
GlyNAC (Glycine and N-Acetylcysteine) Supplementation in Mice Increases Length of Life by Correcting Glutathione Deficiency, Oxidative Stress, Mitochondrial Dysfunction, Abnormalities in Mitophagy and Nutrient Sensing, and Genomic Damage


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Studies
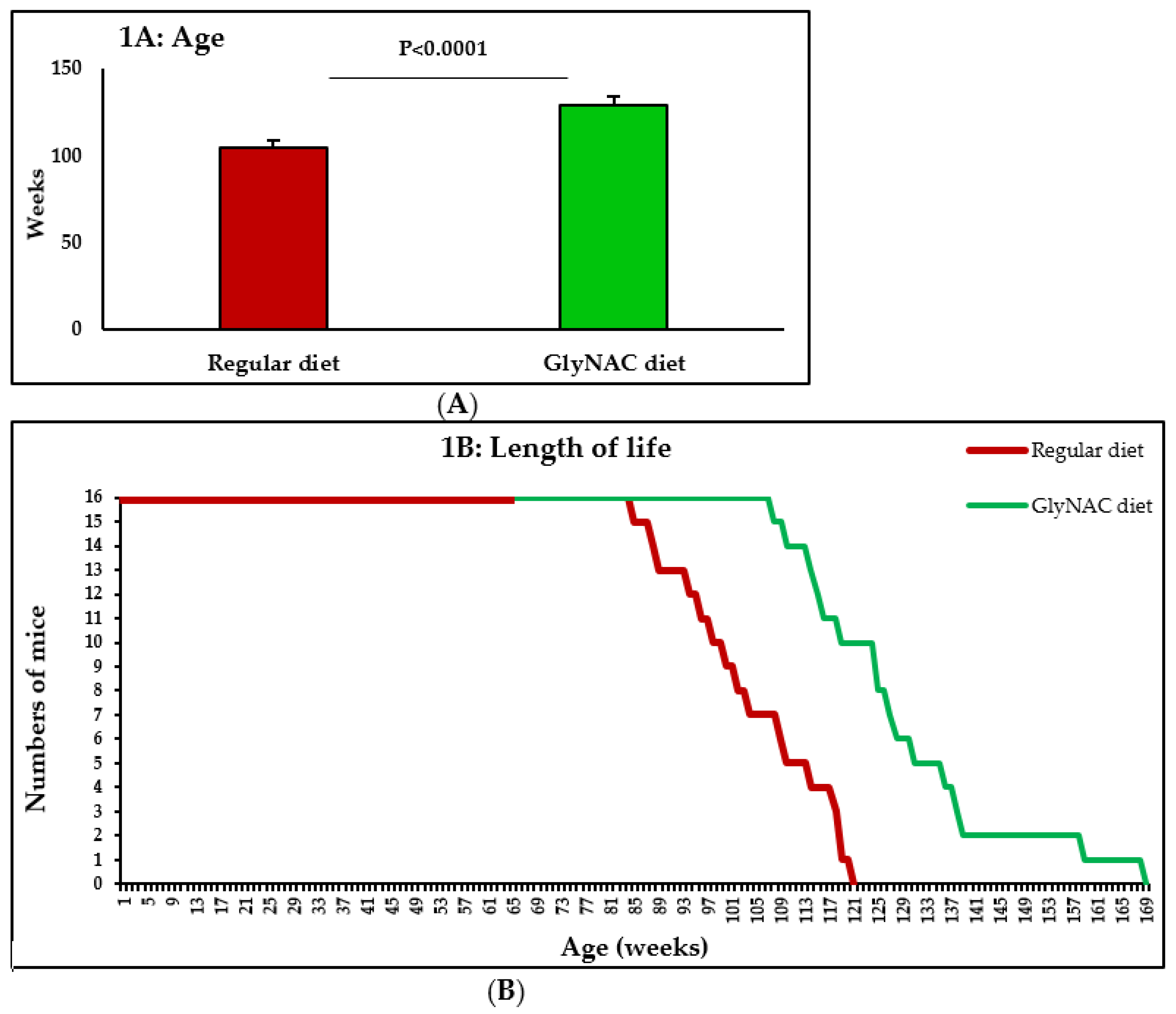
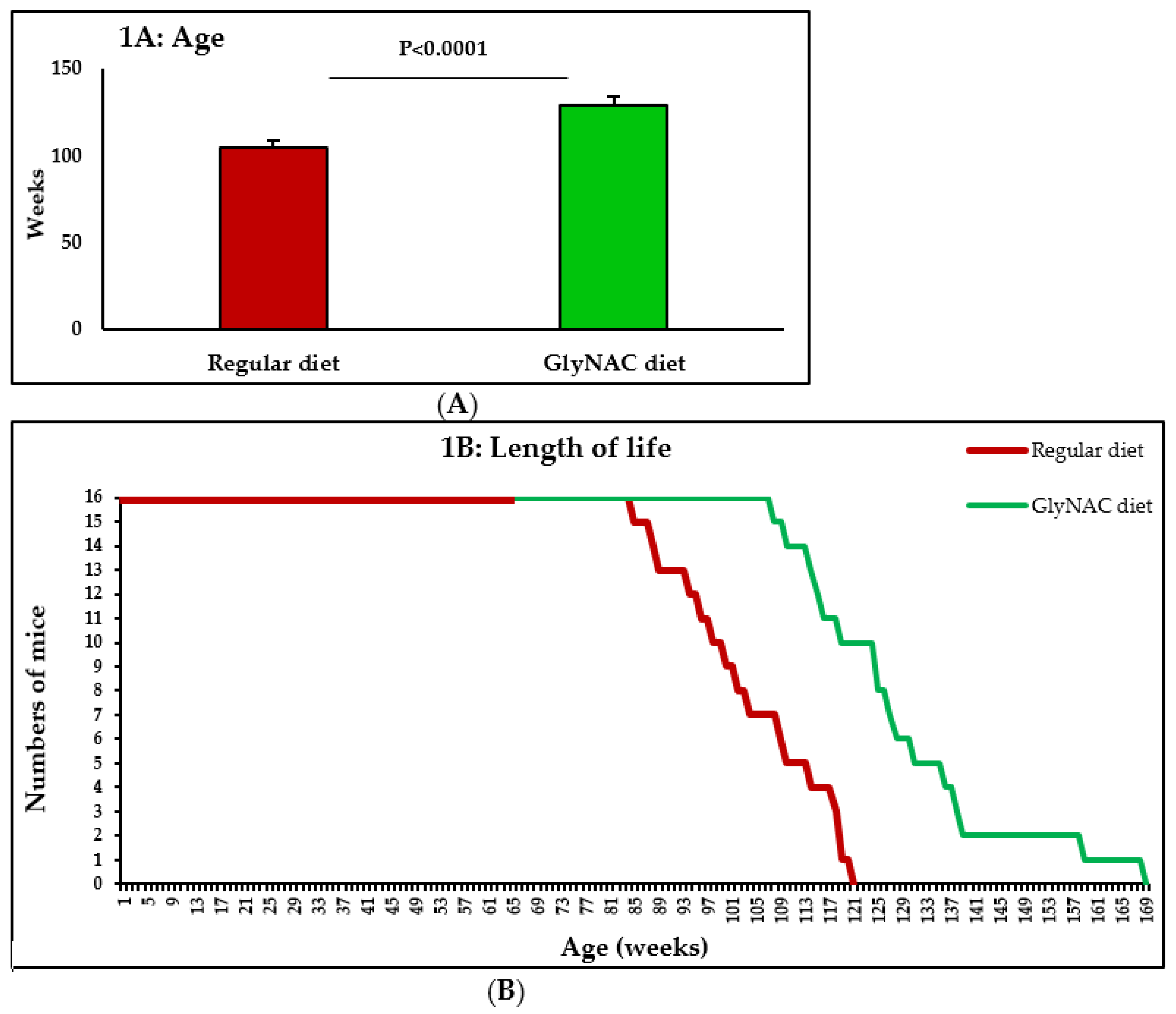
2.2. Study 1: The Effect of GlyNAC Supplementation on Longevity
2.3. Study 2: The Effect of GlyNAC Supplementation on Key Age-Associated Defects in the Heart, Liver and Kidneys
2.4. Outcome Measures
2.4.1. Glutathione Concentrations, Oxidative Stress and Oxidant Damage in the Heart, Liver and Kidneys
2.4.2. Protein Isolation and Immunoblot Analyses
2.5. Statistical Analyses
3. Results
3.1. Study 1: The Effect of GlyNAC Supplementation on Longevity
3.2. Study 2: The Effect of GlyNAC Supplementation on GSH, OxS, Mitochondrial Dysfunction, Mitophagy, Nutrient Sensing and Genomic Damage in the Heart, Liver and Kidneys of Old Mice
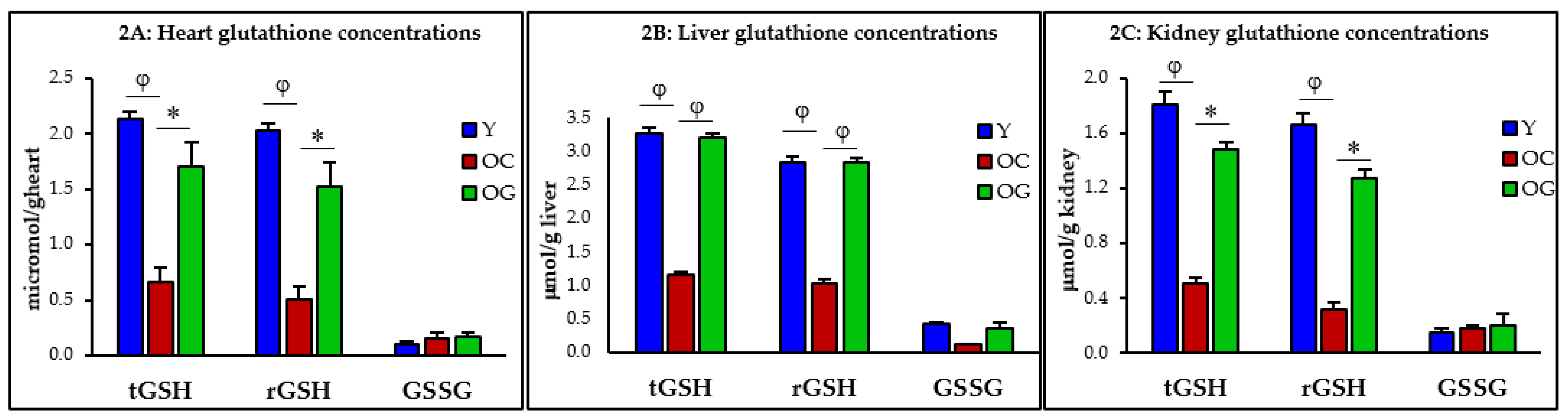
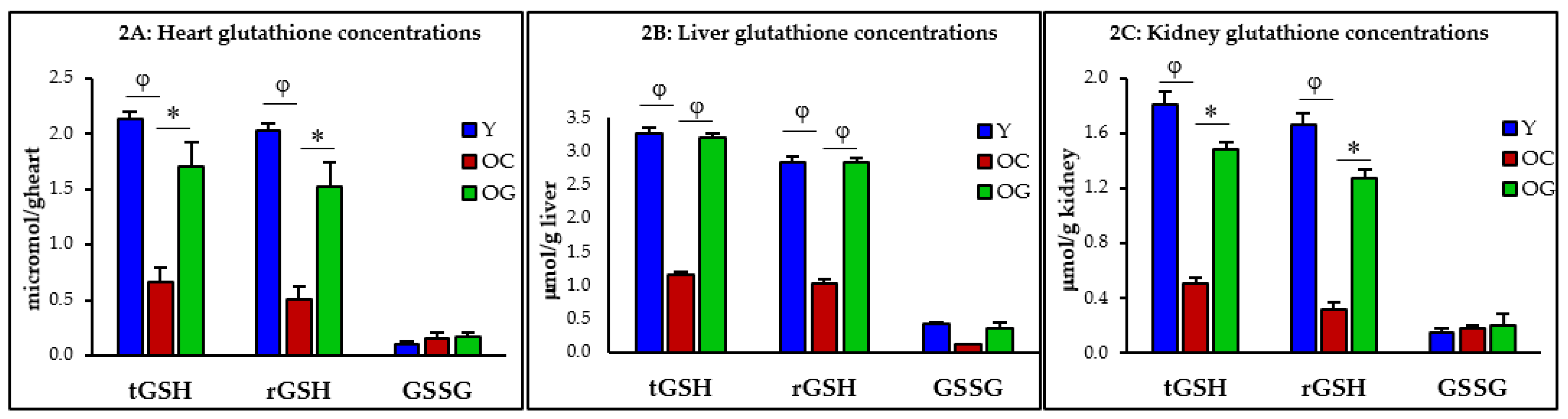
3.2.1. GSH Concentrations
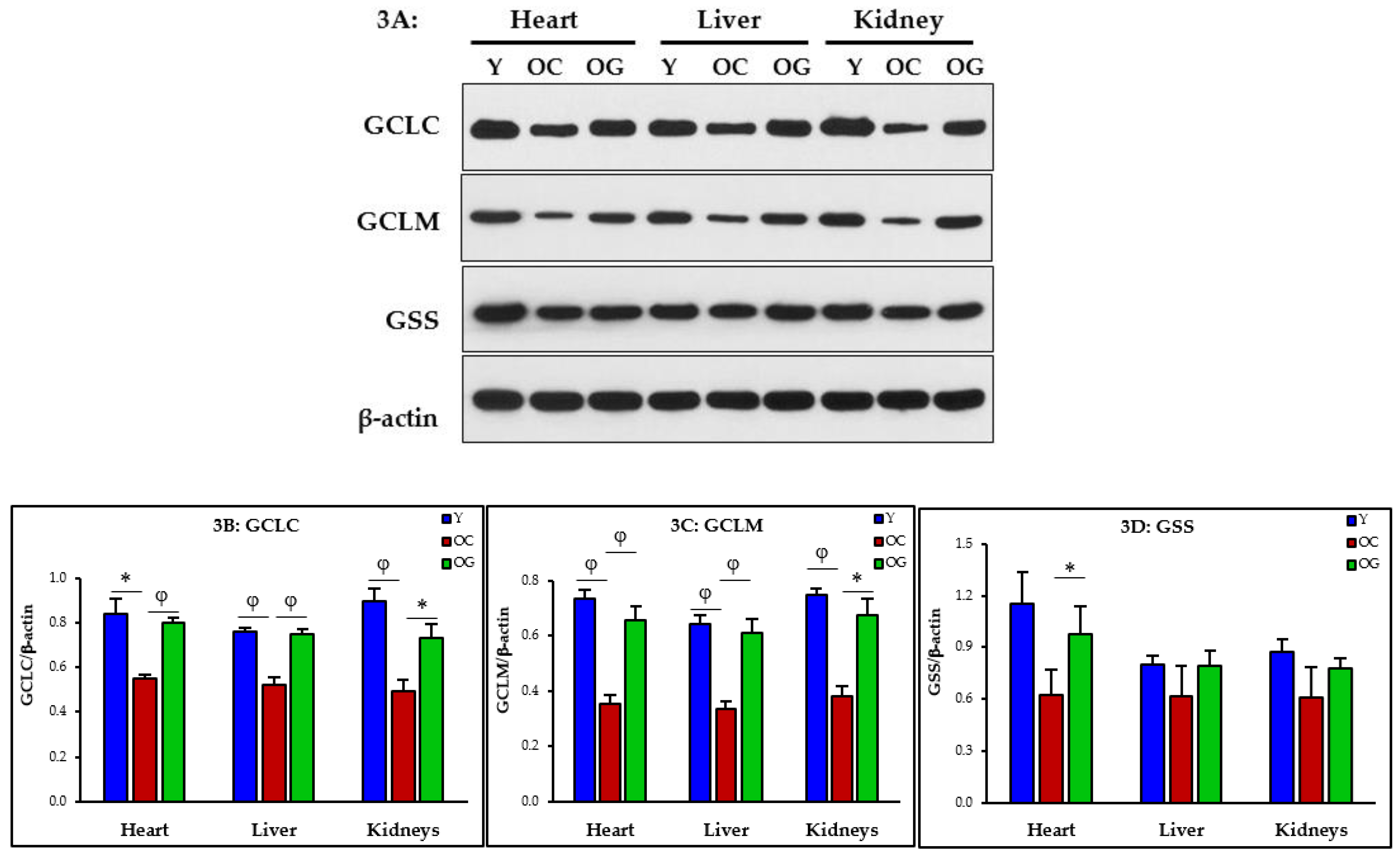
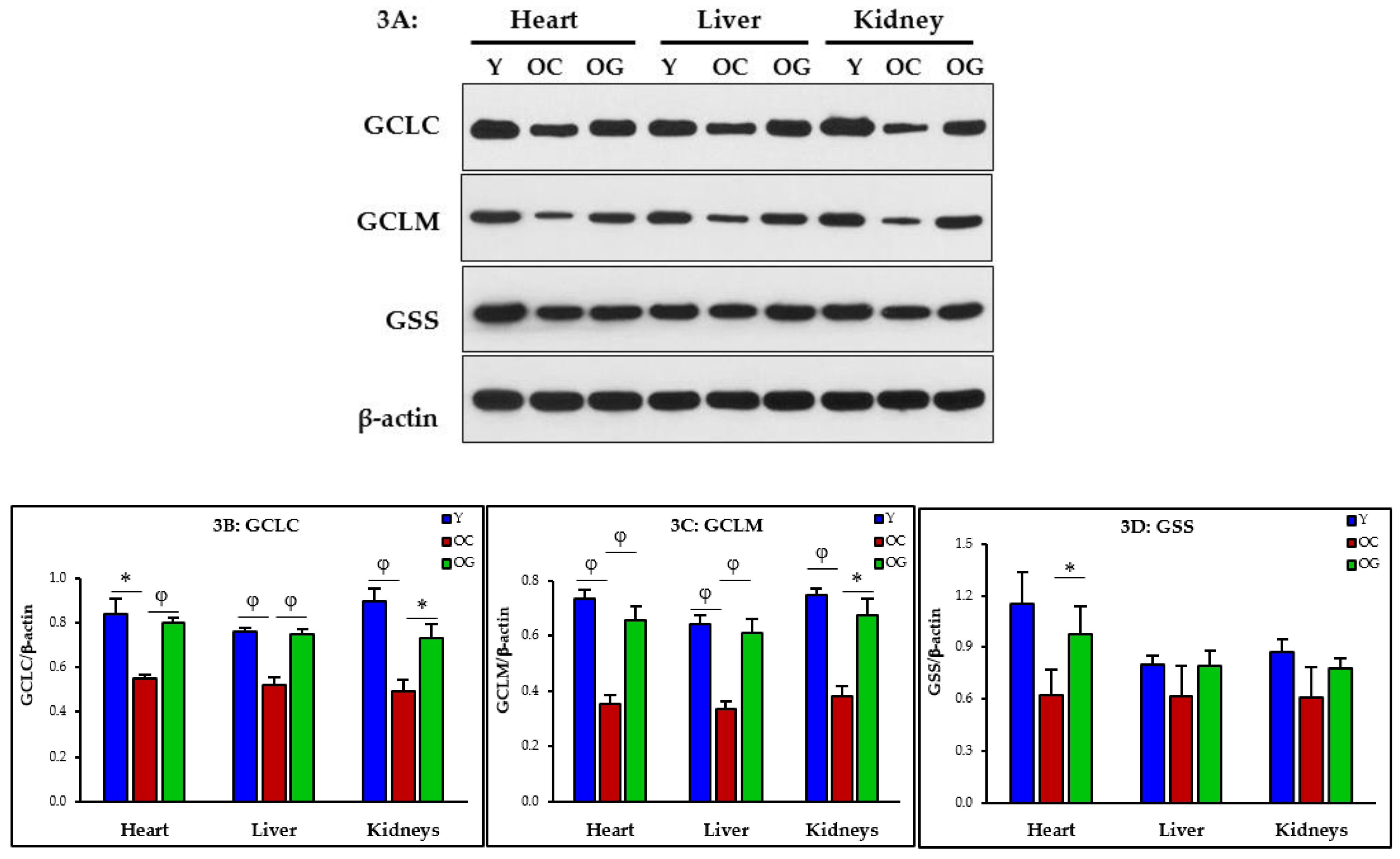
3.2.2. GSH Synthesis
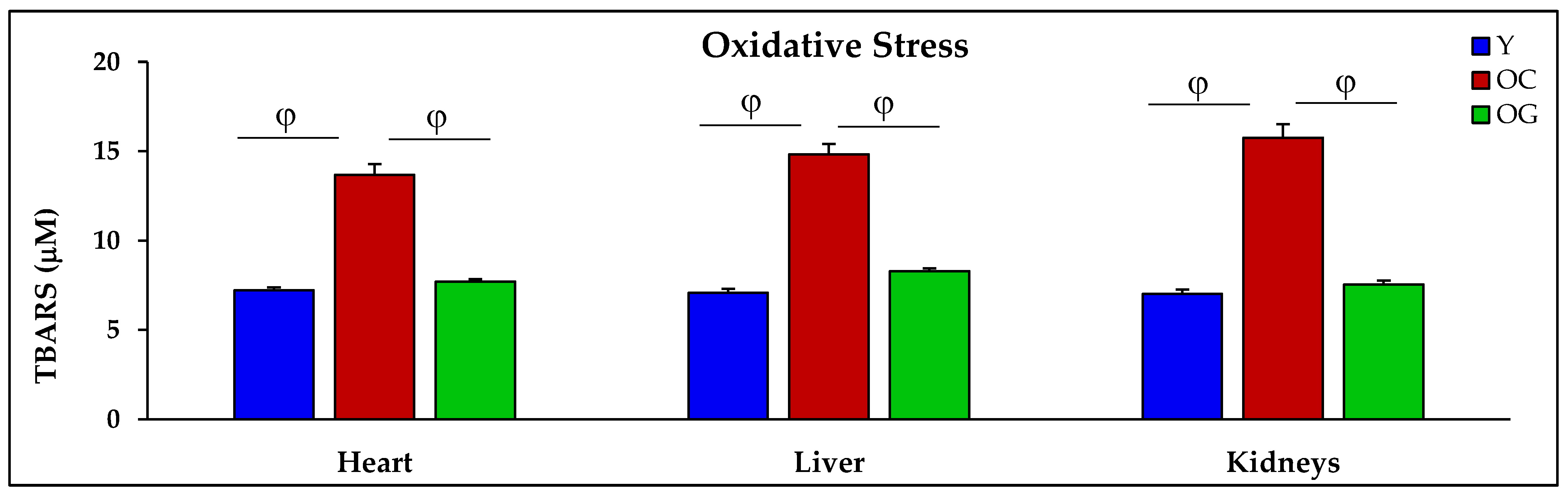
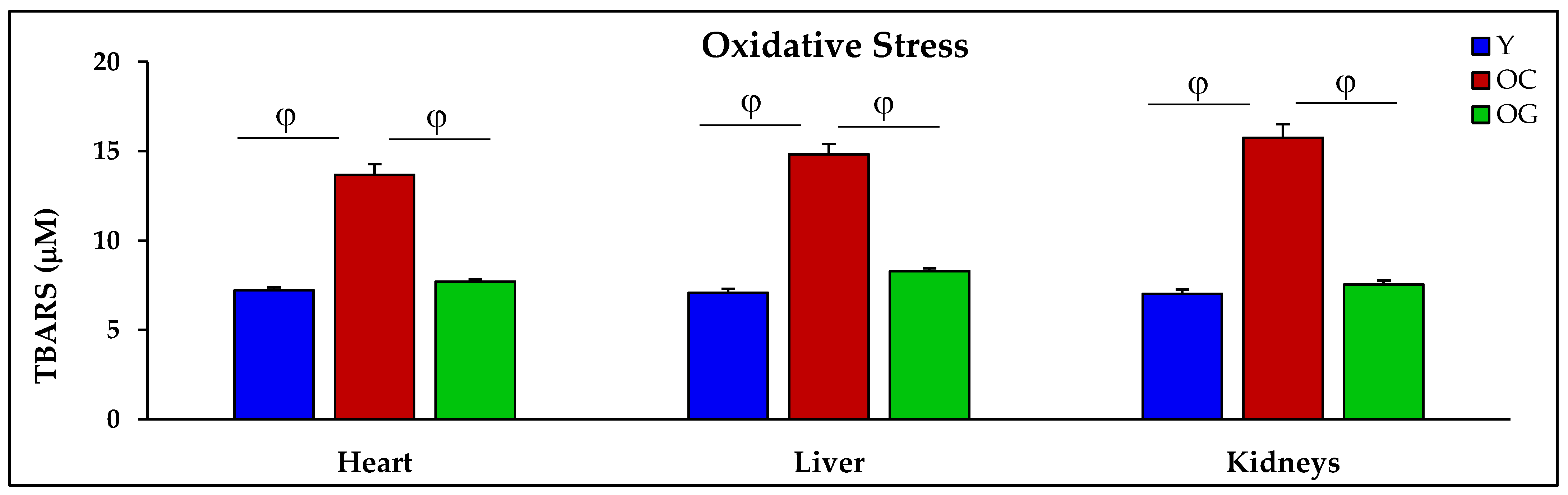
3.2.3. Oxidative Stress
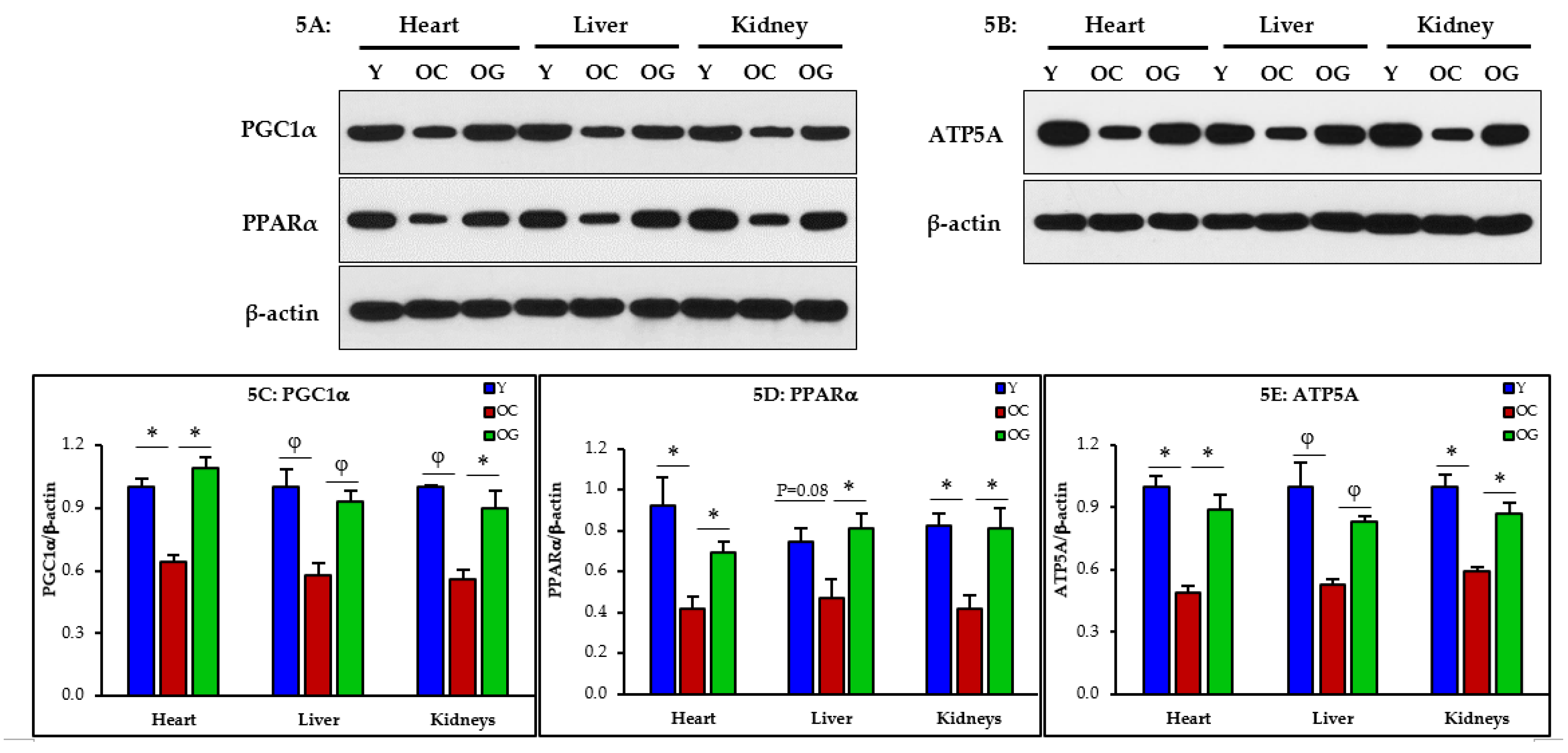
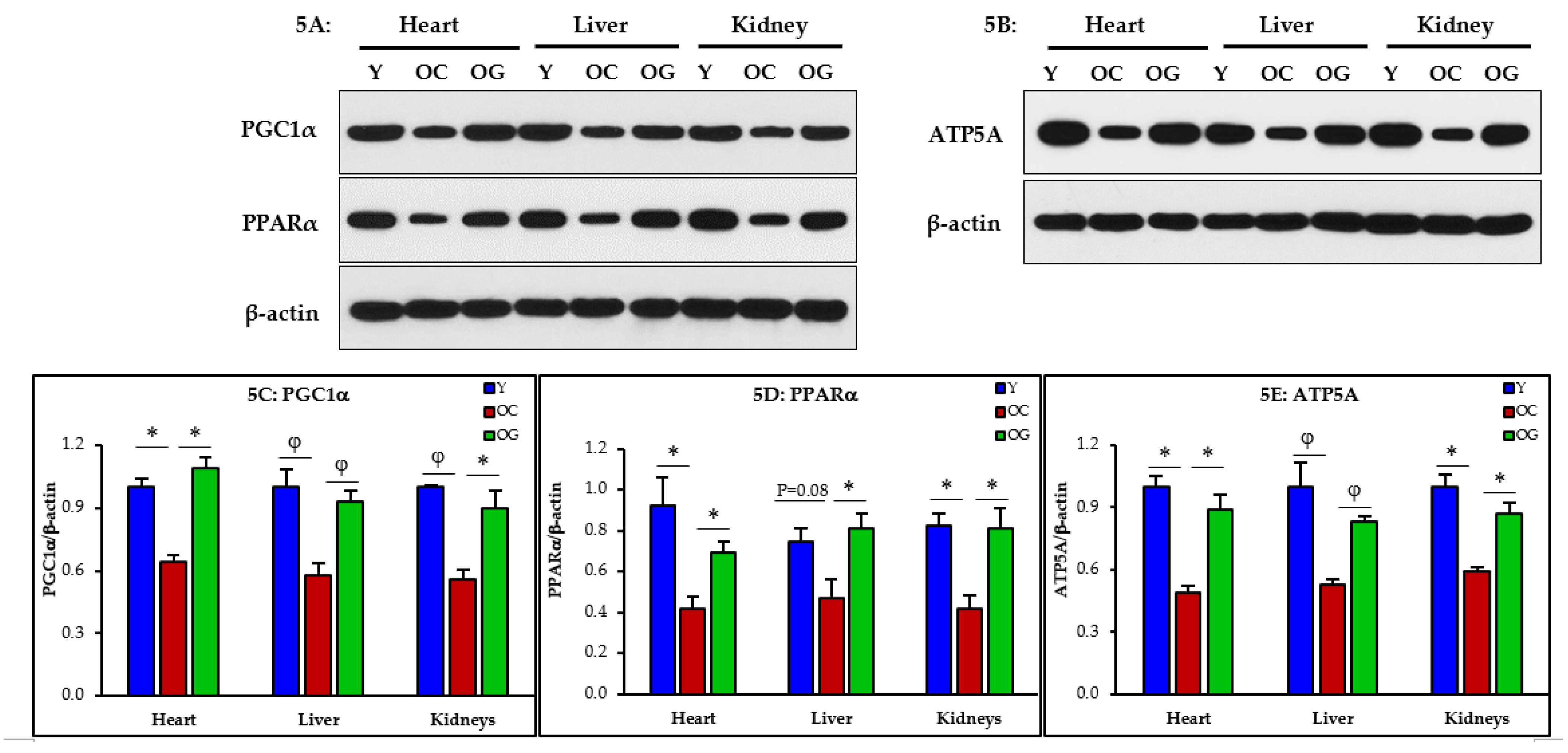
3.2.4. Mitochondrial Dysfunction
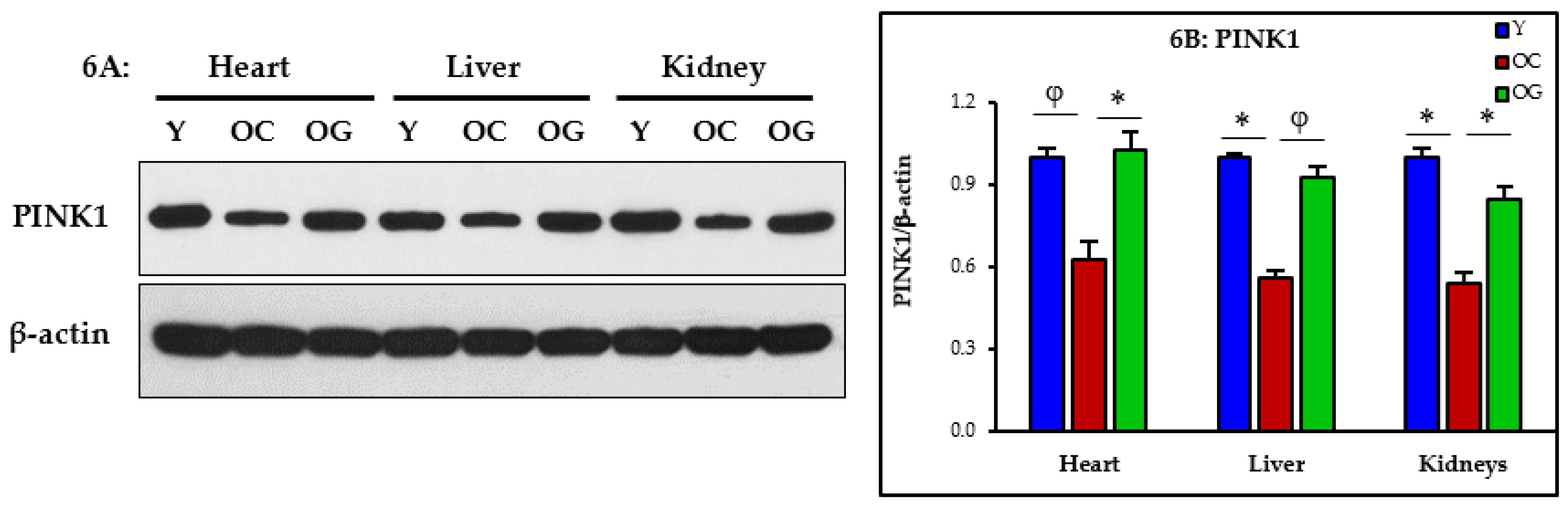
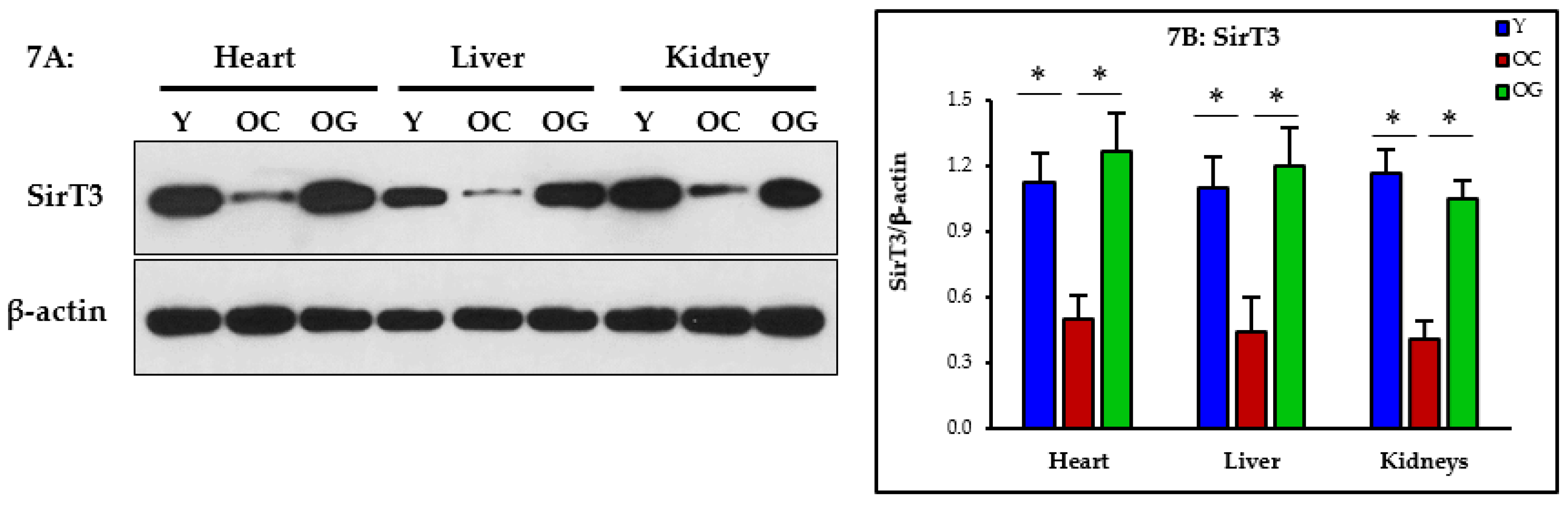
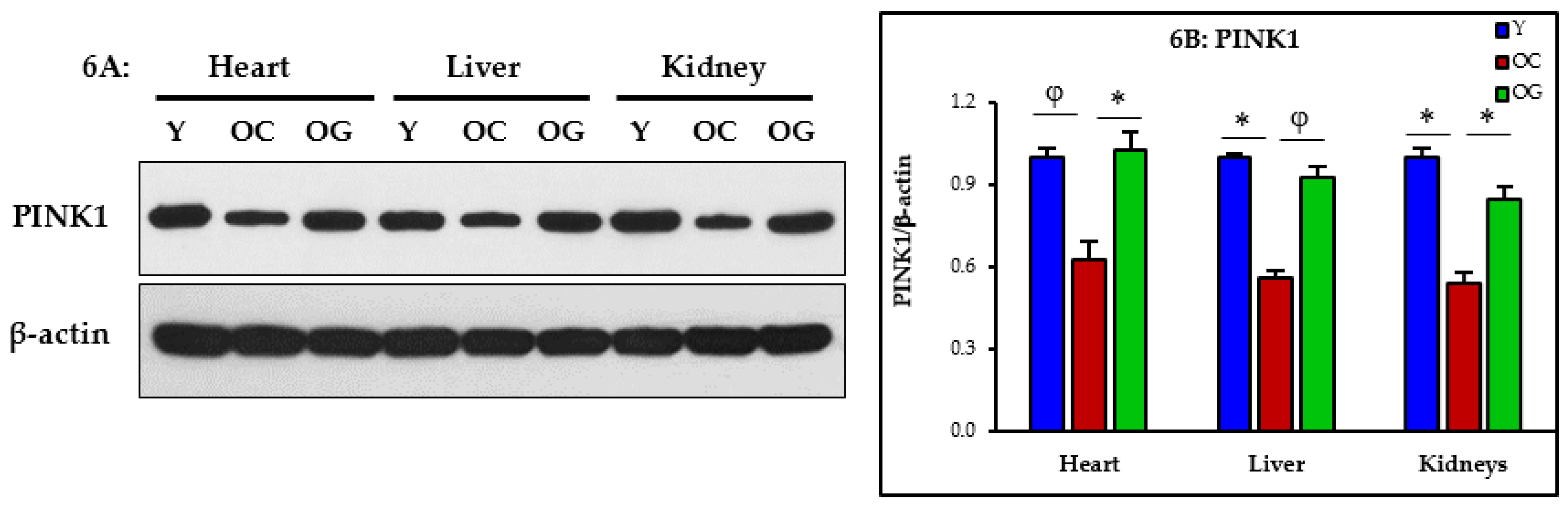
3.2.5. Mitophagy
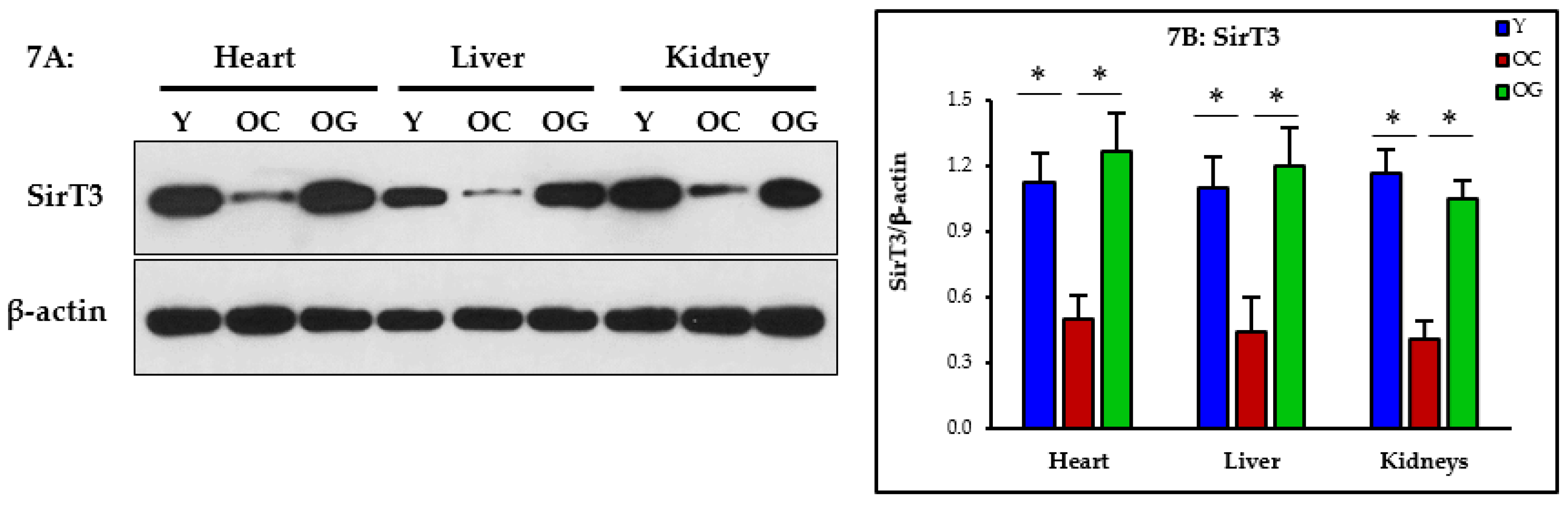
3.2.6. Nutrient Sensing
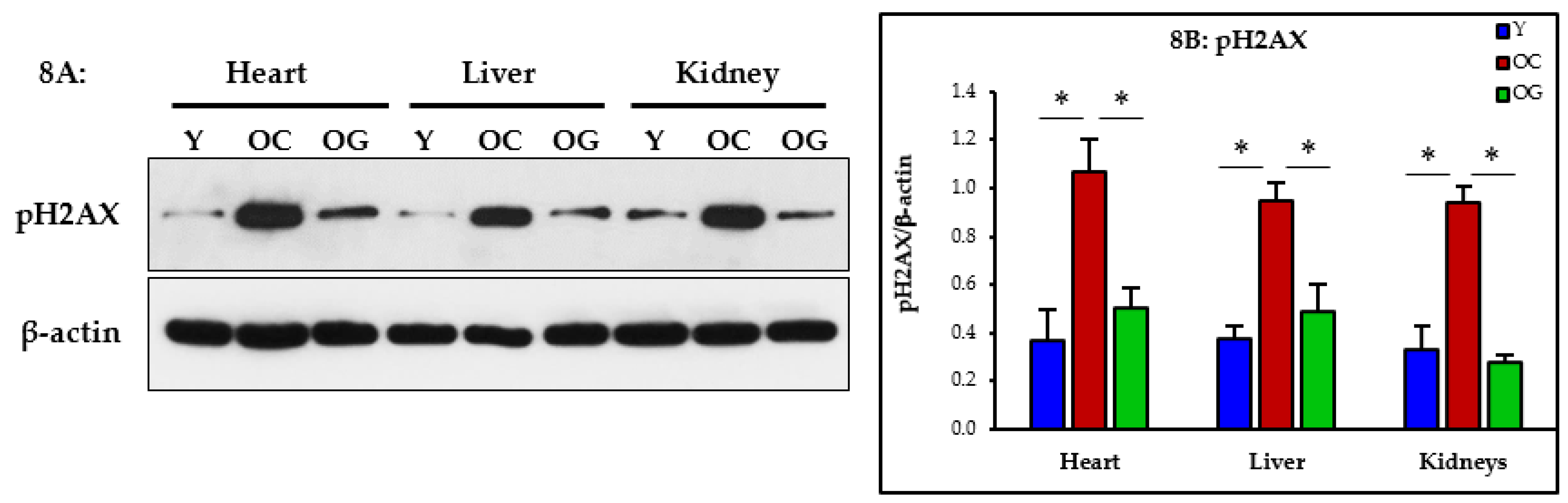
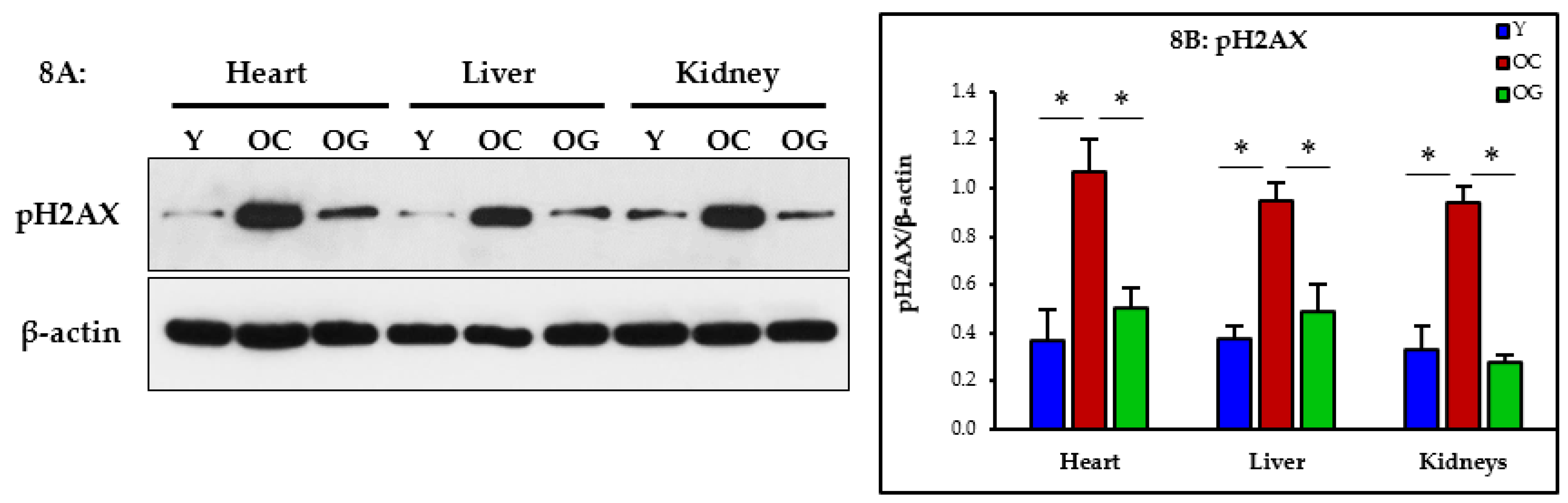
3.2.7. Genomic Damage
4. Discussion
4.1. GlyNAC Supplementation Increases Longevity
4.2. GlyNAC Supplementation Corrects Age-Associated Glutathione Deficiency, Oxidative Stress and Oxidant Damage
4.2.1. Impaired GSH Synthesis and GSH Deficiency in Aging
4.2.2. Oxidative Stress and Oxidant Damage in Aging
4.3. GlyNAC Supplementation Corrects Mitochondrial Dysfunction and Impaired Mitophagy
4.4. GlyNAC Supplementation Improves Nutrient Sensing
4.5. GlyNAC Supplementation Reverses Genomic Damage
4.6. Connecting the Dots
4.7. Why GlyNAC Works
4.8. GlyNAC Is Not the Same as NAC-Alone or GSH-Alone
4.9. Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Partridge, L.; Fuentealba, M.; Kennedy, B.K. The quest to slow ageing through drug discovery. Nat. Rev. Drug Discov. 2020, 19, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef] [PubMed]
- Kibel, A.; Lukinac, A.M.; Dambic, V.; Juric, I.; Selthofer-Relatic, K. Oxidative stress in ischemic heart disease. Oxid. Med. Cell Longev. 2020, 2020, 6627144. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Sánchez-Valle, V.; Chávez-Tapia, N.C.; Uribe, M.; Méndez-Sánchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant mechanisms in renal injury and disease. AntiOxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative stress in chronic kidney disease. Iran J. Kidney Dis. 2015, 9, 165–179. [Google Scholar] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Osahon, O.; Vides, D.B.; Hanania, N.; Minard, C.G.; Sekhar, R.V. Severe glutathione deficiency, oxidative stress and oxidant damage in adults hospitalized with COVID-19: Implications for GlyNAC (Glycine and N-Acetylcysteine) supplementation. Antioxidants 2021, 11, 50. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Van der Vliet, A.; Janssen-Heininger, Y.M.W.; Anathy, V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol. Aspects Med. 2018, 63, 59–69. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; D’Aurelio, M.; Pich, M.M.; Genova, M.L.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta 2000, 1459, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Quirós, P.M.; Langer, T.; López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martinez, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a marker for human disease. Adv. Clin. Chem. 2018, 87, 141–159. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Samson, S.L.; Reddy, V.T.; Gonzalez, E.V.; Sekhar, R.V. Impaired mitochondrial fatty acid oxidation and insulin resistance in aging: Novel protective role of glutathione. Aging Cell 2013, 12, 415–425. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, C.; Suliburk, J.W.; Minard, C.G.; Muthupillai, R.; Chacko, S.; Hsu, J.W.; Jahoor, F.; Sekhar, R.V. Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV patients improves oxidative stress, mitochondrial dysfunction, inflammation, endothelial dysfunction, insulin resistance, genotoxicity, strength, and cognition: Results of an open-label clinical trial. Biomedicines 2020, 8, 390. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, C.; Hsu, J.W.; Chacko, S.; Minard, C.; Jahoor, F.; Sekhar, R.V. Glycine and N-acetylcysteine (GlyNAC) supplementation in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, insulin resistance, endothelial dysfunction, genotoxicity, muscle strength, and cognition: Results of a pilot clinical trial. Clin. Transl Med. 2021, 11, e372. [Google Scholar] [CrossRef]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Wang, X.Q.; Oveisi, F.; Rad, B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertension 2000, 36, 142–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokce, G.; Ozsarlak-Sozer, G.; Oktay, G.; Kirkali, G.; Jaruga, P.; Dizdaroglu, M.; Kerry, Z. Glutathione depletion by buthionine sulfoximine induces oxidative damage to DNA in organs of rabbits in vivo. Biochemistry 2009, 48, 4980–4987. [Google Scholar] [CrossRef]
- Li, X.; Kazgan, N. Mammalian sirtuins and energy metabolism. Int. J. Biol. Sci. 2011, 7, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S. The NAD World: A new systemic regulatory network for metabolism and aging--Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem. Biophys. 2009, 53, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic control by sirtuins and other enzymes that sense NAD+, NADH, or their ratio. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 991–998. [Google Scholar] [CrossRef]
- Newman, J.C.; He, W.; Verdin, E. Mitochondrial protein acylation and intermediary metabolism: Regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 2012, 287, 42436–42443. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Fakouri, N.B.; Hou, Y.; Demarest, T.G.; Christiansen, L.S.; Okur, M.N.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 2019, 286, 1058–1073. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.; Robbins, P.D. Nuclear genomic instability and aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Schöttker, B.; Saum, K.U.; Jansen, E.H.; Boffetta, P.; Trichopoulou, A.; Holleczek, B.; Dieffenbach, A.K.; Brenner, H. Oxidative stress markers and all-cause mortality at older age: A population-based cohort study. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 518–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Yuan, J.Q.; Lv, Y.B.; Gao, X.; Yin, Z.X.; Kraus, V.B.; Luo, J.S.; Chei, C.L.; Matchar, D.B.; Zeng, Y.; et al. Associations between superoxide dismutase, malondialdehyde and all-cause mortality in older adults: A community-based cohort study. BMC Geriatr. 2019, 19, 104. [Google Scholar] [CrossRef]
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Da Fonseca, R.R.; Johnson, W.E.; O’Brien, S.J.; Vasconcelos, V.; Antunes, A. Molecular evolution and the role of oxidative stress in the expansion and functional diversification of cytosolic glutathione transferases. BMC Evol. Biol. 2010, 10, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Bae, Y.A.; Cai, G.B.; Kim, S.H.; Zo, Y.G.; Kong, Y. Modular evolution of glutathione peroxidase genes in association with different biochemical properties of their encoded proteins in invertebrate animals. BMC Evol. Biol. 2009, 9, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieslik, K.A.; Sekhar, R.V.; Granillo, A.; Reddy, A.; Medrano, G.; Heredia, C.P.; Entman, M.L.; Hamilton, D.J.; Li, S.; Reineke, E.; et al. Improved cardiovascular function in old mice after N-Acetyl cysteine and glycine supplemented diet: Inflammation and mitochondrial factors. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1167–1177. [Google Scholar] [CrossRef]
- Sekhar, R. Supplementing glycine and N-acetylcysteine (GlyNAC) rapidly improves health-related quality of life and lowers perception of fatigue in patients with HIV. AIDS 2021, 35, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Hsu, J.W.; Jahoor, F.; Sekhar, R.V. Effect of increasing glutathione with cysteine and glycine supplementation on mitochondrial fuel oxidation, insulin sensitivity, and body composition in older HIV-infected patients. J. Clin. Endocrinol. Metab. 2014, 99, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011, 34, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Dröge, W. Oxidative stress and ageing: Is ageing a cysteine deficiency syndrome? Philos. Trans. R Soc. Lond. B Biol. Sci. 2005, 360, 2355–2372. [Google Scholar] [CrossRef]
- McCarty, M.F.; O’Keefe, J.H.; DiNicolantonio, J.J. Dietary glycine is rate-limiting for glutathione synthesis and may have broad potential for health protection. Ochsner. J. 2018, 18, 81–87. [Google Scholar]
- Sekhar, R.V. GlyNAC Supplementation improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, aging hallmarks, metabolic defects, muscle strength, cognitive decline, and body composition: Implications for healthy aging. J. Nutr. 2021, 151, 3606–3616. [Google Scholar] [CrossRef]
- Genestra, M. Oxyl radicals, redox-sensitive signaling cascades and antioxidants. Cell Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef]
- Zhang, H.; Limphong, P.; Pieper, J.; Liu, Q.; Rodesch, C.K.; Christians, E.; Benjamin, I.J. Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 2012, 26, 1442–1451. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.X.; Li, C.Y.; Tao, R.; Wang, X.P.; Yan, L.J. Reductive stress-induced mitochondrial dysfunction and cardiomyopathy. Oxid. Med. Cell Longev. 2020, 2020, 5136957. [Google Scholar] [CrossRef]
- Gusarov, I.; Shamovsky, I.; Pani, B.; Gautier, L.; Eremina, S.; Katkova-Zhukotskaya, O.; Mironov, A.; Makarov, A.A.; Nudler, E. Dietary thiols accelerate aging of C. elegans. Nat. Commun. 2021, 12, 4336. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Y: Heart | OC: Heart Y vs. OC | OG: Heart OC vs. OG | Y: Liver | OC: Liver Y vs. OC | OG: Liver OC vs. OG | Y: Kidney | OC: Kidney Y vs. OC | OG: Kidney OC vs. OG | |
|---|---|---|---|---|---|---|---|---|---|
| t-GSH (μmol/kg) | 2.1 ± 0.1 | 0.7 ± 0.1 p < 0.001 | 1.7 ± 0.2 p < 0.05 | 3.3 ± 0.1 | 1.2 ± 0.0 p < 0.0001 | 3.2 ± 0.1 p < 0.0001 | 1.8 ± 0.1 | 0.5 ± 0.2 p < 0.01 | 1.5 ± 0.2 p < 0.05 |
| r-GSH (μmol/kg) | 2.0 ± 0.1 | 0.5 ± 0.1 p < 0.01 | 1.6 ± 0.1 p < 0.05 | 2.9 ± 0.1 | 1.0 ± 0.1 p < 0.0001 | 2.8 ± 0.1 p < 0.0001 | 1.7 ± 0.1 | 0.3 ± 0.1 p < 0.01 | 1.3 ± 0.3 p < 0.05 |
| GSSG (μmol/kg) | 0.1 ± 0.0 | 0.2 ± 0.0 p = 0.5 | 0.2 ± 0.0 p = 0.9 | 0.4 ± 0.0 | 0.1 ± 0.0 p < 0.001 | 0.4 ± 0.1 p = 0.08 | 0.1 ± 0.0 | 0.2 ± 0.0 p = 0.7 | 0.2 ± 0.0 p = 0.9 |
| TBARS (μM) | 7.2 ± 0.2 | 13.7 ± 0.6 p < 0.01 | 7.7 ± 0.1 p < 0.01 | 7.1 ± 0.2 | 14.8 ± 0.6 p < 0.001 | 8.3 ± 0.2 p < 0.001 | 7.0 ± 0.2 | 15.8 ± 0.8 p < 0.001 | 7.5 ± 0.2 p < 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Osahon, O.W.; Sekhar, R.V. GlyNAC (Glycine and N-Acetylcysteine) Supplementation in Mice Increases Length of Life by Correcting Glutathione Deficiency, Oxidative Stress, Mitochondrial Dysfunction, Abnormalities in Mitophagy and Nutrient Sensing, and Genomic Damage. Nutrients 2022, 14, 1114. https://doi.org/10.3390/nu14051114
Kumar P, Osahon OW, Sekhar RV. GlyNAC (Glycine and N-Acetylcysteine) Supplementation in Mice Increases Length of Life by Correcting Glutathione Deficiency, Oxidative Stress, Mitochondrial Dysfunction, Abnormalities in Mitophagy and Nutrient Sensing, and Genomic Damage. Nutrients. 2022; 14(5):1114. https://doi.org/10.3390/nu14051114
Chicago/Turabian StyleKumar, Premranjan, Ob W. Osahon, and Rajagopal V. Sekhar. 2022. "GlyNAC (Glycine and N-Acetylcysteine) Supplementation in Mice Increases Length of Life by Correcting Glutathione Deficiency, Oxidative Stress, Mitochondrial Dysfunction, Abnormalities in Mitophagy and Nutrient Sensing, and Genomic Damage" Nutrients 14, no. 5: 1114. https://doi.org/10.3390/nu14051114
APA StyleKumar, P., Osahon, O. W., & Sekhar, R. V. (2022). GlyNAC (Glycine and N-Acetylcysteine) Supplementation in Mice Increases Length of Life by Correcting Glutathione Deficiency, Oxidative Stress, Mitochondrial Dysfunction, Abnormalities in Mitophagy and Nutrient Sensing, and Genomic Damage. Nutrients, 14(5), 1114. https://doi.org/10.3390/nu14051114