
Elimination of Vitamin D Signaling Causes Increased Mortality in a Model of Overactivation of the Insulin Receptor: Role of Lipid Metabolism

 ,
,  , , , ,
, , , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Animal Models
2.3. Glucose Metabolism Analysis
2.4. 3H Glucose Detection
2.5. Glucose Intraventricular Delivery in the CNS and Osmotic Implantation
2.6. Serum and Urine Biochemistry
2.7. Histopathology Analysis
2.8. Hepatic Glycogen Detection
2.9. RNA Isolation and Quantitative Reverse Transcription PCR (qRT-PCR)
2.10. Western Blot Analysis
2.11. Statistical Analyses
3. Results
3.1. Lack of VDR Reduces Lifespan in PTEN Knockout Mice
3.2. Physiological and Biochemical Parameters
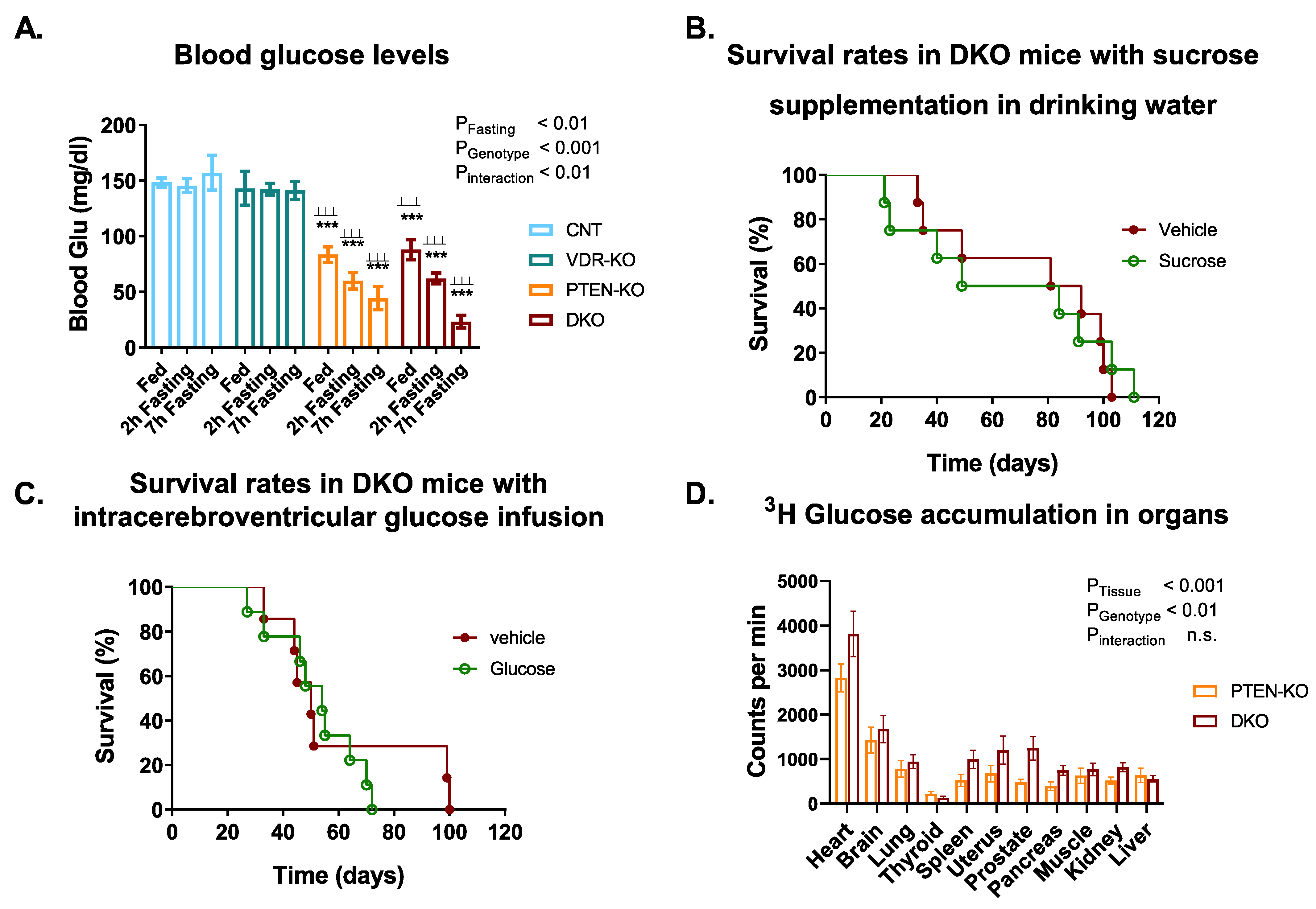
3.3. Blood Glucose Tests Reveal a Disruption of Glucose Metabolism in PTEN-KO and DKO Mice
3.4. Glucose Supplementation Did Not Increase Survival in DKO Mice
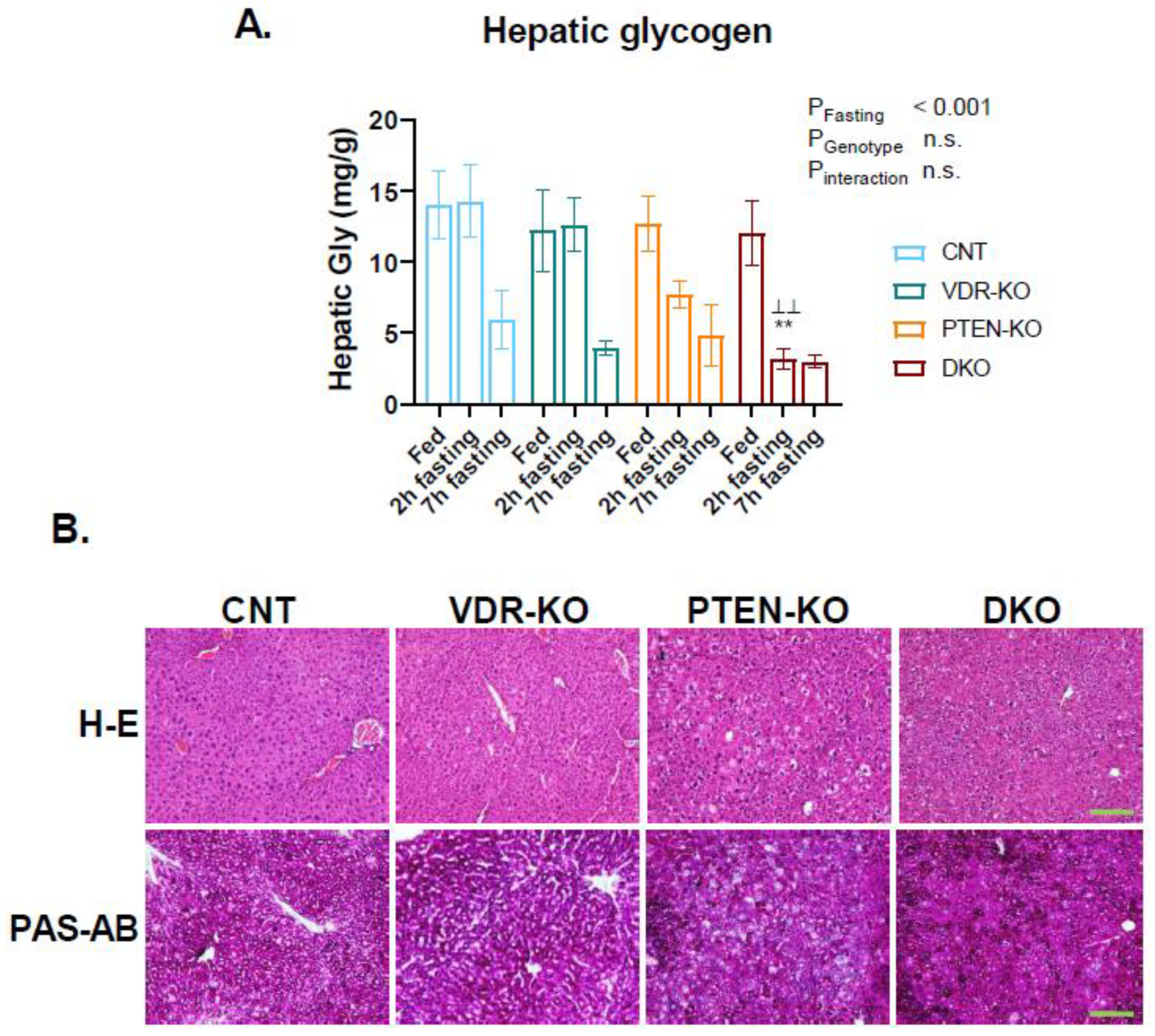
3.5. Faster Reduction in Glycogen Pool in Fasting DKO Mice
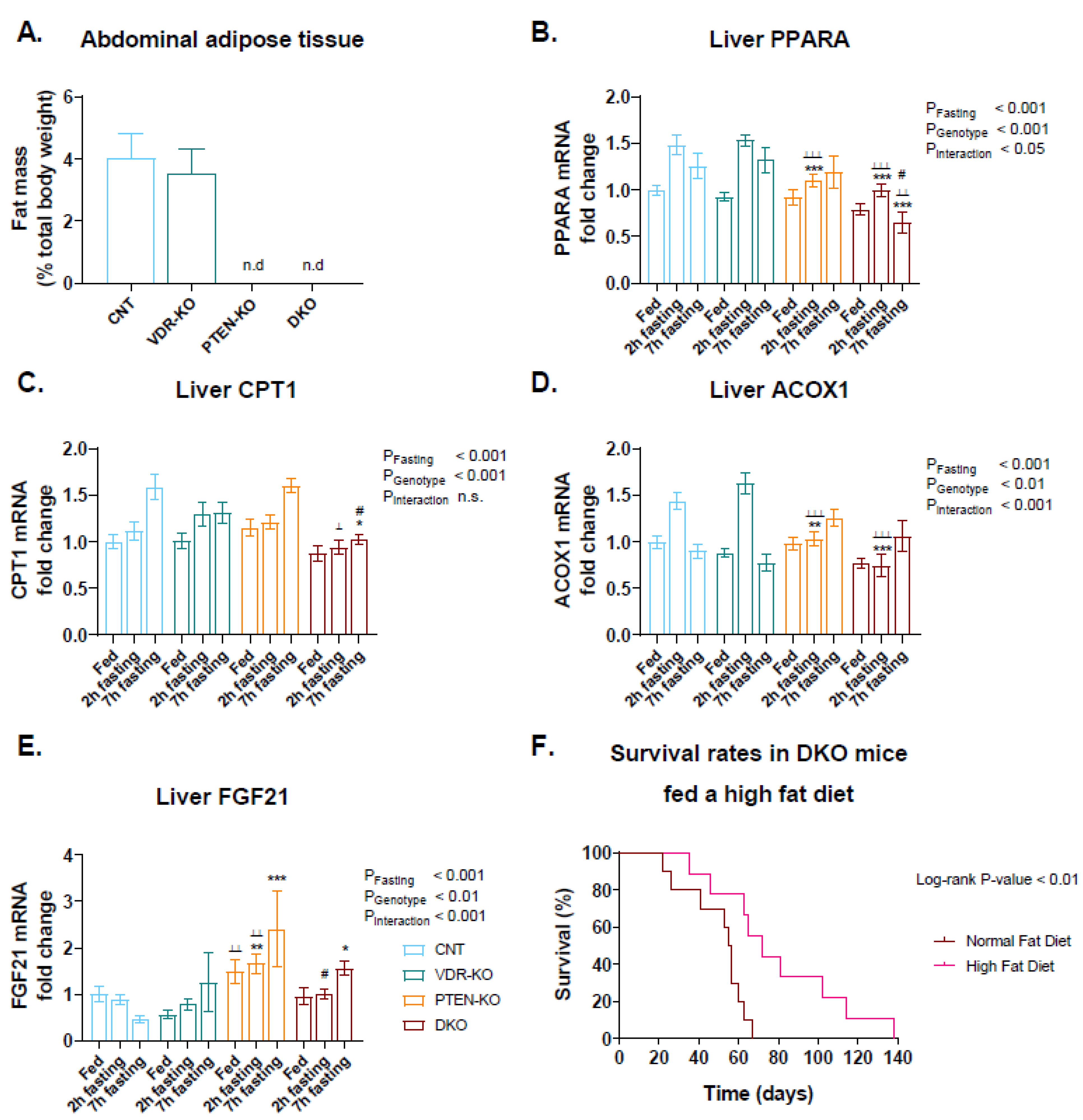
3.6. Delayed Gluconeogenesis in PTEN-KO and DKO Mice
3.7. High-Fat Diet Increased DKO Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C.; Diabetes Control and Complications Trial Research Group. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Effect of Intensive Diabetes Treatment on the Development and Progression of Long-Term Complications in Adolescents with Insulin-Dependent Diabetes Mellitus: Diabetes Control and Complications Trial. Diabetes Control and Complica-tions Trial Research Group. J. Pediatr. 1994, 125, 177–188. [CrossRef]
- Spiller, H.A.; Borys, D.J.; Ryan, M.L.; Sawyer, T.S.; Wilson, B.L. Unintentional Therapeutic Errors Involving Insulin in the Ambulatory Setting Reported to Poison Centers. Ann. Pharm. 2011, 45, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Cryer, P.E. The Barrier of Hypoglycemia in Diabetes. Diabetes 2008, 57, 3169–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M. Human Insulin Receptor and Its Relationship to the Tyrosine Kinase Family of Oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. The PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2015, 7, a026609. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The Functions and Regulation of the PTEN Tumour Suppressor: New Modes and Pro-spects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Pal, A.; Barber, T.M.; Van de Bunt, M.; Rudge, S.A.; Zhang, Q.; Lachlan, K.L.; Cooper, N.S.; Linden, H.; Levy, J.C.; Wakelam, M.J.O.; et al. PTEN Mutations as a Cause of Constitutive Insulin Sensitivity and Obesity. N. Engl. J. Med. 2012, 367, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Aranow, C. Vitamin D and the Immune System. J. Investig. Med. 2011, 59, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Bozic, M.; Álvarez, Á.; de Pablo, C.; Sanchez-Niño, M.-D.; Ortiz, A.; Dolcet, X.; Encinas, M.; Fernandez, E.; Valdivielso, J.M. Impaired Vitamin D Signaling in Endothelial Cell Leads to an Enhanced Leukocyte-Endothelium Interplay: Implications for Atherosclerosis Development. PLoS ONE 2015, 10, e0136863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozic, M.; Guzmán, C.; Benet, M.; Sánchez-Campos, S.; García-Monzón, C.; Gari, E.; Gatius, S.; Valdivielso, J.M.; Jover, R. Hepatocyte Vitamin D Receptor Regulates Lipid Metabolism and Mediates Experimental Diet-Induced Steatosis. J. Hepatol. 2016, 65, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Finklea, J.D.; Grossmann, R.E.; Tangpricha, V. Vitamin D and Chronic Lung Disease: A Review of Molecular Mechanisms and Clinical Studies. Adv. Nutr. 2011, 2, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Luk, J.; Torrealday, S.; Neal Perry, G.; Pal, L. Relevance of Vitamin D in Reproduction. Hum. Reprod. 2012, 27, 3015–3027. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Bolívar, V.; García-Fontana, B.; García-Fontana, C.; Muñoz-Torres, M. Mechanisms Involved in the Relationship between Vitamin D and Insulin Resistance: Impact on Clinical Practice. Nutrients 2021, 13, 3491. [Google Scholar] [CrossRef]
- Mathieu, C.; Badenhoop, K. Vitamin D and Type 1 Diabetes Mellitus: State of the Art. Trends Endocrinol. Metab. 2005, 16, 261–266. [Google Scholar] [CrossRef]
- Joergensen, C.; Hovind, P.; Schmedes, A.; Parving, H.-H.; Rossing, P. Vitamin D Levels, Microvascular Complications, and Mortality in Type 1 Diabetes. Diabetes Care 2011, 34, 1081–1085. [Google Scholar] [CrossRef] [Green Version]
- Mirantes, C.; Eritja, N.; Dosil, M.A.; Santacana, M.; Pallares, J.; Gatius, S.; Bergadà, L.; Maiques, O.; Matias-Guiu, X.; Dolcet, X. An Inducible Knockout Mouse to Model the Cell-Autonomous Role of PTEN in Initiating Endometrial, Prostate and Thyroid Neoplasias. Dis. Model. Mech. 2013, 6, 710–720. [Google Scholar] [CrossRef] [Green Version]
- Van Cromphaut, S.J.; Dewerchin, M.; Hoenderop, J.G.; Stockmans, I.; Van Herck, E.; Kato, S.; Bindels, R.J.; Collen, D.; Car-meliet, P.; Bouillon, R.; et al. Duodenal Calcium Absorption in Vitamin D Receptor-Knockout Mice: Functional and Mo-lecular Aspects. Proc. Natl. Acad. Sci. USA 2001, 98, 13324–13329. [Google Scholar] [CrossRef] [Green Version]
- Lesche, R.; Groszer, M.; Gao, J.; Wang, Y.; Messing, A.; Sun, H.; Liu, X.; Wu, H. Cre/LoxP-Mediated Inactivation of the Mu-rine Pten Tumor Suppressor Gene. Genesis 2002, 32, 148–149. [Google Scholar] [CrossRef]
- Hayashi, S.; McMahon, A.P. Efficient Recombination in Diverse Tissues by a Tamoxifen-Inducible Form of Cre: A Tool for Temporally Regulated Gene Activation/Inactivation in the Mouse. Dev. Biol. 2002, 244, 305–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boini, K.M.; Hennige, A.M.; Huang, D.Y.; Friedrich, B.; Palmada, M.; Boehmer, C.; Grahammer, F.; Artunc, F.; Ullrich, S.; Avram, D.; et al. Serum- and Glucocorticoid-Inducible Kinase 1 Mediates Salt Sensitivity of Glucose Tolerance. Diabetes 2006, 55, 2059–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVos, S.L.; Miller, T.M. Direct Intraventricular Delivery of Drugs to the Rodent Central Nervous System. J. Vis. Exp. 2013, e50326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozic, M.; de Rooij, J.; Parisi, E.; Ortega, M.R.; Fernandez, E.; Valdivielso, J.M. Glutamatergic Signaling Maintains the Epi-thelial Phenotype of Proximal Tubular Cells. J. Am. Soc. Nephrol. 2011, 22, 1099–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Bouillon, R.; Eelen, G.; Verlinden, L.; Mathieu, C.; Carmeliet, G.; Verstuyf, A. Vitamin D and Cancer. J. Steroid. Biochem. Mol. Biol. 2006, 102, 156–162. [Google Scholar] [CrossRef]
- Peyrou, M.; Bourgoin, L.; Poher, A.-L.; Altirriba, J.; Maeder, C.; Caillon, A.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F.; Foti, M. Hepatic PTEN Deficiency Improves Muscle Insulin Sensitivity and Decreases Adiposity in Mice. J. Hepatol. 2015, 62, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.J.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic Elevation of PTEN Induces a Tumor-Suppressive Metabolic State. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.; Erben, R.G. Differences in Triglyceride and Cholesterol Metabolism and Resistance to Obesity in Male and Female Vitamin D Receptor Knockout Mice. J. Anim. Physiol. Anim. Nutr. 2013, 97, 675–683. [Google Scholar] [CrossRef]
- Narvaez, C.J.; Matthews, D.; Broun, E.; Chan, M.; Welsh, J. Lean Phenotype and Resistance to Diet-Induced Obesity in Vit-amin D Receptor Knockout Mice Correlates with Induction of Uncoupling Protein-1 in White Adipose Tissue. Endocri.-Nology 2009, 150, 651–661. [Google Scholar] [CrossRef]
- Crespo-Masip, M.; Pérez-Gómez, A.; Guzmán, C.; Rayego, S.; Doladé, N.; García-Carrasco, A.; Jover, R.; Valdivielso, J.M. PTEN Deletion in Adult Mice Induces Hypoinsulinemia with Concomitant Low Glucose Levels. Front. Endocrinol. 2022, 13, 850214. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W.; Frankel, J.B.; Heldt, A.M.; Grodsky, G.M. Vitamin D Deficiency Inhibits Pancreatic Secretion of Insulin. Science 1980, 209, 823–825. [Google Scholar] [CrossRef] [PubMed]
- Chonchol, M.; Scragg, R. 25-Hydroxyvitamin D, Insulin Resistance, and Kidney Function in the Third National Health and Nutrition Examination Survey. Kidney Int. 2007, 71, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitz, U.; Weber, K.; Soegiarto, D.W.; Wolf, E.; Balling, R.; Erben, R.G. Impaired Insulin Secretory Capacity in Mice Lacking a Functional Vitamin D Receptor. FASEB J. 2003, 17, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Villalta, S.A.; Agrawal, D.K. FOXO1 Mediates Vitamin D Deficiency-Induced Insulin Resistance in Skeletal Mus-cle. J. Bone. Min. Res. 2016, 31, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Ougorets, I.; Caronna, J.J. Chapter 62 Coma. In Critical Care Medicine, 3rd ed.; Parrillo, J.E., Dellinger, R.P., Eds.; Mosby: Philadelphia, PA, USA, 2008; ISBN 978-0-323-04841-5. [Google Scholar]
- Reno, C.M.; Skinner, A.; Bayles, J.; Chen, Y.S.; Daphna-Iken, D.; Fisher, S.J. Severe Hypoglycemia-Induced Sudden Death Is Mediated by Both Cardiac Arrhythmias and Seizures. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E240–E249. [Google Scholar] [CrossRef] [Green Version]
- Shan, R.; Ning, Y.; Ma, Y.; Liu, S.; Wu, J.; Fan, X.; Lv, J.; Wang, B.; Li, S.; Li, L. Prevalence and Risk Factors of Atrioventricu-lar Block among 15 Million Chinese Health Examination Participants in 2018: A Nation-Wide Cross-Sectional Study. BMC Cardiovasc. Disord. 2021, 21, 289. [Google Scholar] [CrossRef]
- Lee, H.-J.; Lee, S.-R.; Choi, E.-K.; Han, K.-D.; Oh, S. Low Lipid Levels and High Variability Are Associated With the Risk of New-Onset Atrial Fibrillation. J. Am. Heart. Assoc. 2019, 8, e012771. [Google Scholar] [CrossRef]
- Iguchi, Y.; Kimura, K.; Aoki, J.; Kobayashi, K.; Terasawa, Y.; Sakai, K.; Shibazaki, K. Prevalence of Atrial Fibrillation in Community-Dwelling Japanese Aged 40 Years or Older in Japan: Analysis of 41,436 Non-Employee Residents in Kurashi-ki-City. Circ. J. 2008, 72, 909–913. [Google Scholar] [CrossRef] [Green Version]
- Lopez, F.L.; Agarwal, S.K.; Maclehose, R.F.; Soliman, E.Z.; Sharrett, A.R.; Huxley, R.R.; Konety, S.; Ballantyne, C.M.; Alonso, A. Blood Lipid Levels, Lipid-Lowering Medications, and the Incidence of Atrial Fibrillation: The Atherosclerosis Risk in Communities Study. Circ. Arrhythm. Electrophysiol. 2012, 5, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gao, L.; Wang, Z.; Guan, B.; Guan, X.; Wang, B.; Han, X.; Xiao, X.; Waleed, K.B.; Chandran, C.; et al. Lipid Profile and Incidence of Atrial Fibrillation: A Prospective Cohort Study in China. Clin. Cardiol. 2018, 41, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourtzinis, G.; Kahan, T.; Bengtsson Boström, K.; Schiöler, L.; Cedstrand Wallin, L.; Hjerpe, P.; Hasselström, J.; Manhem, K. Relation Between Lipid Profile and New-Onset Atrial Fibrillation in Patients With Systemic Hypertension (From the Swedish Primary Care Cardiovascular Database [SPCCD]). Am. J. Cardiol. 2018, 122, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Tanabe, N.; Yagihara, N.; Watanabe, T.; Aizawa, Y.; Kodama, M. Association between Lipid Profile and Risk of Atrial Fibrillation. Circ. J. 2011, 75, 2767–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Yin, X.; Roetker, N.S.; Magnani, J.W.; Kronmal, R.A.; Ellinor, P.T.; Chen, L.Y.; Lubitz, S.A.; McClelland, R.L.; McManus, D.D.; et al. Blood Lipids and the Incidence of Atrial Fibrillation: The Multi-Ethnic Study of Atherosclerosis and the Framingham Heart Study. J. Am. Heart Assoc. 2014, 3, e001211. [Google Scholar] [CrossRef] [Green Version]
- Annoura, M.; Ogawa, M.; Kumagai, K.; Zhang, B.; Saku, K.; Arakawa, K. Cholesterol Paradox in Patients with Paroxysmal Atrial Fibrillation. Cardiology 1999, 92, 21–27. [Google Scholar] [CrossRef]
- Neely, J.R.; Rovetto, M.J.; Oram, J.F. Myocardial Utilization of Carbohydrate and Lipids. Prog. Cardiovasc. Dis. 1972, 15, 289–329. [Google Scholar] [CrossRef]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.-J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-Specific Deletion of Negative Regulator Pten Results in Fatty Liver and Insulin Hypersensitivity. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular Mechanism of PPARα Action and Its Impact on Lipid Metabolism, Inflamma-tion and Fibrosis in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARalpha and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [Green Version]
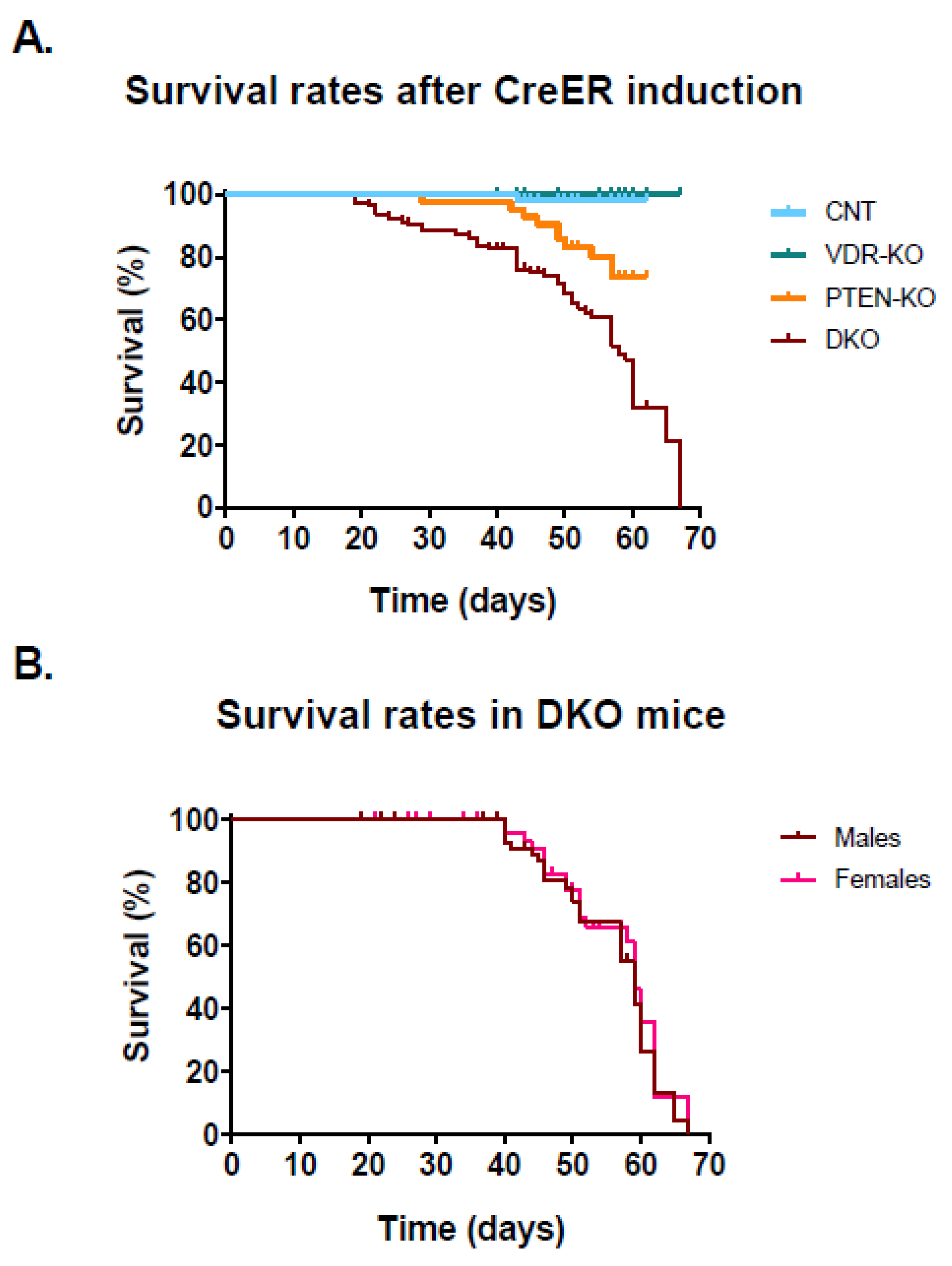




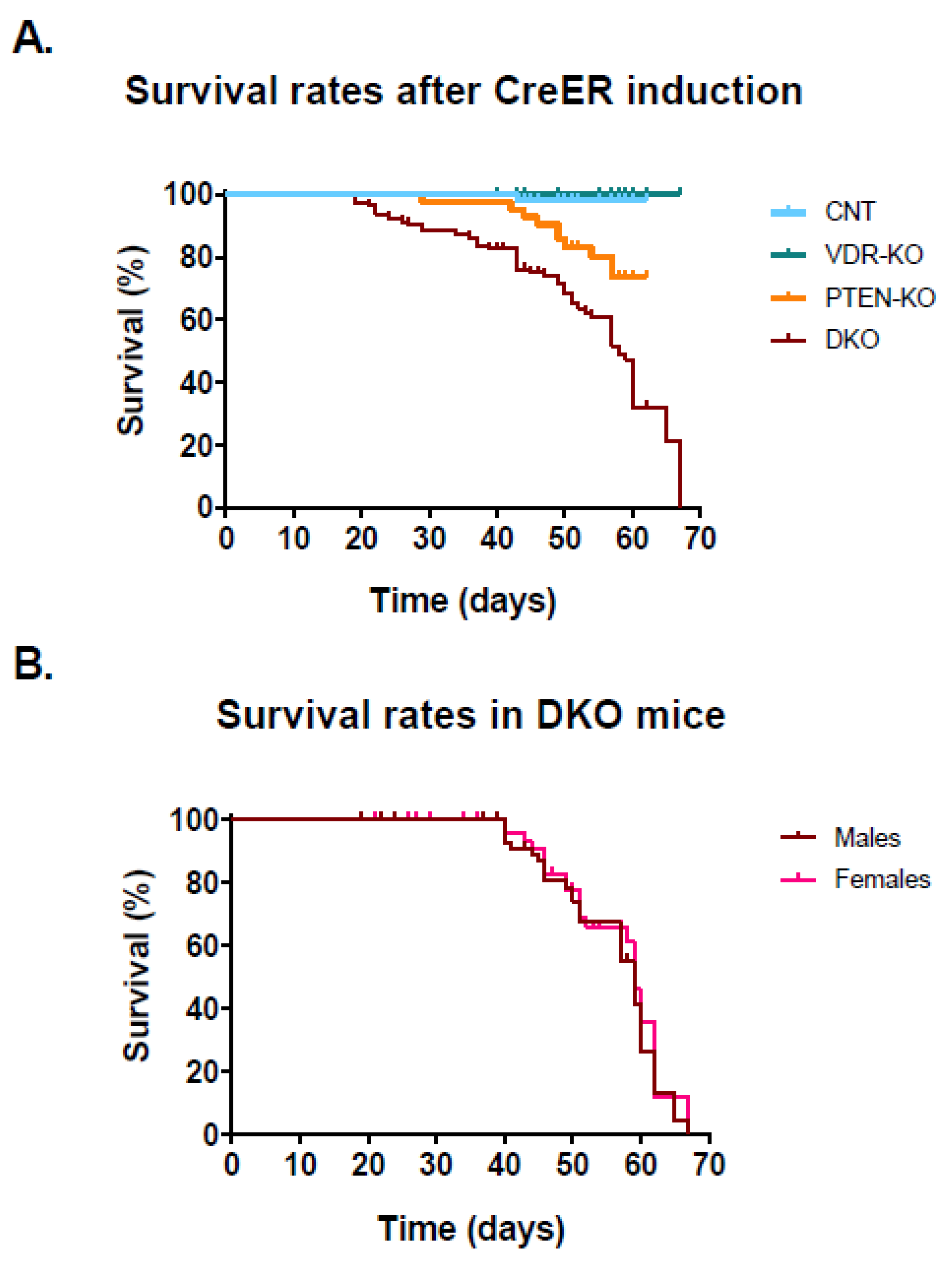{kind=link}
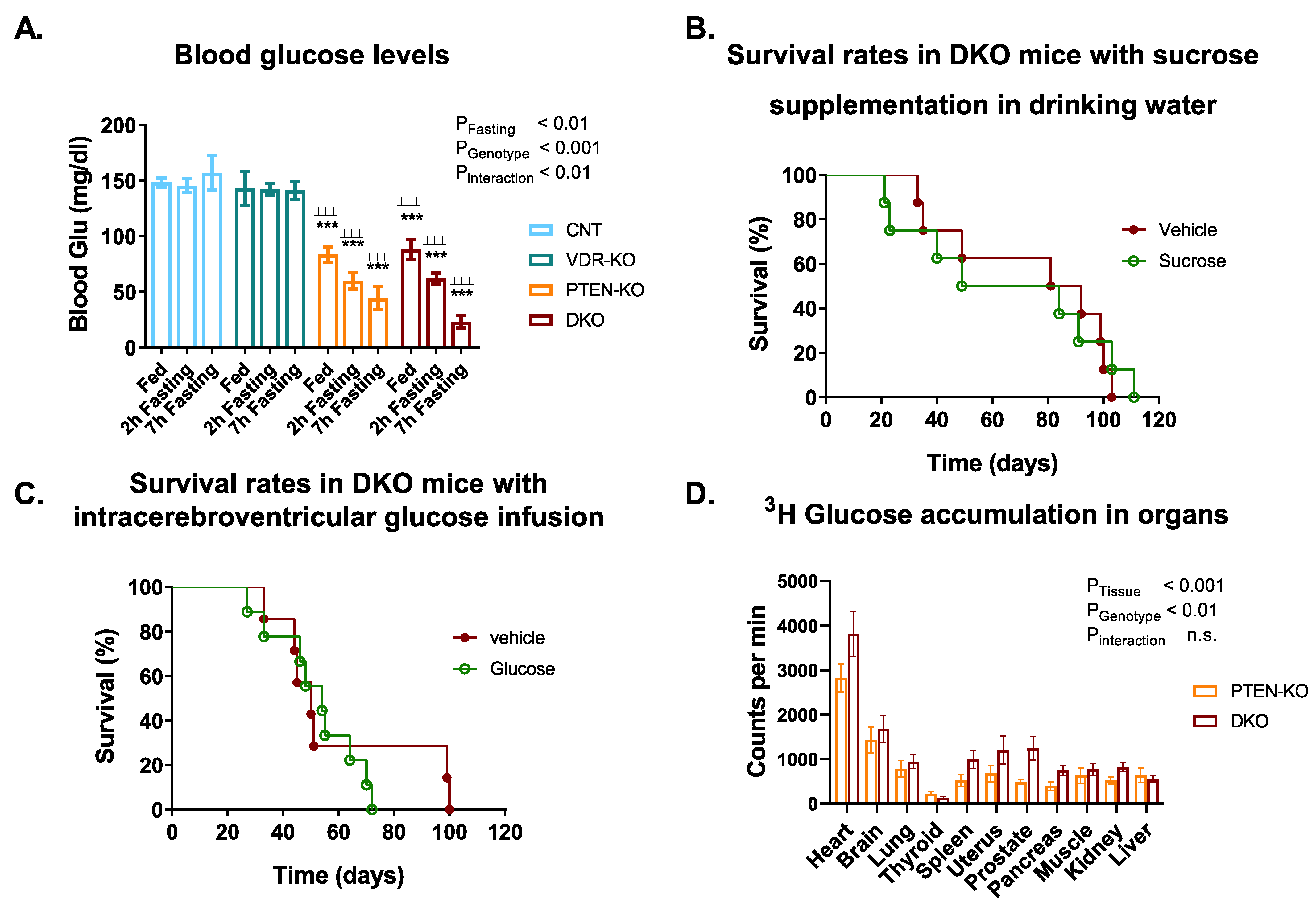{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CNT (n = 11) | VDR-KO (n = 9) | PTEN-KO (n = 8) | DKO (n = 13) | |
|---|---|---|---|---|
| A. Physiological parameters | ||||
| Food intake (g/24 h) | 3.03 ± 0.28 | 3.87 ± 0.29 | 5.02 ± 0.35 ** | 5.26 ± 0.31 *** |
| Total body weight (g) | 27.0 ± 0.81 | 25.0 ± 1.01 | 22.7 ± 0.50 *** | 22.6 ± 0.58 *** |
| B. Circulating parameters | ||||
| BUN (mg/24 h) | 20.6 ± 2.74 | 19.4 ± 1.83 | 20.4 ± 1.45 | 20.6 ± 2.41 |
| Peptide-C (pM) | 175.7 ± 25.3 | 140.9 ± 16.1 | 64.0 ± 3.61 *** ⊥⊥ | 70.0 ± 6.50 *** ⊥⊥ |
| Blood glucose (mg/dL) | 148.4 ± 4.11 | 143.1 ± 15.2 | 83.5 ± 7.12 *** ⊥⊥⊥ | 87.9 ± 9.03 *** ⊥⊥⊥ |
| 25(OH)D3 (ng/mL) | 74.7 ± 10.2 | 65.04 ± 18.9 | 17.8 ± 3.15 *** ⊥⊥ | 26.1 ± 7.05 ** |
| 1,25(OH)2D3 (pmol/L) | 124.7 ± 39.7 | 376.3 ± 67.5 *** | 70.10 ± 40.4 ⊥⊥⊥ | 188.0 ± 41.5 ## |
| Total cholesterol (mg/dL) | 121.3 ± 6.05 | 142.0 ± 13.0 | 110.1 ± 6.44 ⊥ | 101.3 ± 3.85 ⊥⊥ |
| LDL cholesterol (mg/dL) | 16.9 ± 2.43 | 16.7 ± 3.77 | 16.0 ± 4.45 | 9.23 ± 2.50 |
| HDL cholesterol (mg/dL) | 92.0 ± 4.86 | 111.4 ± 9.70 | 83.71 ± 5.55 ⊥ | 79.4 ± 3.52 ⊥⊥ |
| Triglycerides (mg/dL) | 88.3 ± 12.7 | 69.4 ± 5.63 | 93.3 ± 9.11 | 64.7 ± 8.41 |
| CNT (n = 11) | VDR-KO (n = 9) | PTEN-KO (n = 8) | DKO (n = 13) | |
|---|---|---|---|---|
| A. Glucose tolerance test | ||||
| Time (min) | ||||
| 0 | 145.4 ± 6.3 | 134.0 ± 5.3 | 61.5 ± 6.7 *** ⊥ ⊥ ⊥ | 68.0 ± 4.7 *** ⊥ ⊥ ⊥ |
| 20 | 190.1 ± 7.3 | 202.2 ± 8.8 | 96.7 ± 17.0 *** ⊥ ⊥ ⊥ | 72,7 ± 6.9 *** ⊥ ⊥ ⊥ |
| 40 | 180.7 ± 7.7 | 197.2 ± 11.8 | 68.2 ± 9.0 *** ⊥ ⊥ ⊥ | 64.8 ± 5.8 *** ⊥ ⊥ ⊥ |
| 60 | 175.0 ± 9.0 | 195.9 ± 11.9 | 58.8 ± 8.6 *** ⊥ ⊥ ⊥ | 65.3 ± 6.7 *** ⊥ ⊥ ⊥ |
| 120 | 148.3 ± 7.5 | 155.5 ± 16.9 | 54.8 ± 7.0 *** ⊥ ⊥ ⊥ | 51.2 ± 6.2 *** ⊥ ⊥ ⊥ |
| B. Pyruvate tolerance test | ||||
| Time (min) | ||||
| 0 | 157.1 ± 7.3 | 149.1 ± 7.2 | 62.1 ± 10.5 *** ⊥ ⊥ ⊥ | 68.4 ± 9.2 *** ⊥ ⊥ ⊥ |
| 20 | 212.3 ± 8.9 | 196.2 ± 14.6 | 74.3 ± 11.2 *** ⊥ ⊥ ⊥ | 78.6 ± 9.9 *** ⊥ ⊥ ⊥ |
| 40 | 204.6 ± 17.2 | 175.0 ± 20.6 | 62.9 ± 11.7 *** ⊥ ⊥ ⊥ | 54.1 ± 6.6 *** ⊥ ⊥ ⊥ |
| 60 | 204.1 ± 18.4 | 180.8 ± 20.0 | 61.5 ± 14.0 *** ⊥ ⊥ ⊥ | 49.8 ± 6.7 *** ⊥ ⊥ ⊥ |
| 120 | 145.3 ± 10.7 | 148.9 ± 19.4 | 59.8 ± 7.9 *** ⊥ ⊥ ⊥ | 60.3 ± 12.9 *** ⊥ ⊥ ⊥ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crespo-Masip, M.; Perez-Gomez, A.; Garcia-Carrasco, A.; Jover, R.; Guzmán, C.; Dolcet, X.; Ibarz, M.; Martínez, C.; Eritja, À.; Diaz-Tocados, J.M.; et al. Elimination of Vitamin D Signaling Causes Increased Mortality in a Model of Overactivation of the Insulin Receptor: Role of Lipid Metabolism. Nutrients 2022, 14, 1516. https://doi.org/10.3390/nu14071516
Crespo-Masip M, Perez-Gomez A, Garcia-Carrasco A, Jover R, Guzmán C, Dolcet X, Ibarz M, Martínez C, Eritja À, Diaz-Tocados JM, et al. Elimination of Vitamin D Signaling Causes Increased Mortality in a Model of Overactivation of the Insulin Receptor: Role of Lipid Metabolism. Nutrients. 2022; 14(7):1516. https://doi.org/10.3390/nu14071516
Chicago/Turabian StyleCrespo-Masip, Maria, Aurora Perez-Gomez, Alicia Garcia-Carrasco, Ramiro Jover, Carla Guzmán, Xavier Dolcet, Mercé Ibarz, Cristina Martínez, Àuria Eritja, Juan Miguel Diaz-Tocados, and et al. 2022. "Elimination of Vitamin D Signaling Causes Increased Mortality in a Model of Overactivation of the Insulin Receptor: Role of Lipid Metabolism" Nutrients 14, no. 7: 1516. https://doi.org/10.3390/nu14071516
APA StyleCrespo-Masip, M., Perez-Gomez, A., Garcia-Carrasco, A., Jover, R., Guzmán, C., Dolcet, X., Ibarz, M., Martínez, C., Eritja, À., Diaz-Tocados, J. M., & Valdivielso, J. M. (2022). Elimination of Vitamin D Signaling Causes Increased Mortality in a Model of Overactivation of the Insulin Receptor: Role of Lipid Metabolism. Nutrients, 14(7), 1516. https://doi.org/10.3390/nu14071516