

Phosphate Dysregulation and Metabolic Syndrome


{kind=link}
{kind=link}
Abstract
1. Introduction
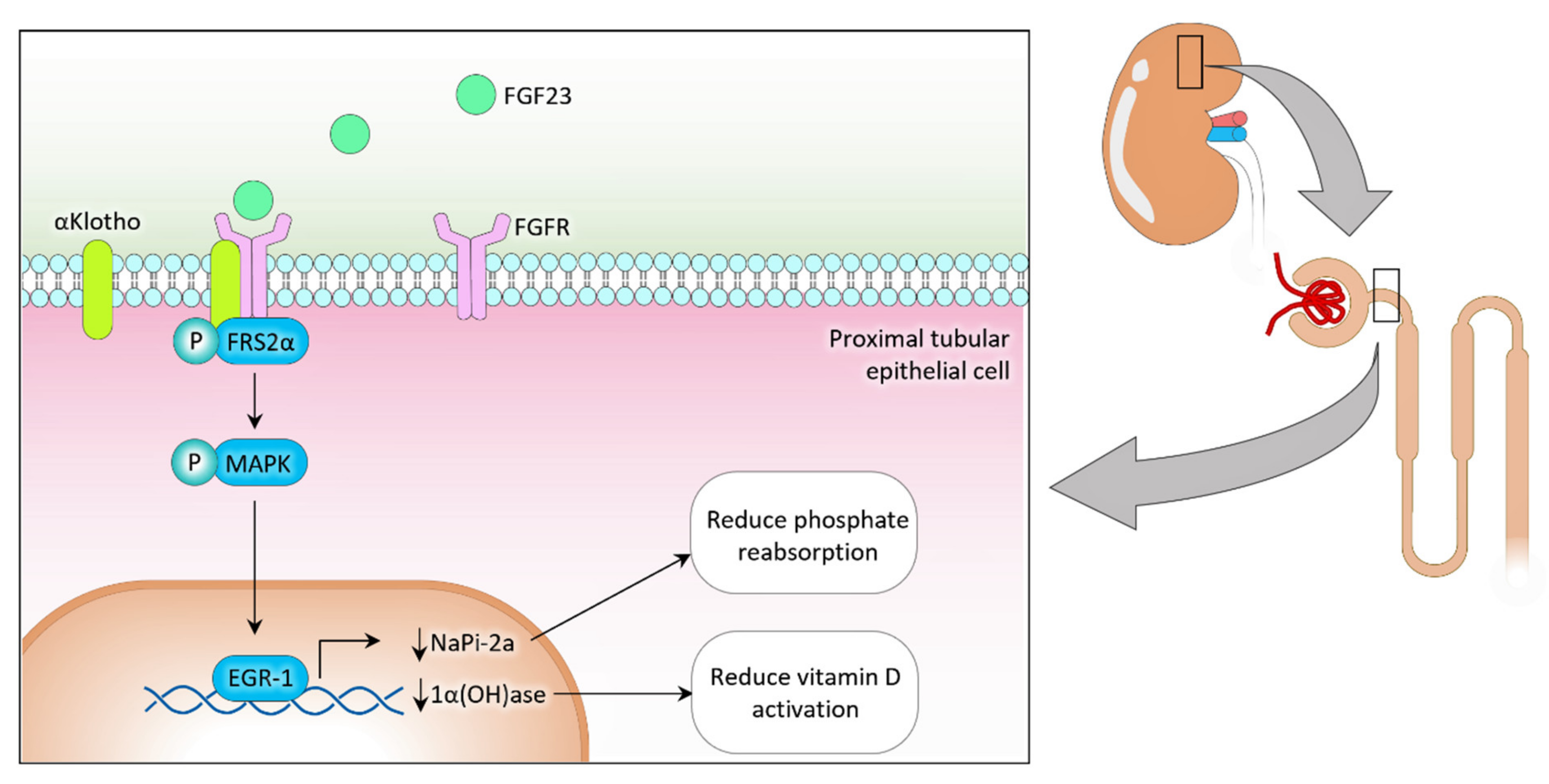
2. Phosphate Regulation
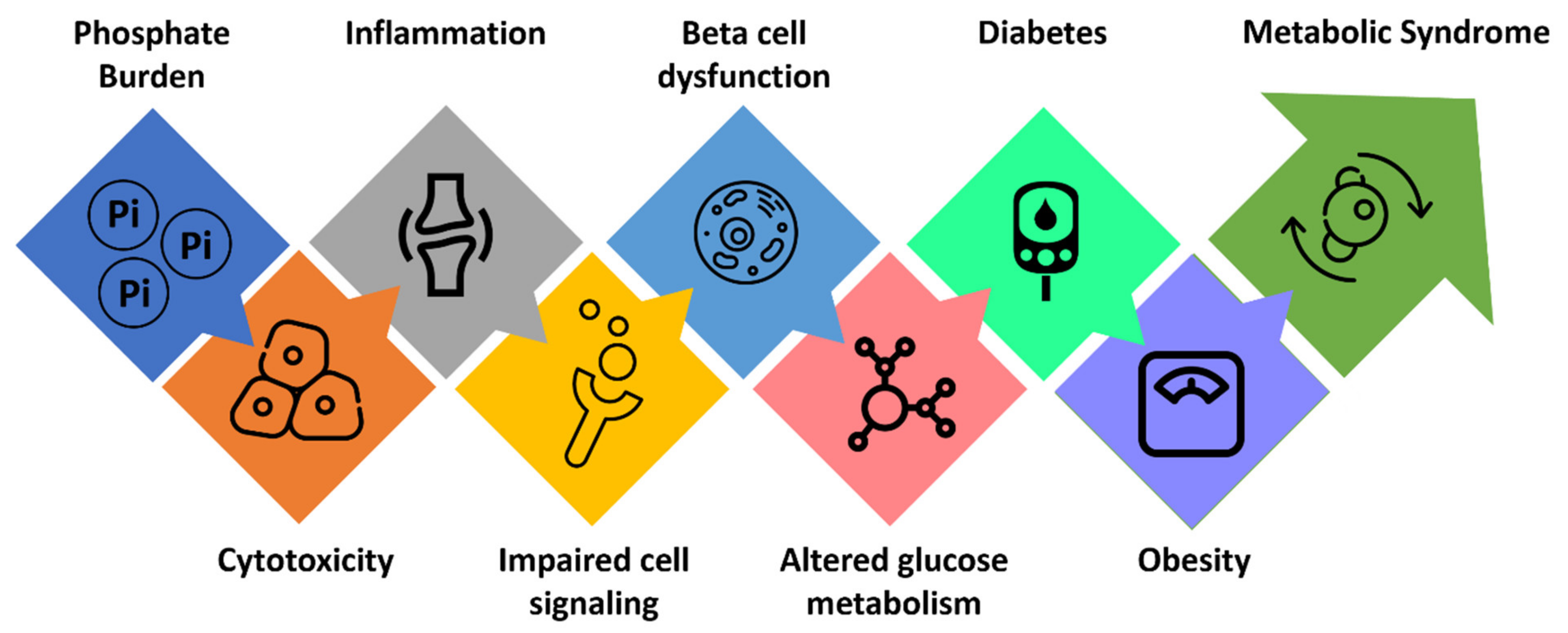
3. Phosphate Burden and Glucose Metabolism
4. Phosphate Burden and Cardiometabolic Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swarup, S.; Goyal, A.; Grigorova, Y.; Zeltser, R. Metabolic Syndrome. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Razzaque, M.S. The FGF23-Klotho axis: Endocrine regulation of phosphate homeostasis. Nat. Rev. Endocrinol. 2009, 5, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S. FGF23-mediated regulation of systemic phosphate homeostasis: Is Klotho an essential player? Am. J. Physiol. Ren. Physiol. 2009, 296, F470–F476. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S. Phosphate toxicity: New insights into an old problem. Clin. Sci. 2011, 120, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Quan, X.; Hwang, K.H.; Xu, S.; Das, R.; Choi, S.K.; Wiederkehr, A.; Wollheim, C.B.; Cha, S.K.; Park, K.S. Mitochondrial oxidative stress mediates high-phosphate-induced secretory defects and apoptosis in insulin-secreting cells. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E933–E941. [Google Scholar] [CrossRef] [PubMed]
- Hernando, N.; Gagnon, K.; Lederer, E. Phosphate Transport in Epithelial and Nonepithelial Tissue. Physiol. Rev. 2021, 101, 1–35. [Google Scholar] [CrossRef]
- D’Alessandro, C.; Piccoli, G.B.; Cupisti, A. The “phosphorus pyramid”: A visual tool for dietary phosphate management in dialysis and CKD patients. BMC Nephrol. 2015, 16, 9. [Google Scholar] [CrossRef]
- Benini, O.; D’Alessandro, C.; Gianfaldoni, D.; Cupisti, A. Extra-phosphate load from food additives in commonly eaten foods: A real and insidious danger for renal patients. J. Ren. Nutr. 2011, 21, 303–308. [Google Scholar] [CrossRef]
- Lorenzo, C.; Hanley, A.J.; Rewers, M.J.; Haffner, S.M. Calcium and phosphate concentrations and future development of type 2 diabetes: The Insulin Resistance Atherosclerosis Study. Diabetologia 2014, 57, 1366–1374. [Google Scholar] [CrossRef]
- Osadnik, K.; Osadnik, T.; Delijewski, M.; Lejawa, M.; Fronczek, M.; Regula, R.; Gasior, M.; Pawlas, N. Calcium and Phosphate Levels are Among Other Factors Associated with Metabolic Syndrome in Patients with Normal Weight. Diabetes Metab. Syndr. Obes. 2020, 13, 1281–1288. [Google Scholar] [CrossRef]
- Chang, A.R.; Lazo, M.; Appel, L.J.; Gutiérrez, O.M.; Grams, M.E. High dietary phosphorus intake is associated with all-cause mortality: Results from NHANES III. Am. J. Clin. Nutr. 2014, 99, 320–327. [Google Scholar] [CrossRef]
- Goodson, J.M.; Shi, P.; Razzaque, M.S. Dietary phosphorus enhances inflammatory response: A study of human gingivitis. J. Steroid Biochem. Mol. Biol. 2019, 188, 166–171. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef]
- Brown, M.A.; Hural, J. Functions of IL-4 and Control of Its Expression. Crit. Rev. Immunol. 2017, 37, 181–212. [Google Scholar] [CrossRef]
- Goodson, J.M.; Shi, P.; Mumena, C.H.; Haq, A.; Razzaque, M.S. Dietary phosphorus burden increases cariogenesis independent of vitamin D uptake. J. Steroid Biochem. Mol. Biol. 2017, 167, 33–38. [Google Scholar] [CrossRef]
- Razzaque, M.S. Salivary phosphate as a biomarker for human diseases. FASEB Bioadv. 2022, 4, 102–108. [Google Scholar] [CrossRef]
- Calo, L.A.; Savica, V.; Piccoli, A.; Fusaro, M.; D’Angelo, A.; Davis, P.A. Reduction of hyperphosphatemia is related with the reduction of C-reactive protein in dialysis patients. Study in sevelamer-resistant dialysis patients treated with chitosan chewing gum as salivary phosphate binder. Ren. Fail. 2011, 33, 11–14. [Google Scholar] [CrossRef]
- Ichikawa, S.; Sorenson, A.H.; Austin, A.M.; Mackenzie, D.S.; Fritz, T.A.; Moh, A.; Hui, S.L.; Econs, M.J. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology 2009, 150, 2543–2550. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Nidumanda Appaiah, H.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef]
- Takeshita, A.; Kawakami, K.; Furushima, K.; Miyajima, M.; Sakaguchi, K. Central role of the proximal tubular alphaKlotho/FGF receptor complex in FGF23-regulated phosphate and vitamin D metabolism. Sci. Rep. 2018, 8, 6917. [Google Scholar] [CrossRef]
- Masuyama, R.; Stockmans, I.; Torrekens, S.; Van Looveren, R.; Maes, C.; Carmeliet, P.; Bouillon, R.; Carmeliet, G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Investig. 2006, 116, 3150–3159. [Google Scholar] [CrossRef]
- Razzaque, M.S. The role of Klotho in energy metabolism. Nat. Rev. Endocrinol. 2012, 8, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Abreu, M.A.; Meyer-Fernandes, J.R. Extracellular Inorganic Phosphate-Induced Release of Reactive Oxygen Species: Roles in Physiological Processes and Disease Development. Int. J. Mol. Sci. 2021, 22, 7768. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.A.; Kowaltowski, A.J. Phosphate increases mitochondrial reactive oxygen species release. Free Radic. Res. 2004, 38, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2017, 127, 1589. [Google Scholar] [CrossRef] [PubMed]
- Mousa, A.; Naderpoor, N.; Teede, H.; Scragg, R.; de Courten, B. Vitamin D supplementation for improvement of chronic low-grade inflammation in patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Nutr. Rev. 2018, 76, 380–394. [Google Scholar] [CrossRef]
- Namazi, N.; Larijani, B.; Azadbakht, L. Dietary Inflammatory Index and its Association with the Risk of Cardiovascular Diseases, Metabolic Syndrome, and Mortality: A Systematic Review and Meta-Analysis. Horm. Metab. Res. 2018, 50, 345–358. [Google Scholar] [CrossRef]
- Khan, M.; Jose, A.; Sharma, S. Physiology, Parathyroid Hormone. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rendina-Ruedy, E.; Rosen, C.J. Parathyroid hormone (PTH) regulation of metabolic homeostasis: An old dog teaches us new tricks. Mol. Metab. 2022, 60, 101480. [Google Scholar] [CrossRef]
- Hetz, R.; Beeler, E.; Janoczkin, A.; Kiers, S.; Li, L.; Willard, B.B.; Razzaque, M.S.; He, P. Excessive Inorganic Phosphate Burden Perturbed Intracellular Signaling: Quantitative Proteomics and Phosphoproteomics Analyses. Front. Nutr. 2021, 8, 765391. [Google Scholar] [CrossRef]
- He, P.; Mann-Collura, O.; Fling, J.; Edara, N.; Hetz, R.; Razzaque, M.S. High phosphate actively induces cytotoxicity by rewiring pro-survival and pro-apoptotic signaling networks in HEK293 and HeLa cells. FASEB J. 2021, 35, e20997. [Google Scholar] [CrossRef]
- Voelkl, J.; Egli-Spichtig, D.; Alesutan, I.; Wagner, C.A. Inflammation: A putative link between phosphate metabolism and cardiovascular disease. Clin. Sci. 2021, 135, 201–227. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, Q.; Chen, X.; Zhang, M.; He, Y.; Ji, Z.; Chen, R.; Zhang, J.; Zhang, H.; Shi, G.; et al. Mineral and bone disorder biomarkers and inflammation indexes in patients with end stage renal disease. Ann. Palliat. Med. 2020, 9, 3938–3946. [Google Scholar] [CrossRef]
- Lane, M.; Howland, G.; West, M.; Hockey, M.; Marx, W.; Loughman, A.; O’Hely, M.; Jacka, F.; Rocks, T. The effect of ultra-processed very low-energy diets on gut microbiota and metabolic outcomes in individuals with obesity: A systematic literature review. Obes. Res. Clin. Pract. 2020, 14, 197–204. [Google Scholar] [CrossRef]
- Chassaing, B.; Gewirtz, A.T. Gut microbiota, low-grade inflammation, and metabolic syndrome. Toxicol. Pathol. 2014, 42, 49–53. [Google Scholar] [CrossRef]
- Ojo, O.; Ojo, O.O.; Zand, N.; Wang, X. The Effect of Dietary Fibre on Gut Microbiota, Lipid Profile, and Inflammatory Markers in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Nutrients 2021, 13, 1805. [Google Scholar] [CrossRef]
- Osuka, S.; Razzaque, M.S. Can features of phosphate toxicity appear in normophosphatemia? J. Bone Miner. Metab. 2012, 30, 10–18. [Google Scholar] [CrossRef]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef]
- Freinkel, N.; Younsi, C.E.; Bonnar, J.; Dawson, R.M. Rapid transient efflux of phosphate ions from pancreatic islets as an early action of insulin secretagogues. J. Clin. Investig. 1974, 54, 1179–1189. [Google Scholar] [CrossRef][Green Version]
- Bukowiecki, L.; Trus, M.; Matschinsky, F.M.; Freinkel, N. Alterations in pancreatic islet phosphate content during secretory stimulation with glucose. Biochim. Biophys. Acta 1979, 583, 370–377. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Quan, X.; Xu, S.; Das, R.; Cha, S.K.; Kong, I.D.; Shong, M.; Wollheim, C.B.; Park, K.S. Intracellular alkalinization by phosphate uptake via type III sodium-phosphate cotransporter participates in high-phosphate-induced mitochondrial oxidative stress and defective insulin secretion. FASEB J. 2016, 30, 3979–3988. [Google Scholar] [CrossRef]
- Koshkin, V.; Bikopoulos, G.; Chan, C.B.; Wheeler, M.B. The characterization of mitochondrial permeability transition in clonal pancreatic beta-cells. Multiple modes and regulation. J. Biol. Chem. 2004, 279, 41368–41376. [Google Scholar] [CrossRef]
- Mahmud, I.; Rahman, Z.; Keka, S.I.; Devnath, S.; Masum, N.; Hossain, S. Hyperphosphataemia is associated with the diabetes-related cardiovascular risk factors. J. Oleo Sci. 2011, 60, 79–85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Haap, M.; Heller, E.; Thamer, C.; Tschritter, O.; Stefan, N.; Fritsche, A. Association of serum phosphate levels with glucose tolerance, insulin sensitivity and insulin secretion in non-diabetic subjects. Eur. J. Clin. Nutr. 2006, 60, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Harrop, G.A.; Benedict, E.M. The participation of inorganic substances in carbohydrate metabolism. J. Biol. Chem. 1924, 59, 683–697. [Google Scholar] [CrossRef]
- Osuna, J.I.; Castillo, M.; Rodriguez, E.; Campillo, J.E.; Osorio, C. Influence of inorganic phosphate on glucose-induced insulin release in vitro. Adv. Exp. Med. Biol. 1986, 208, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Harter, H.R.; Santiago, J.V.; Rutherford, W.E.; Slatopolsky, E.; Klahr, S. The relative roles of calcium, phosphorus, and parathyroid hormone in glucose- and tolbutamide-mediated insulin release. J. Clin. Investig. 1976, 58, 359–367. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Lang, R. Hypophosphatemia and glucose intolerance: Evidence for tissue insensitivity to insulin. N. Engl. J. Med. 1980, 303, 1259–1263. [Google Scholar] [CrossRef]
- Raikou, V.D.; Kyriaki, D.; Gavriil, S. Importance of serum phosphate in elderly patients with diabetes mellitus. World J. Diabetes 2020, 11, 416–424. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Gutekunst, L.; Mehrotra, R.; Kovesdy, C.P.; Bross, R.; Shinaberger, C.S.; Noori, N.; Hirschberg, R.; Benner, D.; Nissenson, A.R.; et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 519–530. [Google Scholar] [CrossRef]
- Sherman, R.A.; Mehta, O. Phosphorus and potassium content of enhanced meat and poultry products: Implications for patients who receive dialysis. Clin. J. Am. Soc. Nephrol. 2009, 4, 1370–1373. [Google Scholar] [CrossRef]
- Sherman, R.A.; Mehta, O. Dietary phosphorus restriction in dialysis patients: Potential impact of processed meat, poultry, and fish products as protein sources. Am. J. Kidney Dis. 2009, 54, 18–23. [Google Scholar] [CrossRef]
- Karp, H.; Ekholm, P.; Kemi, V.; Itkonen, S.; Hirvonen, T.; Närkki, S.; Lamberg-Allardt, C. Differences among total and in vitro digestible phosphorus content of plant foods and beverages. J. Ren. Nutr. 2012, 22, 416–422. [Google Scholar] [CrossRef]
- Jiang, S.; Ma, X.; Li, M.; Yan, S.; Zhao, H.; Pan, Y.; Wang, C.; Yao, Y.; Jin, L.; Li, B. Association between dietary mineral nutrient intake, body mass index, and waist circumference in U.S. adults using quantile regression analysis NHANES 2007–2014. PeerJ 2020, 8, e9127. [Google Scholar] [CrossRef]
- Kim, H.K.; Mizuno, M.; Vongpatanasin, W. Phosphate, the forgotten mineral in hypertension. Curr. Opin. Nephrol. Hypertens. 2019, 28, 345–351. [Google Scholar] [CrossRef]
- Dhingra, R.; Sullivan, L.M.; Fox, C.S.; Wang, T.J.; D’Agostino, R.B., Sr.; Gaziano, J.M.; Vasan, R.S. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch. Intern. Med. 2007, 167, 879–885. [Google Scholar] [CrossRef]
- Ellam, T.; Wilkie, M.; Chamberlain, J.; Crossman, D.; Eastell, R.; Francis, S.; Chico, T.J. Dietary phosphate modulates atherogenesis and insulin resistance in apolipoprotein E knockout mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1988–1990. [Google Scholar] [CrossRef]
- Stephens, B.Y.; Kaur, J.; Vranish, J.R.; Barbosa, T.C.; Blankenship, J.K.; Smith, S.A.; Fadel, P.J. Effect of acute high-phosphate intake on muscle metaboreflex activation and vascular function. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H308–H314. [Google Scholar] [CrossRef]
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Köhler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222. [Google Scholar] [CrossRef]
- Zhu, G.H.; Sun, X.P.; Liu, Z.; Fan, Z.X.; Wang, Y.L.; Tan, J.; Li, J.; Hua, Q. The relation between serum phosphorus levels and long-term mortality in Chinese patients with ST-segment elevation myocardial infarction. J. Geriatr. Cardiol. 2019, 16, 775–781. [Google Scholar] [CrossRef]
- Huang, C.X.; Plantinga, L.C.; Fink, N.E.; Melamed, M.L.; Coresh, J.; Powe, N.R. Phosphate levels and blood pressure in incident hemodialysis patients: A longitudinal study. Adv. Chronic. Kidney Dis. 2008, 15, 321–331. [Google Scholar] [CrossRef][Green Version]
- Yoo, K.D.; Kang, S.; Choi, Y.; Yang, S.H.; Heo, N.J.; Chin, H.J.; Oh, K.H.; Joo, K.W.; Kim, Y.S.; Lee, H. Sex, Age, and the Association of Serum Phosphorus with All-Cause Mortality in Adults With Normal Kidney Function. Am. J. Kidney Dis. 2016, 67, 79–88. [Google Scholar] [CrossRef]
- Cheungpasitporn, W.; Thongprayoon, C.; Mao, M.A.; Kittanamongkolchai, W.; Sakhuja, A.; Erickson, S.B. Admission serum phosphate levels predict hospital mortality. Hosp. Pract. 2018, 46, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Utsugi, T.; Ohno, T.; Ohyama, Y.; Uchiyama, T.; Saito, Y.; Matsumura, Y.; Aizawa, H.; Itoh, H.; Kurabayashi, M.; Kawazu, S.; et al. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism 2000, 49, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Kebler, R.; McDonald, F.D.; Cadnapaphornchai, P. Dynamic changes in serum phosphorus levels in diabetic ketoacidosis. Am. J. Med. 1985, 79, 571–576. [Google Scholar] [CrossRef]
- Håglin, L.M.; Törnkvist, B.; Bäckman, L.O. High serum phosphate and triglyceride levels in smoking women and men with CVD risk and type 2 diabetes. Diabetol. Metab. Syndr. 2014, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Håglin, L.; Törnkvist, B.; Bäckman, L. Obesity, smoking habits, and serum phosphate levels predicts mortality after life-style intervention. PLoS ONE 2020, 15, e0227692. [Google Scholar] [CrossRef]
- Uwitonze, A.M.; Razzaque, M.S. Role of Magnesium in Vitamin D Activation and Function. J. Am. Osteopath. Assoc. 2018, 118, 181–189. [Google Scholar] [CrossRef]
- Razzaque, M.S. COVID-19 pandemic: Can zinc supplementation provide an additional shield against the infection? Comput. Struct. Biotechnol. J. 2021, 19, 1371–1378. [Google Scholar] [CrossRef]
- Amos, A.; Razzaque, M.S. Zinc and its role in vitamin D function. Curr. Res. Physiol. 2022, 5, 203–207. [Google Scholar] [CrossRef]
- Erem, S.; Atfi, A.; Razzaque, M.S. Anabolic effects of vitamin D and magnesium in aging bone. J. Steroid Biochem. Mol. Biol. 2019, 193, 105400. [Google Scholar] [CrossRef]
- Wimalawansa, S.J.; Razzaque, M.S.; Al-Daghri, N.M. Calcium and vitamin D in human health: Hype or real? J. Steroid Biochem. Mol. Biol. 2018, 180, 4–14. [Google Scholar] [CrossRef]
- Geneen, L.J.; Kimber, C.; Doree, C.; Stanworth, S.; Shah, A. Efficacy and Safety of Intravenous Iron Therapy for Treating Anaemia in Critically ill Adults: A Rapid Systematic Review with Meta-Analysis. Transfus. Med. Rev. 2021. [Google Scholar] [CrossRef]
- Shutto, Y.; Shimada, M.; Kitajima, M.; Yamabe, H.; Razzaque, M.S. Lack of awareness among future medical professionals about the risk of consuming hidden phosphate-containing processed food and drinks. PLoS ONE 2011, 6, e29105. [Google Scholar] [CrossRef]
- Shutto, Y.; Shimada, M.; Kitajima, M.; Yamabe, H.; Saitoh, Y.; Saitoh, H.; Razzaque, M.S. Inadequate awareness among chronic kidney disease patients regarding food and drinks containing artificially added phosphate. PLoS ONE 2013, 8, e78660. [Google Scholar] [CrossRef]
- Shires, R.; Teitelbaum, S.L.; Bergfeld, M.A.; Fallon, M.D.; Slatopolsky, E.; Avioli, L.V. The effect of streptozotocin-induced chronic diabetes mellitus on bone and mineral homeostasis in the rat. J. Lab. Clin. Med. 1981, 97, 231–240. [Google Scholar]
- Winiarska, A.; Filipska, I.; Knysak, M.; Stompór, T. Dietary Phosphorus as a Marker of Mineral Metabolism and Progression of Diabetic Kidney Disease. Nutrients 2021, 13, 789. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mironov, N.; Haque, M.; Atfi, A.; Razzaque, M.S. Phosphate Dysregulation and Metabolic Syndrome. Nutrients 2022, 14, 4477. https://doi.org/10.3390/nu14214477
Mironov N, Haque M, Atfi A, Razzaque MS. Phosphate Dysregulation and Metabolic Syndrome. Nutrients. 2022; 14(21):4477. https://doi.org/10.3390/nu14214477
Chicago/Turabian StyleMironov, Nikolay, Mainul Haque, Azeddine Atfi, and Mohammed S. Razzaque. 2022. "Phosphate Dysregulation and Metabolic Syndrome" Nutrients 14, no. 21: 4477. https://doi.org/10.3390/nu14214477
APA StyleMironov, N., Haque, M., Atfi, A., & Razzaque, M. S. (2022). Phosphate Dysregulation and Metabolic Syndrome. Nutrients, 14(21), 4477. https://doi.org/10.3390/nu14214477