
Xanthohumol Interferes with the Activation of TGF-β Signaling in the Process Leading to Intestinal Fibrosis

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cell Culture and Drug Treatment
2.3. Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
2.4. Immunoblot and Immunoprecipitation (IP) Analysis
2.5. Luciferase Assays
2.6. Immunofluorescence
2.7. Chromatin Immunoprecipitation (ChIP)
2.8. Statistical Analysis
3. Results
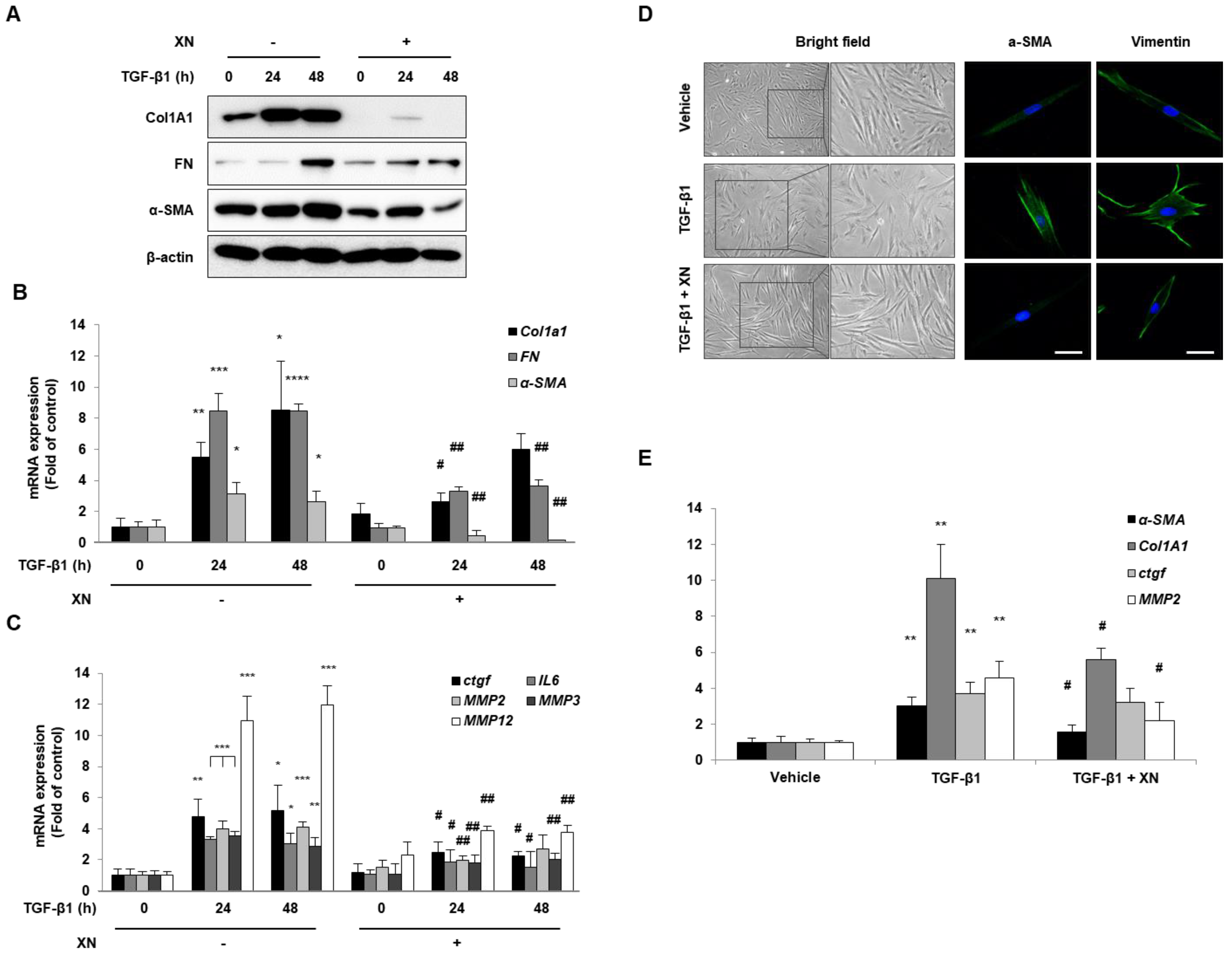
3.1. XN Inhibits Intestinal Fibrosis in Primary HIFs
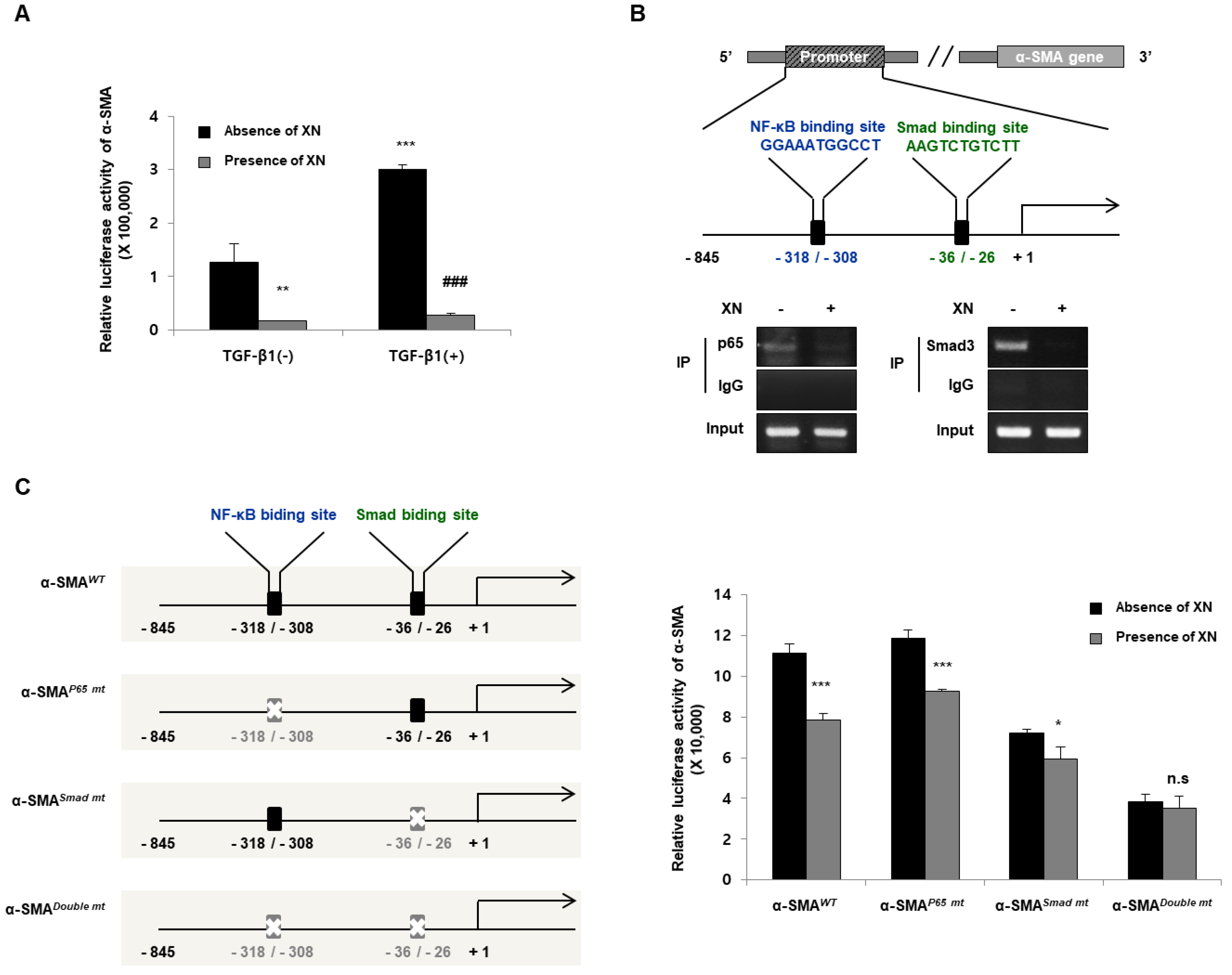
3.2. XN Reduces Fibrotic Responses via Regulation of α-SMA Promoter
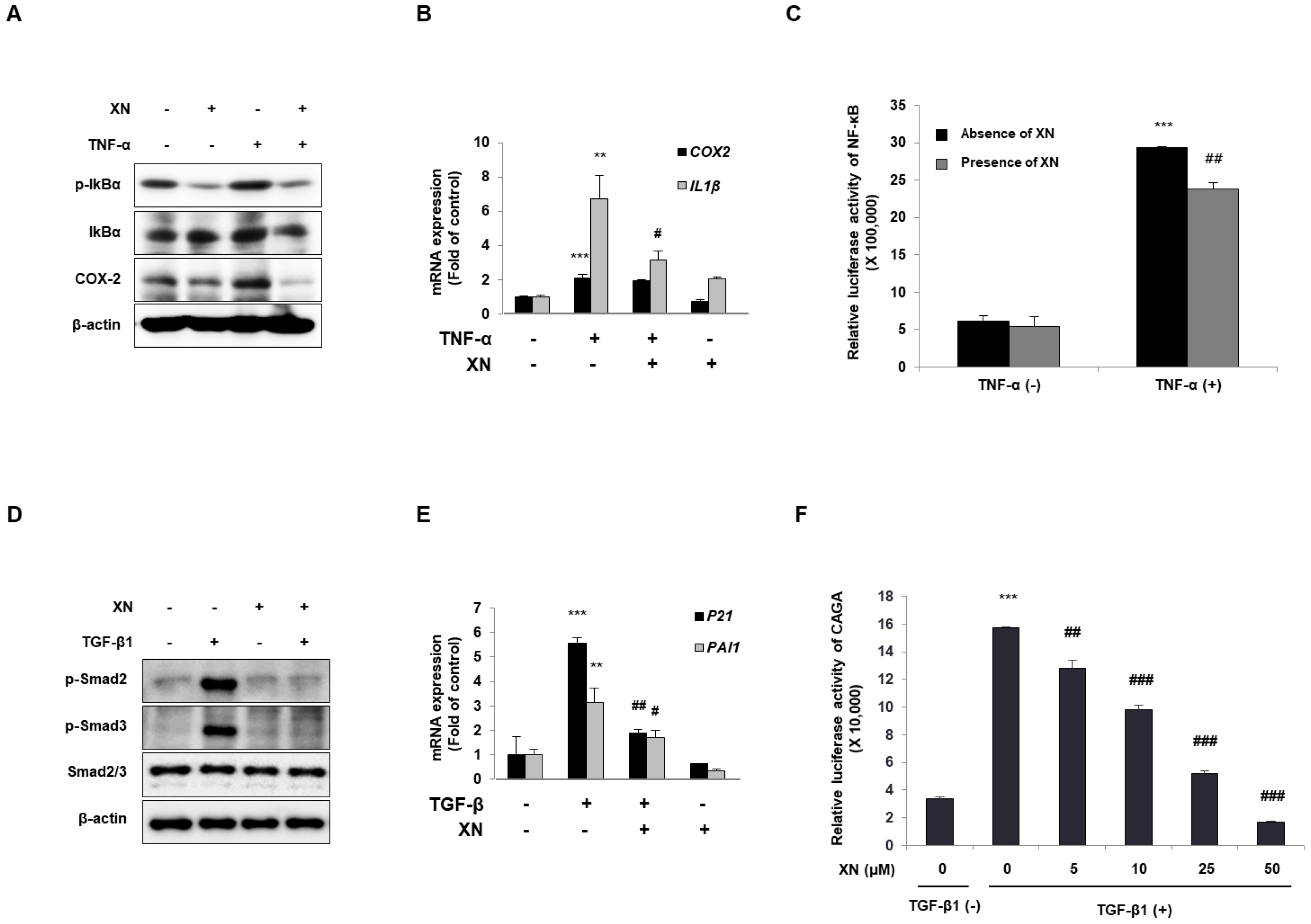
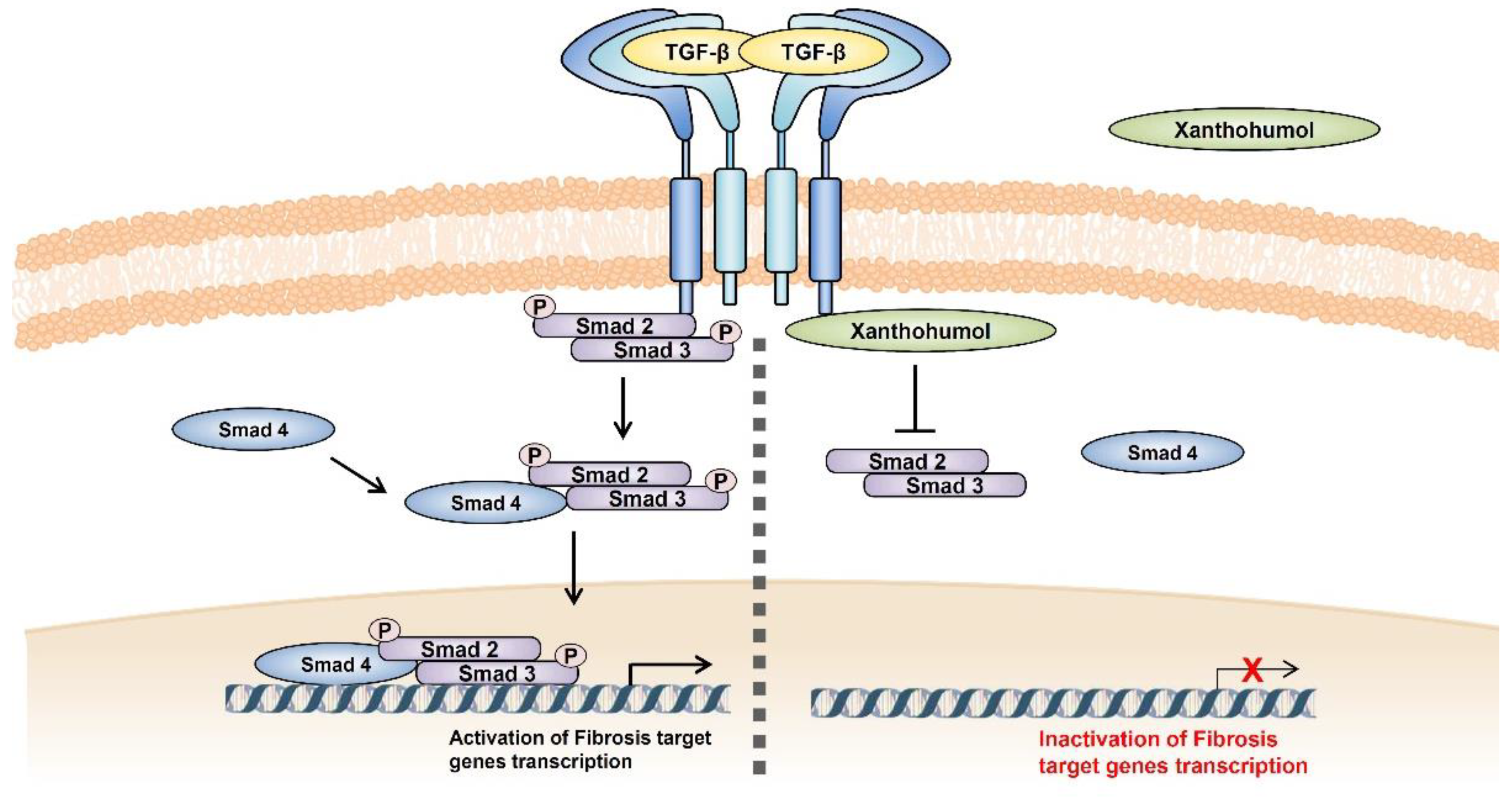
3.3. XN Interrupts Canonical Activation of Both NF-κB and TGF-β Mechanisms
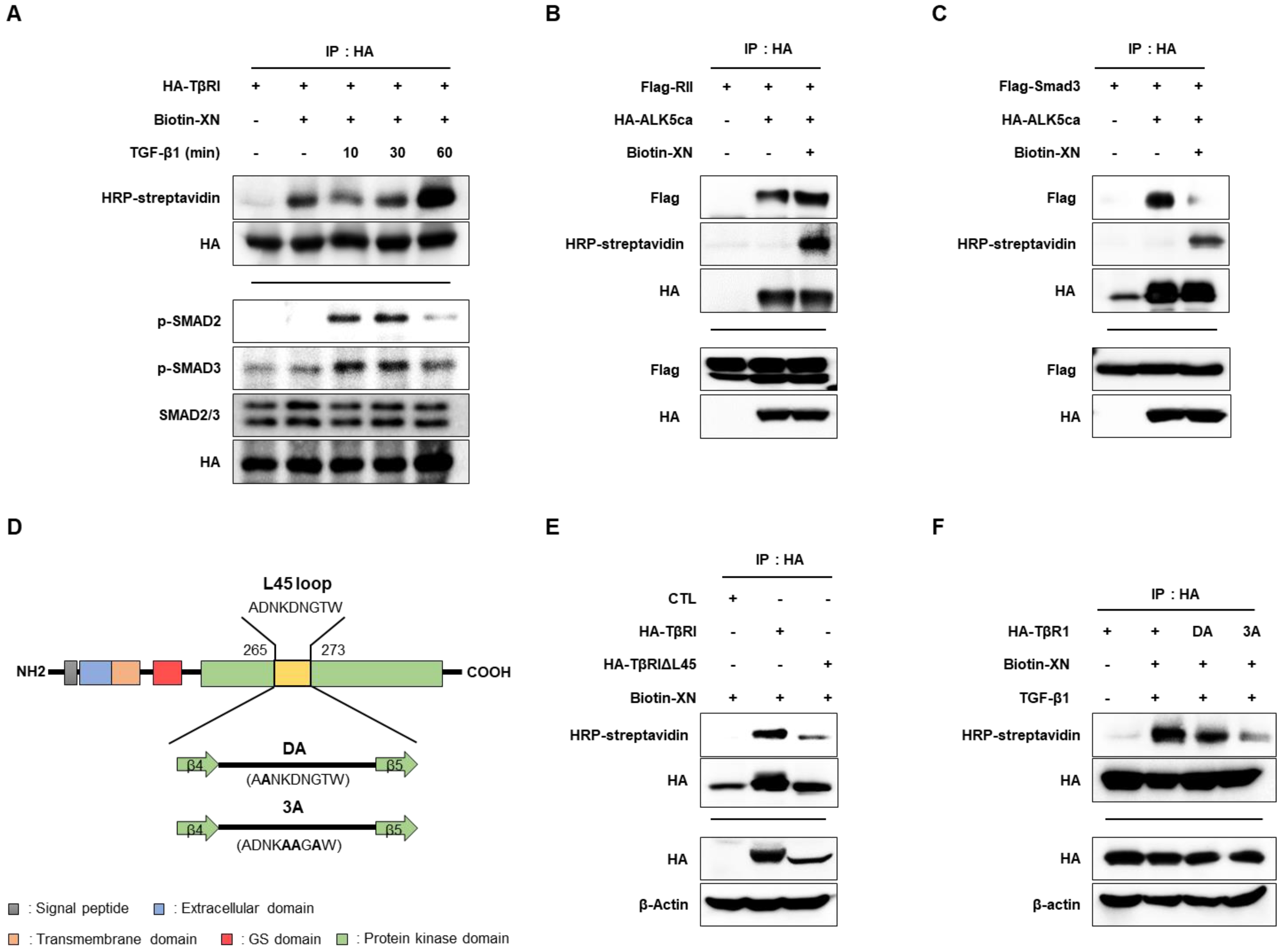
3.4. XN Interacts with TβRI L45 to Regulate TGF-β/Smad3 Signaling in SW620 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pizarro, T.T.; Stappenbeck, T.S.; Rieder, F.; Rosen, M.J.; Colombel, J.-F.; Donowitz, M.; Towne, J.; Mazmanian, S.K.; Faith, J.J.; Hodin, R.A. Challenges in IBD research: Preclinical human IBD mechanisms. Inflamm. Bowel Dis. 2019, 25, S5–S12. [Google Scholar] [CrossRef] [PubMed]
- Lenti, M.V.; Di Sabatino, A. Intestinal fibrosis. Mol. Asp. Med. 2019, 65, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Pariente, B.; Cosnes, J.; Danese, S.; Sandborn, W.J.; Lewin, M.; Fletcher, J.G.; Chowers, Y.; d’Haens, G.; Feagan, B.G.; Hibi, T. Development of the Crohn’s disease digestive damage score, the Lemann score. Inflamm. Bowel Dis. 2011, 17, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F. Toward an antifibrotic therapy for inflammatory bowel disease. United Eur. Gastroenterol. J. 2016, 4, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, G.; Lenti, M.V.; Di Sabatino, A. Therapeutic Targeting of Intestinal Fibrosis in Crohn’s Disease. Cells 2022, 11, 429. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D.A. Mechanisms of fibrogenesis. Exp. Biol. Med. 2008, 233, 109–122. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Meng, X.-m.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Sun, T.-L.; Li, W.-Q.; Tong, X.-L.; Liu, X.-Y.; Zhou, W.-H. Xanthohumol attenuates isoprenaline-induced cardiac hypertrophy and fibrosis through regulating PTEN/AKT/mTOR pathway. Eur. J. Pharmacol. 2021, 891, 173690. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Dorn, C.; Kraus, B.; Motyl, M.; Weiss, T.S.; Gehrig, M.; Schölmerich, J.; Heilmann, J.; Hellerbrand, C. Xanthohumol, a chalcon derived from hops, inhibits hepatic inflammation and fibrosis. Mol. Nutr. Food Res. 2010, 54, S205–S213. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-M.; Yun, S.-M.; Choi, Y.-H.; Heo, J.; Kim, N.-J.; Kim, S.-H.; Kim, E.-H. Xanthohumol prevents dextran sulfate sodium-induced colitis via inhibition of IKKβ/NF-κB signaling in mice. Oncotarget 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Strong, S.A.; Pizarro, T.T.; Klein, J.S.; Cominelli, F.; Fiocchi, C. Proinflammatory cytokines differentially modulate their own expression in human intestinal mucosal mesenchymal cells. Gastroenterology 1998, 114, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-M.; Hahm, K.B.; Park, J.-M.; Hong, S.P.; Kim, E.-H. Paradoxically Augmented Anti-Tumorigenic Action of Proton Pump Inhibitor and Gastrin in APCMin/+ Intestinal Polyposis Model1. Neoplasia 2014, 16, 73-W21. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Itoh, Y.; Koinuma, D.; Omata, C.; Ogami, T.; Motizuki, M.; Yaguchi, S.-i.; Itoh, T.; Miyake, K.; Tsutsumi, S.; Aburatani, H. A comparative analysis of Smad-responsive motifs identifies multiple regulatory inputs for TGF-β transcriptional activation. J. Biol. Chem. 2019, 294, 15466–15479. [Google Scholar] [CrossRef]
- Brodziak-Jarosz, L.; Fujikawa, Y.; Pastor-Flores, D.; Kasikci, S.; Jirásek, P.; Pitzl, S.; Owen, R.W.; Klika, K.D.; Gerhäuser, C.; Amslinger, S. A click chemistry approach identifies target proteins of xanthohumol. Mol. Nutr. Food Res. 2016, 60, 737–748. [Google Scholar] [CrossRef]
- Groppe, J.; Hinck, C.S.; Samavarchi-Tehrani, P.; Zubieta, C.; Schuermann, J.P.; Taylor, A.B.; Schwarz, P.M.; Wrana, J.L.; Hinck, A.P. Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 2008, 29, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N.; Loftus, E.V.; Ng, S.C.; Lakatos, P.L.; Moum, B. Hospitalisations and surgery in Crohn’s disease. Gut 2012, 61, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Speca, S.; Giusti, I.; Rieder, F.; Latella, G. Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. WJG 2012, 18, 3635. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Luke, A.; Sauder, K.; Moons, D.S.; Horowitz, J.C.; Higgins, P.D. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: Impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm. Bowel Dis. 2012, 18, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Vallière, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Pittet, V.; Rogler, G.; Michetti, P.; Fournier, N.; Vader, J.-P.; Schoepfer, A.; Mottet, C.; Burnand, B.; Froehlich, F.; Group, S.I.B.D.C.S. Penetrating or stricturing diseases are the major determinants of time to first and repeat resection surgery in Crohn’s disease. Digestion 2013, 87, 212–221. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-β signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, W.; Wang, Y.-L.; Chen, H.; Bringas Jr, P.; Datto, M.B.; Frederick, J.P.; Wang, X.-F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Pickard, K.M.; Rampton, D.; Kruidenier, L.; Rovedatti, L.; Leakey, N.A.; Corazza, G.R.; Monteleone, G.; MacDonald, T.T. Blockade of transforming growth factor β upregulates T-box transcription factor T-bet, and increases T helper cell type 1 cytokine and matrix metalloproteinase-3 production in the human gut mucosa. Gut 2008, 57, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Babyatsky, M.W.; Rossiter, G.; Podolsky, D.K. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 1996, 110, 975–984. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, B.; Jin, T.; Ocansey, D.K.W.; Jiang, J.; Mao, F. Intestinal Fibrosis in Inflammatory Bowel Disease and the Prospects of Mesenchymal Stem Cell Therapy. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.P.; Mulsow, J.J.; O’keane, C.; Docherty, N.G.; Watson, R.W.G.; O’connell, P.R. Fibrogenesis in Crohn’s disease. Off. J. Am. Coll. Gastroenterol. 2007, 102, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Jackson, C.; Pickard, K.; Buckley, M.; Rovedatti, L.; Leakey, N.; Picariello, L.; Cazzola, P.; Monteleone, G.; Tonelli, F. Transforming growth factor β signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut 2009, 58, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Stallmach, A.; Schuppan, D.; Riese, H.H.; Matthes, H.; Riecken, E.O. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology 1992, 102, 1920–1929. [Google Scholar] [CrossRef]
- Vallance, B.A.; Gunawan, M.I.; Hewlett, B.; Bercik, P.; Van Kampen, C.; Galeazzi, F.; Sime, P.J.; Gauldie, J.; Collins, S.M. TGF-β1 gene transfer to the mouse colon leads to intestinal fibrosis. Am. J. Physiol. -Gastrointest. Liver Physiol. 2005, 289, G116–G128. [Google Scholar] [CrossRef]
- Jiang, C.-H.; Sun, T.-L.; Xiang, D.-X.; Wei, S.-S.; Li, W.-Q. Anticancer activity and mechanism of xanthohumol: A prenylated flavonoid from hops (Humulus lupulus L.). Front. Pharmacol. 2018, 9, 530. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Mahli, A.; Weiskirchen, S.; Hellerbrand, C. The hop constituent xanthohumol exhibits hepatoprotective effects and inhibits the activation of hepatic stellate cells at different levels. Front. Physiol. 2015, 6, 140. [Google Scholar] [CrossRef]
- Dokduang, H.; Yongvanit, P.; Namwat, N.; Pairojkul, C.; Sangkhamanon, S.; Yageta, M.S.; Murakami, Y.; Loilome, W. Xanthohumol inhibits STAT3 activation pathway leading to growth suppression and apoptosis induction in human cholangiocarcinoma cells. Oncol. Rep. 2016, 35, 2065–2072. [Google Scholar] [CrossRef]
- Wang, W.; Chen, Z.; Zheng, T.; Zhang, M. Xanthohumol alleviates T2DM-induced liver steatosis and fibrosis by mediating the NRF2/RAGE/NF-κB signaling pathway. Future Med. Chem. 2021, 13, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Dorn, C.; Heilmann, J.; Hellerbrand, C. Protective effect of xanthohumol on toxin-induced liver inflammation and fibrosis. Int. J. Clin. Exp. Pathol. 2012, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Chen, Y.-G. Controlling TGF-β signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P. Structural studies of the TGF-βs and their receptors–insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Cárcamo, J.; Zentella, A.; Doody, J.; Laiho, M.; Wang, X.-F.; Massague, J. TGFβ signals through a heteromeric protein kinase receptor complex. Cell 1992, 71, 1003–1014. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Itoh, S.; Thorikay, M.; Kowanetz, M.; Moustakas, A.; Itoh, F.; Heldin, C.-H.; ten Dijke, P. Elucidation of Smad requirement in transforming growth factor-β type I receptor-induced responses. J. Biol. Chem. 2003, 278, 3751–3761. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Cat. No. | Company |
|---|---|---|
| α-SMA | ab7817 | Abcam |
| β-actin | sc-47778 | Santa Cruz Biotechnology |
| COL1A1 | SP1D8 | Development Studies Hybridoma Bank |
| COX-2 | RB-9072-P1 | Cayman chemical |
| FN | sc-8422 | Santa Cruz Biotechnology |
| HA | sc-7392 | Santa Cruz Biotechnology |
| Flag | F1804 | Sigma |
| IkBα | #9242 | Cell Signaling Technology |
| p-IkBα | #2859 | Cell Signaling Technology |
| p65 | #8242 | Cell Signaling Technology |
| p-Smad2 | #3108 | Cell Signaling Technology |
| p-Smad3 | #9520 | Cell Signaling Technology |
| Smad2/3 | #5678 | Cell Signaling Technology |
| streptavidin | SA-5004 | Vector Laboratories |
| Species | Gene | Primer Sequence | |
|---|---|---|---|
| Human (qRT-PCR) | 18S rRNA | Forward | GCAATTATTCCCCATGAACG |
| Reverse | GGCCTCACTAAACCATCCAA | ||
| Col1a1 | Forward | GATTCCCTGGACCTAAAGGTGC | |
| Reverse | AGCCTCTCCATCTTTGCCAGCA | ||
| FN | Forward | GAACTATGATGCCGACCAGAA | |
| Reverse | GGTTGTGCAGATTTCCTCGT | ||
| α-SMA | Forward | GCAAACAGGAATACGATGAAGCC | |
| Reverse | AACACATAGGTAACGAGTCAGAGC | ||
| MMP-2 | Forward | AGCGAGTGGATGCCGCCTTTAA | |
| Reverse | CATTCCAGGCATCTGCGATGAG | ||
| MMP-3 | Forward | CACTCACAGACCTGACTCGGTT | |
| Reverse | AAGCAGGATCACAGTTGGCTGG | ||
| MMP-12 | Forward | GATGCTGTCACTACCGTGGGAA | |
| Reverse | CAATGCCAGATGGCAAGGTTGG | ||
| CTGF | Forward | CTTGCGAAGCTGACCTGGAAGA | |
| Reverse | CCGTCGGTACATACTCCACAGA | ||
| IL-6 | Forward | AGGGCTCTTCGGCAAATGTA | |
| Reverse | GAAGGAATGCCCATTAACAACAA | ||
| IL-1β | Forward | TTAAAGCCCGCCTGACAGA | |
| Reverse | GCGAATGACAGAGGGTTTCTT | ||
| COX-2 | Forward | TGCATTCTTTGCCCAGCACT | |
| Reverse | AAAGGCGCAGTTTACGCTGT | ||
| PAI-1 | Forward | CTCATCAGCCACTGGAAAGGCA | |
| Reverse | GACTCGTGAAGTCAGCCTGAAAC | ||
| p21 | Forward | AGGTGGACCTGGAGACTCTCAG | |
| Reverse | TCCTCTTGGAGAAGATCAGCCG | ||
| Human (ChIP) | p65 | Foward | TTCTTCTTTGCATGCTACCG |
| Reverse | ATGGTTTGCACATTCCACAG | ||
| Smad3 | Foward | CAGTGGAATGCAGTGGAAGA | |
| Reverse | AGGGAAGCTGAAAGCTGAAG | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, S.-M.; Han, Y.-M.; Song, M.-Y.; Lee, D.-Y.; Kim, H.S.; Kim, S.-H.; Kim, E.-H. Xanthohumol Interferes with the Activation of TGF-β Signaling in the Process Leading to Intestinal Fibrosis. Nutrients 2023, 15, 99. https://doi.org/10.3390/nu15010099
Yun S-M, Han Y-M, Song M-Y, Lee D-Y, Kim HS, Kim S-H, Kim E-H. Xanthohumol Interferes with the Activation of TGF-β Signaling in the Process Leading to Intestinal Fibrosis. Nutrients. 2023; 15(1):99. https://doi.org/10.3390/nu15010099
Chicago/Turabian StyleYun, Sun-Mi, Young-Min Han, Moon-Young Song, Da-Young Lee, Hyun Su Kim, Seok-Ho Kim, and Eun-Hee Kim. 2023. "Xanthohumol Interferes with the Activation of TGF-β Signaling in the Process Leading to Intestinal Fibrosis" Nutrients 15, no. 1: 99. https://doi.org/10.3390/nu15010099
APA StyleYun, S.-M., Han, Y.-M., Song, M.-Y., Lee, D.-Y., Kim, H. S., Kim, S.-H., & Kim, E.-H. (2023). Xanthohumol Interferes with the Activation of TGF-β Signaling in the Process Leading to Intestinal Fibrosis. Nutrients, 15(1), 99. https://doi.org/10.3390/nu15010099