
The Association between Accumulation of Toxic Advanced Glycation End-Products and Cytotoxic Effect in MC3T3-E1 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Cell Viability
2.4. Slot Blot Analysis
2.5. Western Blot Analysis
2.6. Real-Time Quantitative PCR (qPCR)
2.7. Immunofluorescence Staining
2.8. Statistical Analysis
3. Results
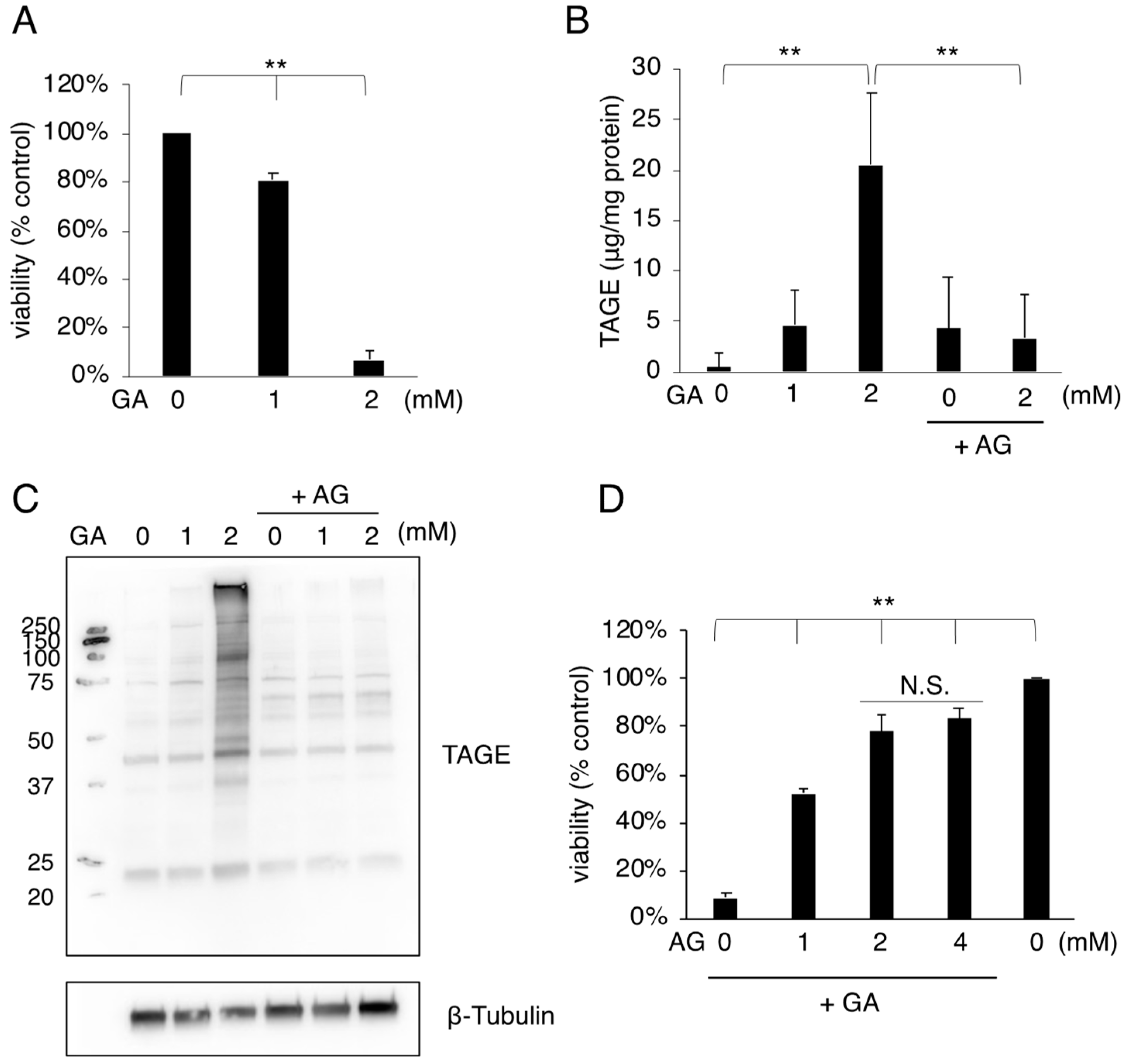
3.1. MC3T3-E1 Cell Death Associated with the Accumulation of TAGE
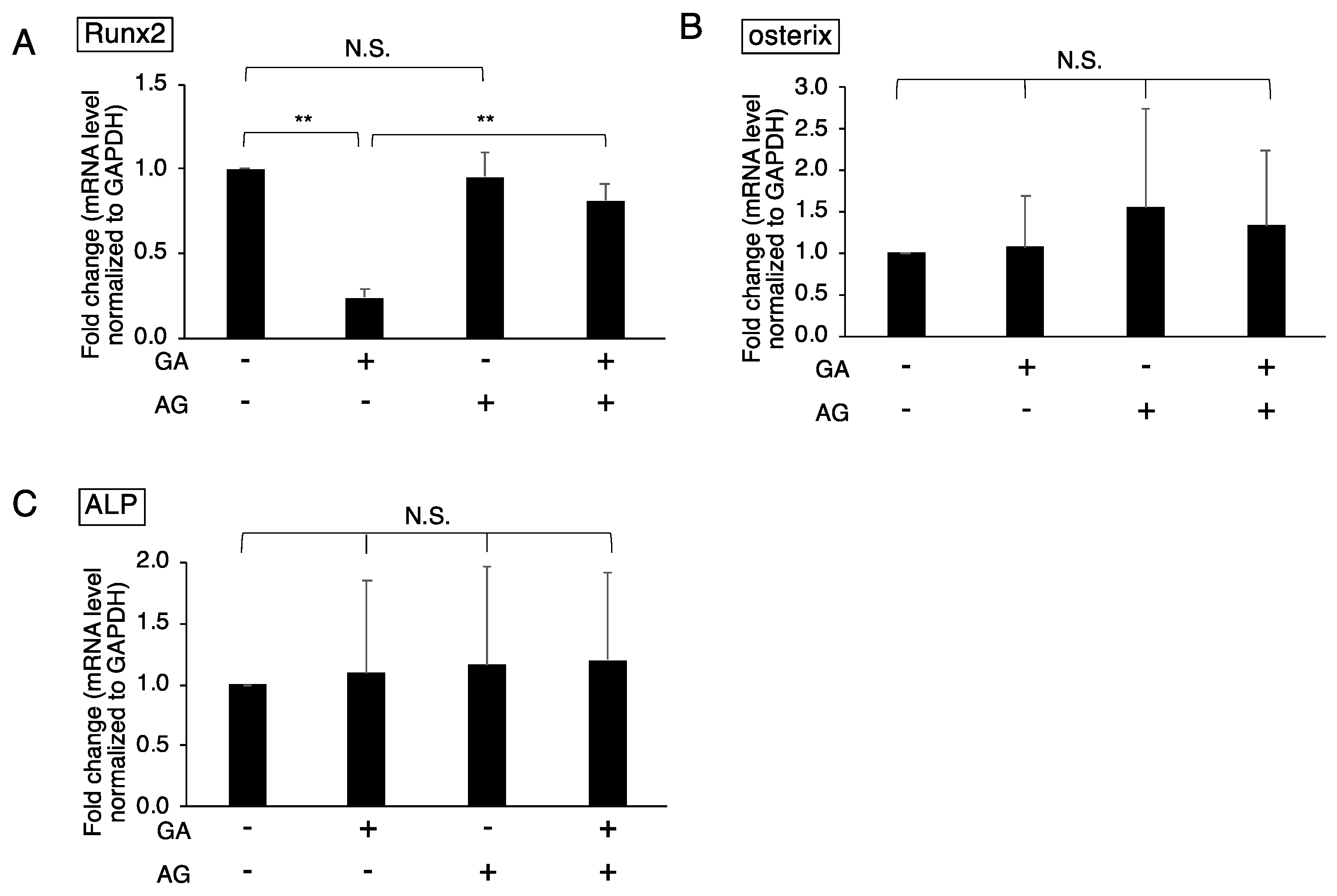
3.2. Downregulated Expression of Runx2 in GA-Treated MC3T3-E1 Cells
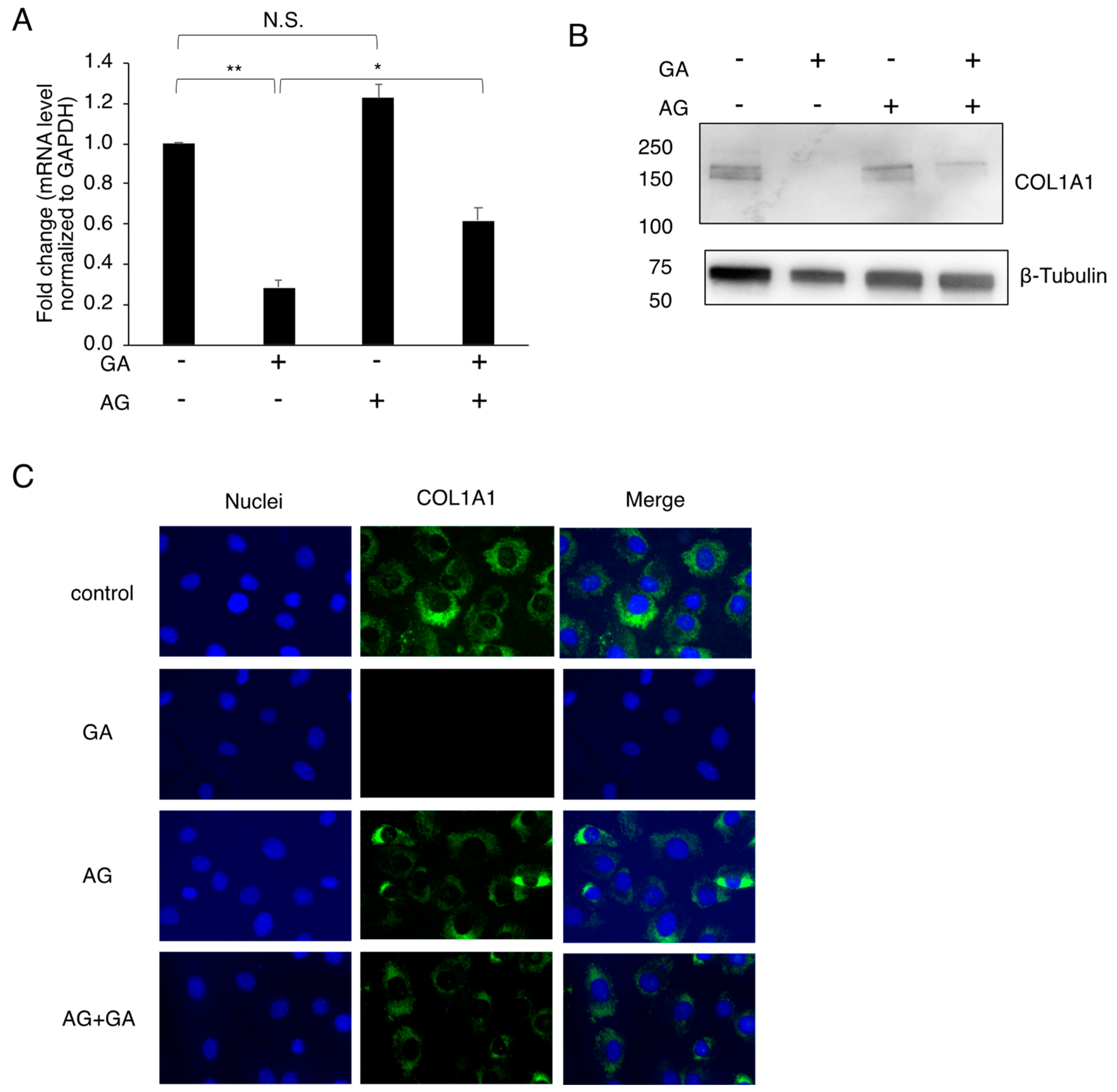
3.3. Effects of the GA Treatment on COL1A1 in MC3T3-E1 Cells
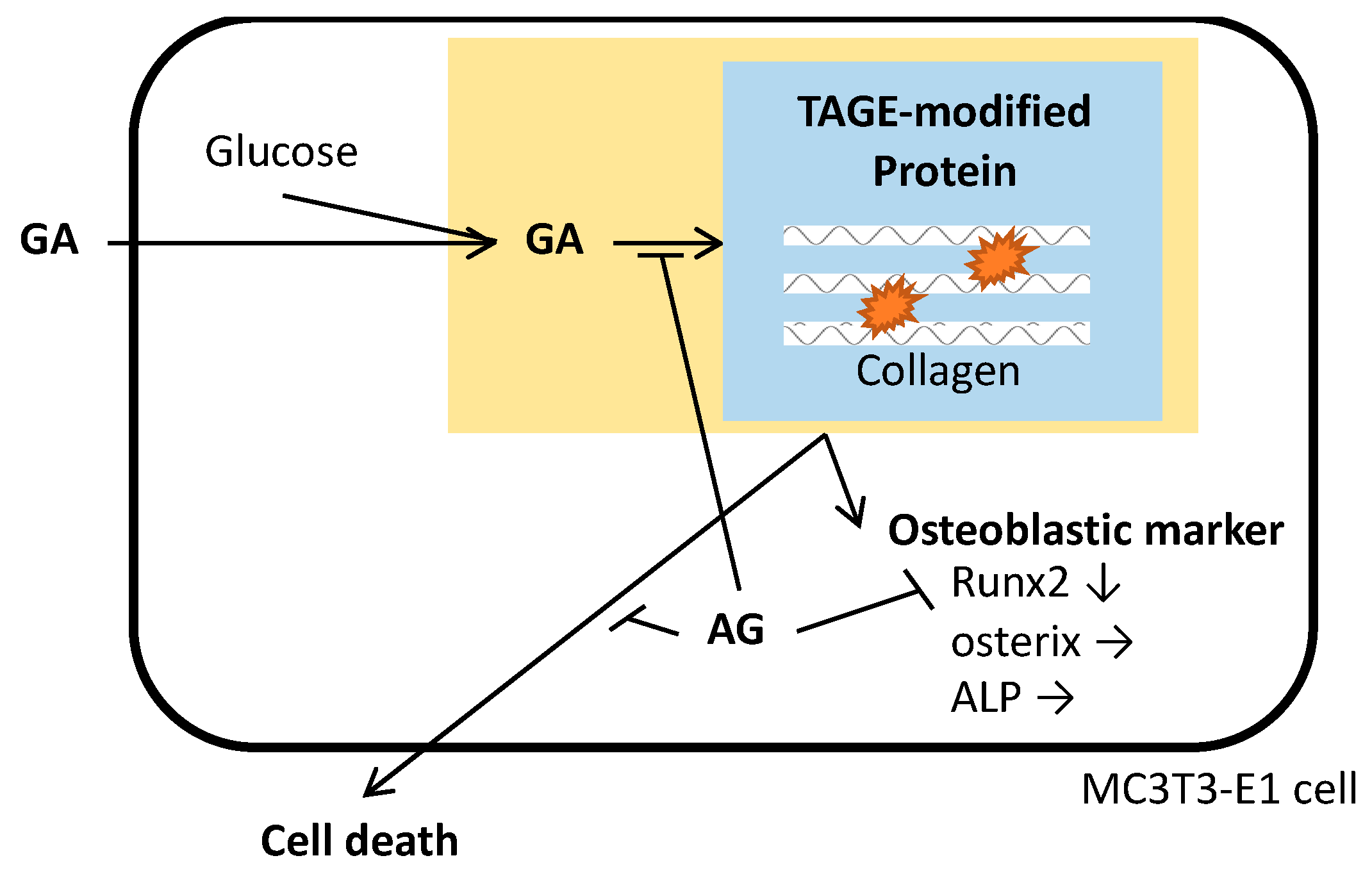
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta-analysis. Osteoporos. Int. 2007, 18, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Romero-Díaz, C.; Duarte-Montero, D.; Gutiérrez-Romero, S.A.; Mendivil, C.O. Diabetes and bone fragility. Diabetes Ther. 2021, 12, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Johnell, O.; Kanis, J.A.; Odén, A.; Sernbo, I.; Redlund-Johnell, I.; Petterson, C.; De Laet, C.; Jönsson, B. Mortality after osteoporotic fractures. Osteoporos. Int. 2004, 15, 38–42. [Google Scholar] [CrossRef]
- Asadipooya, K.; Uy, E.M. Advanced glycation end products (AGEs), receptor for AGEs, diabetes, and bone: Review of the literature. J. Endocr. Soc. 2019, 3, 1799–1818. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Fujii, K.; Mori, Y.; Marumo, K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos. Int. 2006, 17, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Farlay, D.; Armas, L.A.; Gineyts, E.; Akhter, M.P.; Recker, R.R.; Boivin, G. Nonenzymatic glycation and degree of mineralization are higher in bone from fractured patients with type 1 diabetes mellitus. J. Bone Miner. Res. 2016, 31, 190–195. [Google Scholar] [CrossRef]
- Mitome, J.; Yamamoto, H.; Saito, M.; Yokoyama, K.; Marumo, K.; Hosoya, T. Nonenzymatic cross-linking pentosidine increase in bone collagen and are associated with disorders of bone mineralization in dialysis patients. Calcif. Tissue Int. 2011, 88, 521–529. [Google Scholar] [CrossRef]
- Sanguineti, R.; Puddu, A.; Mach, F.; Montecucco, F.; Viviani, G.L. Advanced glycation end products play adverse proinflammatory activities in osteoporosis. Mediat. Inflamm. 2014, 2014, 975872. [Google Scholar] [CrossRef]
- Alikhani, M.; Alikhani, Z.; Boyd, C.; MacLellan, C.M.; Raptis, M.; Liu, R.; Pischon, N.; Trackman, P.C.; Gerstenfeld, L.; Graves, D.T. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone 2007, 40, 345–353. [Google Scholar] [CrossRef]
- Okazaki, K.; Yamaguchi, T.; Tanaka, K.; Notsu, M.; Ogawa, N.; Yano, S.; Sugimoto, T. Advanced glycation end products (AGEs), but not high glucose, inhibit the osteoblastic differentiation of mouse stromal ST2 cells through the suppression of osterix expression, and inhibit cell growth and increasing cell apoptosis. Calcif. Tissue Int. 2012, 91, 286–296. [Google Scholar] [CrossRef]
- Notsu, M.; Yamaguchi, T.; Okazaki, K.; Tanaka, K.; Ogawa, N.; Kanazawa, I.; Sugimoto, T. Advanced glycation end product 3 (AGE3) suppresses the mineralization of mouse stromal ST2 cells and human mesenchymal stem cells by increasing TGF-β expression and secretion. Endocrinology 2014, 155, 2402–2410. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.; van Hinsbergh, V.W. Fructose-mediated non-enzymatic glycation: Sweet coupling or bad modification. Diabetes Metab. Res. Rev. 2004, 20, 369–382. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T. Molecular mechanisms of aldehyde toxicity: A chemical perspective. Chem. Res. Toxicol. 2014, 27, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, H.; Dong, K.; Liu, S.; Zhou, W.; Zhang, J.; Meng, L.; Rausch-Fan, X.; Xu, X. Different concentrations of glucose regulate proliferation and osteogenic differentiation of osteoblasts via the PI3 kinase/Akt pathway. Implant Dent. 2015, 24, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J. Uncarboxylated osteocalcin inhibits high glucose-induced ROS production and stimulates osteoblastic differentiation by preventing the activation of PI3K/Akt in MC3T3-E1 cells. Int. J. Mol. Med. 2016, 37, 173–181. [Google Scholar] [CrossRef]
- Yamabe, S.; Hirose, J.; Uehara, Y.; Okada, T.; Okamoto, N.; Oka, K.; Taniwaki, T.; Mizuta, H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 2013, 280, 1617–1629. [Google Scholar] [CrossRef]
- Suzuki, R.; Fujiwara, Y.; Saito, M.; Arakawa, S.; Shirakawa, J.; Yamanaka, M.; Komohara, Y.; Marumo, K.; Nagai, R. Intracellular accumulation of advanced glycation end products induces osteoblast apoptosis via endoplasmic reticulum stress. J. Bone Miner. Res. 2020, 35, 1992–2003. [Google Scholar] [CrossRef]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol. Med. 2000, 6, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Hyase, F. Isolation and identification of the 3-hydroxy-5-hydroxymethyl-pyridinium compound as a novel advanced glycation end product on glyceraldehyde-related Maillard reaction. Biosci. Biotechnol. Biochem. 2003, 67, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Tessier, F.J.; Monnier, V.M.; Sayre, L.M.; Kornfield, J.A. Triosidines: Novel Maillard reaction products and cross-links from the reaction of triose sugars with lysine and arginine residues. Biochem. J. 2003, 369, 705–719. [Google Scholar] [CrossRef]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef]
- Takeuchi, M. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef]
- Takeuchi, M. Toxic AGEs (TAGE) theory: A new concept for preventing the development of diseases related to lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular toxic AGEs (TAGE) triggers numerous types of cell damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Yamagishi, S.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 1112–1119. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. The relevance of toxic AGEs (TAGE) cytotoxicity to NASH pathogenesis: A mini-review. Nutrients 2019, 11, 462. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products promote the production of reactive oxygen species in HepG2 cells. Int. J. Mol. Sci. 2020, 21, 4861. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy metabolism of the osteoblast: Implications for osteoporosis. Endocr. Rev. 2017, 38, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kazachkov, M.; Chen, K.; Babiy, S.; Yu, P.H. Evidence for in vivo scavenging by aminoguanidine of formaldehyde produced via semicarbazide-sensitive amine oxidase-mediated deamination. J. Pharmacol. Exp. Ther. 2007, 322, 1201–1207. [Google Scholar] [CrossRef]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced glycation end products (AGEs): Biochemistry, signaling, analytical methods, and epigenetic effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef]
- Hie, M.; Tsukamoto, I. Increased expression of the receptor for activation of NF-kappaB and decreased runt-related transcription factor 2 expression in bone of rats with streptozotocin-induced diabetes. Int. J. Mol. Med. 2010, 26, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, M.; Wang, S.; Xiao, Z.; Xiong, Y.; Wang, G. Recent advances of osterix transcription factor in osteoblast differentiation and bone formation. Front. Cell Dev. Biol. 2020, 8, 601224. [Google Scholar] [CrossRef] [PubMed]
- Avery, N.C.; Bailey, A.J. The effects of the Maillard reaction on the physical properties and cell interactions of collagen. Pathol. Biol. 2006, 54, 387–395. [Google Scholar] [CrossRef]
- Kumar, P.A.; Chitra, P.S.; Reddy, G.B. Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomol. Concepts 2016, 7, 293–309. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. The Association between Accumulation of Toxic Advanced Glycation End-Products and Cytotoxic Effect in MC3T3-E1 Cells. Nutrients 2022, 14, 990. https://doi.org/10.3390/nu14050990
Sakasai-Sakai A, Takata T, Takeuchi M. The Association between Accumulation of Toxic Advanced Glycation End-Products and Cytotoxic Effect in MC3T3-E1 Cells. Nutrients. 2022; 14(5):990. https://doi.org/10.3390/nu14050990
Chicago/Turabian StyleSakasai-Sakai, Akiko, Takanobu Takata, and Masayoshi Takeuchi. 2022. "The Association between Accumulation of Toxic Advanced Glycation End-Products and Cytotoxic Effect in MC3T3-E1 Cells" Nutrients 14, no. 5: 990. https://doi.org/10.3390/nu14050990
APA StyleSakasai-Sakai, A., Takata, T., & Takeuchi, M. (2022). The Association between Accumulation of Toxic Advanced Glycation End-Products and Cytotoxic Effect in MC3T3-E1 Cells. Nutrients, 14(5), 990. https://doi.org/10.3390/nu14050990