9939609 SNP of FTO together with homozygosity for the C allele in rs7566605 SNP of INSIG2 is associated with decreased BMI reduction | |
| Zlatohlavek et al., Clin. Biochem., 2013 [73] | Homozygosity for the G allele in rs17817449 SNP of FTO is associated with greater BMI reduction both individually and in synergy with homozygosity for the C allele in rs17782313 SNP of MC4R | |
| Hagman et al., Pediatr. Diabetes, 2018 [54] | Homozygosity for the A allele in rs8050136 SNP is correlated with greater BMI reduction | |
| Heitkamp et al., JAMA Pediatr., 2020 [55] | ETS2 | Homozygosity for the C allele in rs2836754 SNP is associated with greater body weight and BMI reduction |
| Heitkamp et al., JAMA Pediatr., 2020 [55] | KAT8 | The A allele in rs9925964 SNP is associated with greater BMI-SDS reduction |
| Moleres et al., J. Pediatr., 2012 [61] | TMEM1 | The G allele in rs7561317 SNP is associated with greater BMI-SDS and fat mass reduction individually and as a part of a GRS |
| Hollensted et al., Obesity, 2018 [56], Scherag et al., Obesity, 2011 [69], Zlatohlavek et al., Med. Sci. Monit., 2018 [74] | The rs4854349, rs4854344, and rs11127485 SNPs are not associated with BMI or body composition change | |
| Moleres et al., J. Pediatr., 2012 [61] | PPARγ | The G allele of rs1801282 SNP is associated with a greater BMI and fat mass reduction as part of a GRS |
| Scherag et al., Obesity, 2011 [69] | SDCCAG8 | Homozygosity for the T allele in rs10926984 SNP, the T allele in rs12145833 SNP and the C allele in rs2783963 SNP is associated with reduced BMI change |
| Heitkamp et al., JAMA Pediatr., 2020 [55] | CPNE8 | The A allele in rs11170468 SNP is associated with resistance to BMI-SDS reduction |
| Adipose tissue metabolism | ||
| Moleres et al., Nutr. Hosp., 2014 [62] | APOA1 | The A allele in rs670 SNP is associated with greater weight and BMI reduction, while combined analyses with the A allele in rs1800777 SNP explains up to 24% of BMI-SDS amelioration |
| Moleres et al., Nutr. Hosp., 2014 [62] | CETP | The A allele in rs1800777 is associated with greater weight and BMI reduction, while combined analyses with the A allele in rs670 SNP explains up to 24% of BMI-SDS amelioration |
| Gao et al., Exp. Physiolm., 2015 [53] | LPL | Homozygosity for the G allele in rs283 SNP is associated with greater body fat reduction |
| Reinehr et al., Diabetes, 2008 [65], Reinehr et al., Arch. Dis. Child., 2009 [66] | INSIG2 | Homozygosity for the C alle in rs7566605 SNP is associated with lower BMI and BMI-SDS reduction |
| Reinehr et al., Arch. Dis. Child., 2009 [66] | Homozygosity for the C allele in rs7566605 SNP of INSIG2 together with homozygosity for the A allele in rs9939609 SNP of FTO is associated with decreased BMI reduction | |
| Deram et al., J. Clin. Endocrinol. Metab., 2008 [50] | PLIN1 | The T allele in rs1052700 is associated with greater BMI and body composition change |
| Moleres et al., J. Pediatr., 2012 [61] | ADIPOQ | The C allele in rs822395, the G allele in rs2241766, and the T allele in rs1501299 SNPs are associated with a greater BMI and fat mass reduction as part of a GRS |
| Gajewska et al., 2016, Nutrients [52] | The rs266729 and rs1686119 SNPs are not associated with BMI or body composition change | |
| Adipose tissue inflammation and obesity | ||
| Heitkamp et al., JAMA Pediatr., 2020 [55] | IFNGR1 | Homozygosity for the G allele of the rs13201877 SNP is associated with greater weight and BMI reduction |
| Heitkamp et al., JAMA Pediatr., 2020 [55] | SLC39A8 | The T allele in rs13107325 SNP is associated with decreased BMI-SDS reduction |
| Moleres et al., J. Pediatr., 2012 [61] | IL6 | The G allele in rs1800795 SNP is associated with a greater BMI and fat mass reduction as part of a GRS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vourdoumpa, A.; Paltoglou, G.; Charmandari, E. The Genetic Basis of Childhood Obesity: A Systematic Review. Nutrients 2023, 15, 1416. https://doi.org/10.3390/nu15061416
Vourdoumpa A, Paltoglou G, Charmandari E. The Genetic Basis of Childhood Obesity: A Systematic Review. Nutrients. 2023; 15(6):1416. https://doi.org/10.3390/nu15061416
Chicago/Turabian StyleVourdoumpa, Aikaterini, George Paltoglou, and Evangelia Charmandari. 2023. "The Genetic Basis of Childhood Obesity: A Systematic Review" Nutrients 15, no. 6: 1416. https://doi.org/10.3390/nu15061416
APA StyleVourdoumpa, A., Paltoglou, G., & Charmandari, E. (2023). The Genetic Basis of Childhood Obesity: A Systematic Review. Nutrients, 15(6), 1416. https://doi.org/10.3390/nu15061416