


Glutamine Supplementation Preserves Glutamatergic Neuronal Activity in the Infralimbic Cortex, Which Delays the Onset of Mild Cognitive Impairment in 3×Tg-AD Female Mice

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
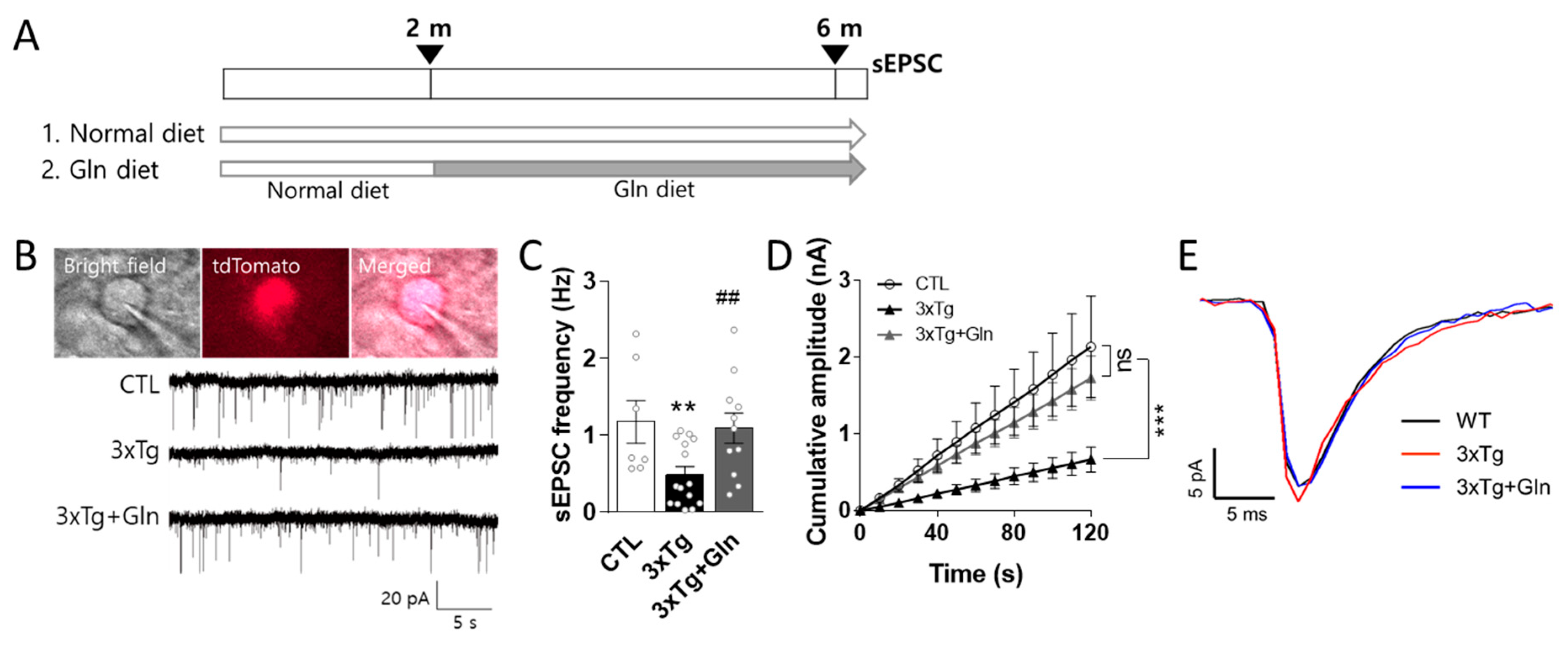
2.2. Glutamatergic Neurotransmission Activity Analysis
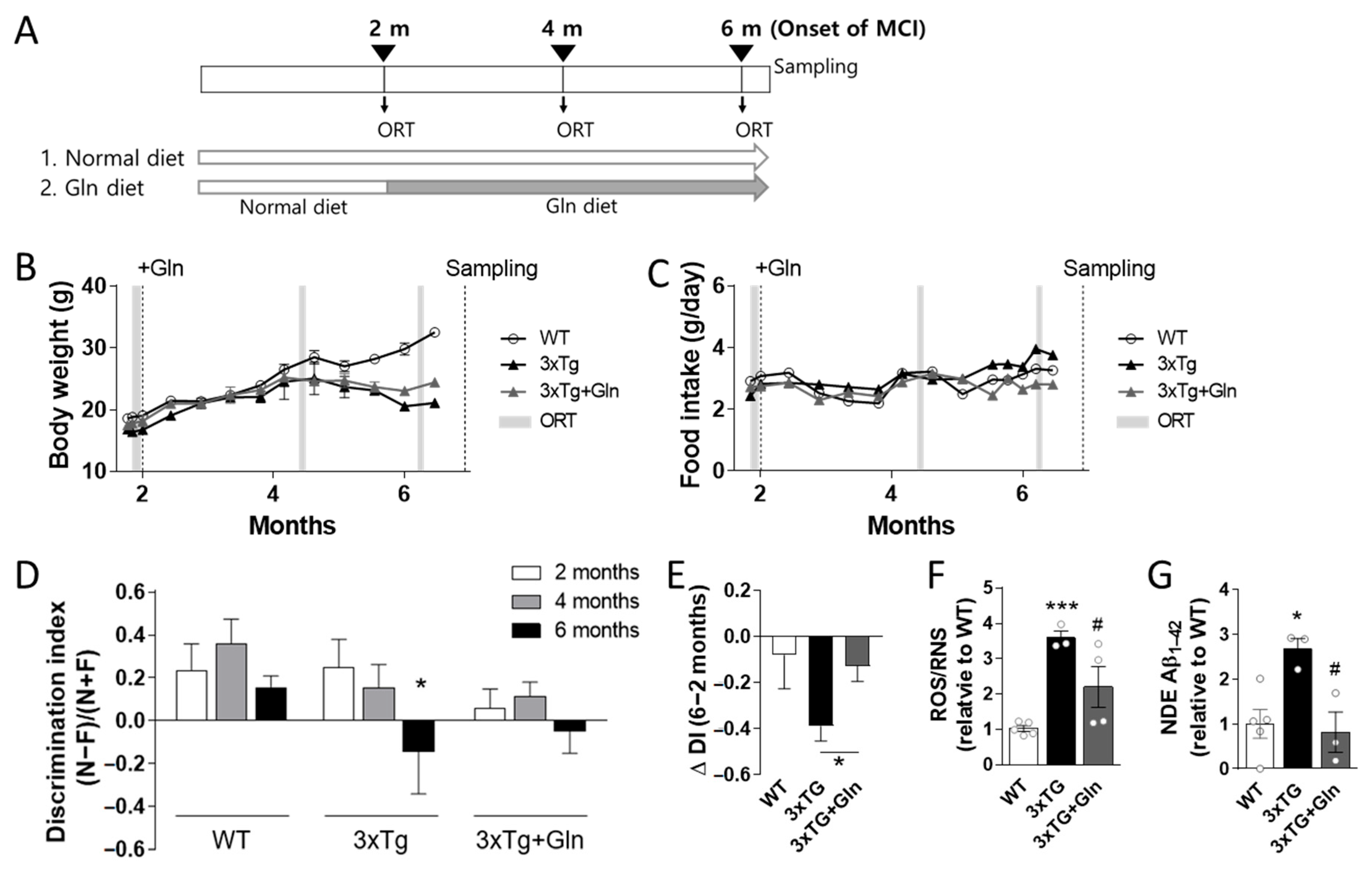
2.3. Object Recognition Test (ORT)
2.4. Total ROS/RNS Assay
2.5. Neuron-Derived Exosome (NDE) Isolation and Aβ1–42 Enzyme-Linked Immunosorbent Assay (ELISA)
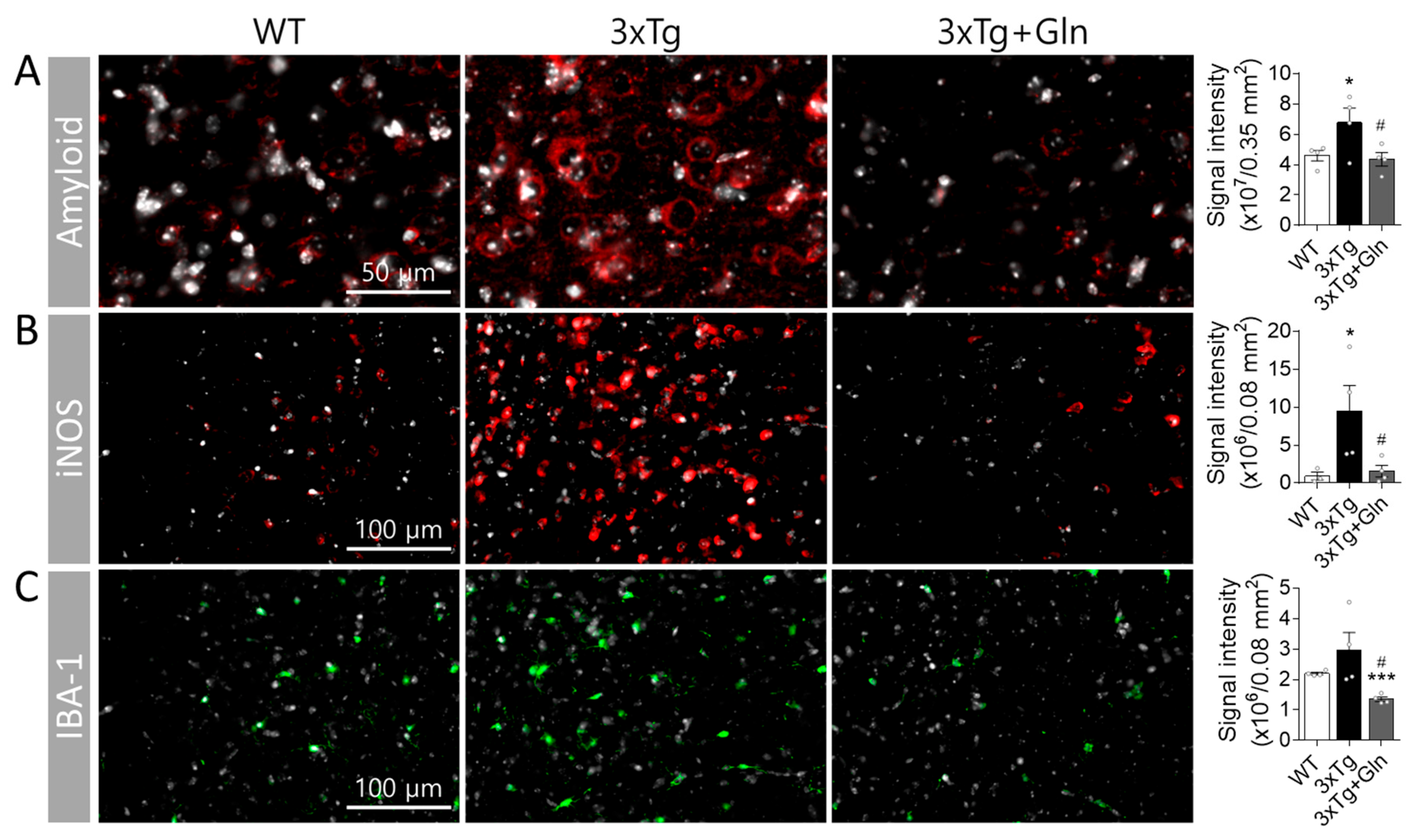
2.6. Immunohistochemistry (IHC)
2.7. Statistical Analysis
3. Results
3.1. Gln Supplementation Maintained Glutamatergic Neurotransmission Activity in the IL
3.2. Gln Supplementation Prevented MCI Onset in 3×Tg Mice
3.3. Gln Prevented Increases in Amyloid, iNOS, and IBA-1 Contents in 3×Tg Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild cognitive impairment: Clinical characterization and outcome. Arch. Neurol. 1999, 56, 303–308. [Google Scholar] [CrossRef]
- Winblad, B.; Palmer, K.; Kivipelto, M.; Jelic, V.; Fratiglioni, L.; Wahlund, L.O.; Nordberg, A.; Backman, L.; Albert, M.; Almkvist, O.; et al. Mild cognitive impairment--beyond controversies, towards a consensus: Report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 2004, 256, 240–246. [Google Scholar] [CrossRef]
- Petersen, R.C.; Morris, J.C. Mild cognitive impairment as a clinical entity and treatment target. Arch. Neurol. 2005, 62, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Baek, J.H.; Jung, S.; Chung, H.J.; Lee, D.K.; Kim, H.J. Ingestion of Bis(2-ethylhexyl) phthalate (DEHP) during adolescence causes depressive-like behaviors through hypoactive glutamatergic signaling in the medial prefrontal cortex. Environ. Pollut. 2021, 289, 117978. [Google Scholar] [CrossRef]
- Lee, Y.; Son, H.; Kim, G.; Kim, S.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Glutamine deficiency in the prefrontal cortex increases depressive-like behaviours in male mice. J. Psychiatry Neurosci. 2013, 38, 183–191. [Google Scholar] [CrossRef]
- Son, H.; Baek, J.H.; Go, B.S.; Jung, D.H.; Sontakke, S.B.; Chung, H.J.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; et al. Glutamine has antidepressive effects through increments of glutamate and glutamine levels and glutamatergic activity in the medial prefrontal cortex. Neuropharmacology 2018, 143, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2012, 13, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Baek, J.H.; Vignesh, A.; Son, H.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Glutamine Supplementation Ameliorates Chronic Stress-induced Reductions in Glutamate and Glutamine Transporters in the Mouse Prefrontal Cortex. Exp. Neurobiol. 2019, 28, 270–278. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Gauthier, J.; Araki, Y.; Lin, D.T.; Yoshizawa, Y.; Higashi, K.; Park, A.R.; Spiegelman, D.; Dobrzeniecka, S.; Piton, A.; et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011, 88, 306–316. [Google Scholar] [CrossRef]
- Rahn, K.A.; Slusher, B.S.; Kaplin, A.I. Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr. Med. Chem. 2012, 19, 1335–1345. [Google Scholar] [CrossRef]
- Son, H.; Kim, S.; Jung, D.H.; Baek, J.H.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Lee, D.K.; et al. Insufficient glutamine synthetase activity during synaptogenesis causes spatial memory impairment in adult mice. Sci. Rep. 2019, 9, 252. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Wei, J.; Liu, W.; Zhong, P.; Li, X.; Yan, Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 2012, 73, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Jung, S.; Son, H.; Kang, J.S.; Kim, H.J. Glutamine Supplementation Prevents Chronic Stress-Induced Mild Cognitive Impairment. Nutrients 2020, 12, 910. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Jung, S.; Shin, J.H.; Kang, M.J.; Kim, H.J. Anti-Stress and Anti-Depressive Effects of Spinach Extracts on a Chronic Stress-Induced Depression Mouse Model through Lowering Blood Corticosterone and Increasing Brain Glutamate and Glutamine Levels. J. Clin. Med. 2018, 7, 406. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic. Biol. Med. 2011, 51, 171–178. [Google Scholar] [CrossRef]
- Butterfield, D.A. Oxidative Stress in Brain in Amnestic Mild Cognitive Impairment. Antioxidants 2023, 12, 462. [Google Scholar] [CrossRef]
- Fontella, F.U.; Siqueira, I.R.; Vasconcellos, A.P.; Tabajara, A.S.; Netto, C.A.; Dalmaz, C. Repeated restraint stress induces oxidative damage in rat hippocampus. Neurochem. Res. 2005, 30, 105–111. [Google Scholar] [CrossRef]
- Peng, Y.L.; Liu, Y.N.; Liu, L.; Wang, X.; Jiang, C.L.; Wang, Y.X. Inducible nitric oxide synthase is involved in the modulation of depressive behaviors induced by unpredictable chronic mild stress. J. Neuroinflamm. 2012, 9, 75. [Google Scholar] [CrossRef]
- Gimenez-Llort, L.; Blazquez, G.; Canete, T.; Johansson, B.; Oddo, S.; Tobena, A.; LaFerla, F.M.; Fernandez-Teruel, A. Modeling behavioral and neuronal symptoms of Alzheimer’s disease in mice: A role for intraneuronal amyloid. Neurosci. Biobehav. Rev. 2007, 31, 125–147. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef]
- Sun, B.; Dalvi, P.; Abadjian, L.; Tang, N.; Pulliam, L. Blood neuron-derived exosomes as biomarkers of cognitive impairment in HIV. AIDS 2017, 31, F9–F17. [Google Scholar] [CrossRef]
- Lee, E.E.; Winston-Gray, C.; Barlow, J.W.; Rissman, R.A.; Jeste, D.V. Plasma Levels of Neuron- and Astrocyte-Derived Exosomal Amyloid Beta1–42, Amyloid Beta1–40, and Phosphorylated Tau Levels in Schizophrenia Patients and Non-psychiatric Comparison Subjects: Relationships with Cognitive Functioning and Psychopathology. Front. Psychiatry 2020, 11, 532624. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Mu, D.; Ma, X.; Wang, D.; Zhong, J.; Gao, J.; Yu, S.; Qiu, L. Review on the roles of specific cell-derived exosomes in Alzheimer’s disease. Front. Neurosci. 2022, 16, 936760. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Knight, E.M.; Verkhratsky, A.; Luckman, S.M.; Allan, S.M.; Lawrence, C.B. Hypermetabolism in a triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 187–193. [Google Scholar] [CrossRef]
- Sancheti, H.; Patil, I.; Kanamori, K.; Díaz Brinton, R.; Zhang, W.; Lin, A.L.; Cadenas, E. Hypermetabolic state in the 7-month-old triple transgenic mouse model of Alzheimer’s disease and the effect of lipoic acid: A 13C-NMR study. J. Cereb. Blood Flow Metab. 2014, 34, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 2011, 6, 55. [Google Scholar] [CrossRef]
- Lee, K.W.; Lee, S.H.; Kim, H.; Song, J.S.; Yang, S.D.; Paik, S.G.; Han, P.L. Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6 inbred mice expressing transgenic amyloid precursor protein. J. Neurosci. Res. 2004, 76, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.S.; Hamlett, E.D.; Stone, T.D.; Sims-Robinson, C. Neuronally derived extracellular vesicles: An emerging tool for understanding Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 22. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e601. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Carlson, O.D.; Mustapic, M.; Kapogiannis, D. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 769–773. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Lopez Sanchez, M.I.G.; van Wijngaarden, P.; Trounce, I.A. Amyloid precursor protein-mediated mitochondrial regulation and Alzheimer’s disease. Br. J. Pharmacol. 2019, 176, 3464–3474. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J. Neurochem. 2006, 96, 1–13. [Google Scholar] [CrossRef]
- Xia, Y.; Zweier, J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958. [Google Scholar] [CrossRef]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-beta peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef] [PubMed]
- Tiiman, A.; Palumaa, P.; Tougu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378. [Google Scholar] [CrossRef]
- Mates, J.M.; Perez-Gomez, C.; Nunez de Castro, I.; Asenjo, M.; Marquez, J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int. J. Biochem. Cell Biol. 2002, 34, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Houdijk, A.P.; Visser, J.J.; Rijnsburger, E.R.; Teerlink, T.; van Leeuwen, P.A. Dietary glutamine supplementation reduces plasma nitrate levels in rats. Clin. Nutr. 1998, 17, 11–14. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, J.H.; Kang, J.S.; Song, M.; Lee, D.K.; Kim, H.J. Glutamine Supplementation Preserves Glutamatergic Neuronal Activity in the Infralimbic Cortex, Which Delays the Onset of Mild Cognitive Impairment in 3×Tg-AD Female Mice. Nutrients 2023, 15, 2794. https://doi.org/10.3390/nu15122794
Baek JH, Kang JS, Song M, Lee DK, Kim HJ. Glutamine Supplementation Preserves Glutamatergic Neuronal Activity in the Infralimbic Cortex, Which Delays the Onset of Mild Cognitive Impairment in 3×Tg-AD Female Mice. Nutrients. 2023; 15(12):2794. https://doi.org/10.3390/nu15122794
Chicago/Turabian StyleBaek, Ji Hyeong, Jae Soon Kang, Miyoung Song, Dong Kun Lee, and Hyun Joon Kim. 2023. "Glutamine Supplementation Preserves Glutamatergic Neuronal Activity in the Infralimbic Cortex, Which Delays the Onset of Mild Cognitive Impairment in 3×Tg-AD Female Mice" Nutrients 15, no. 12: 2794. https://doi.org/10.3390/nu15122794
APA StyleBaek, J. H., Kang, J. S., Song, M., Lee, D. K., & Kim, H. J. (2023). Glutamine Supplementation Preserves Glutamatergic Neuronal Activity in the Infralimbic Cortex, Which Delays the Onset of Mild Cognitive Impairment in 3×Tg-AD Female Mice. Nutrients, 15(12), 2794. https://doi.org/10.3390/nu15122794