
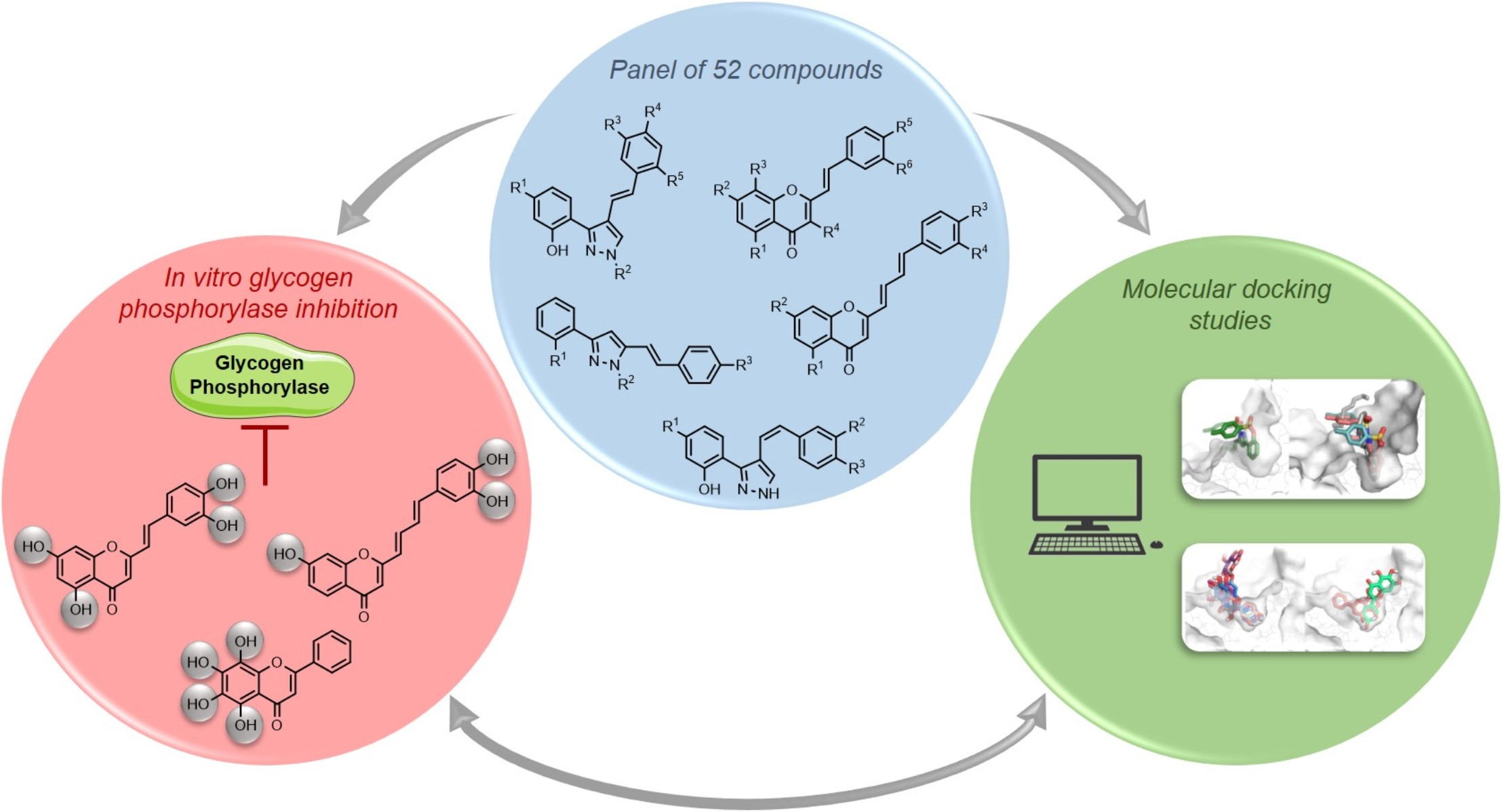
An In Silico and an In Vitro Inhibition Analysis of Glycogen Phosphorylase by Flavonoids, Styrylchromones, and Pyrazoles

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
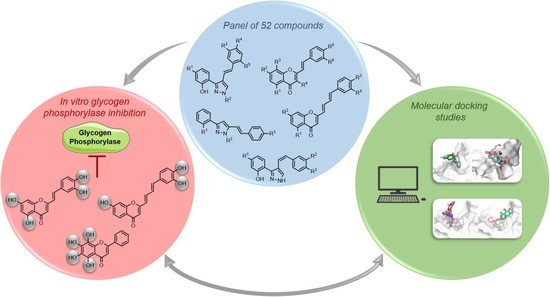
:
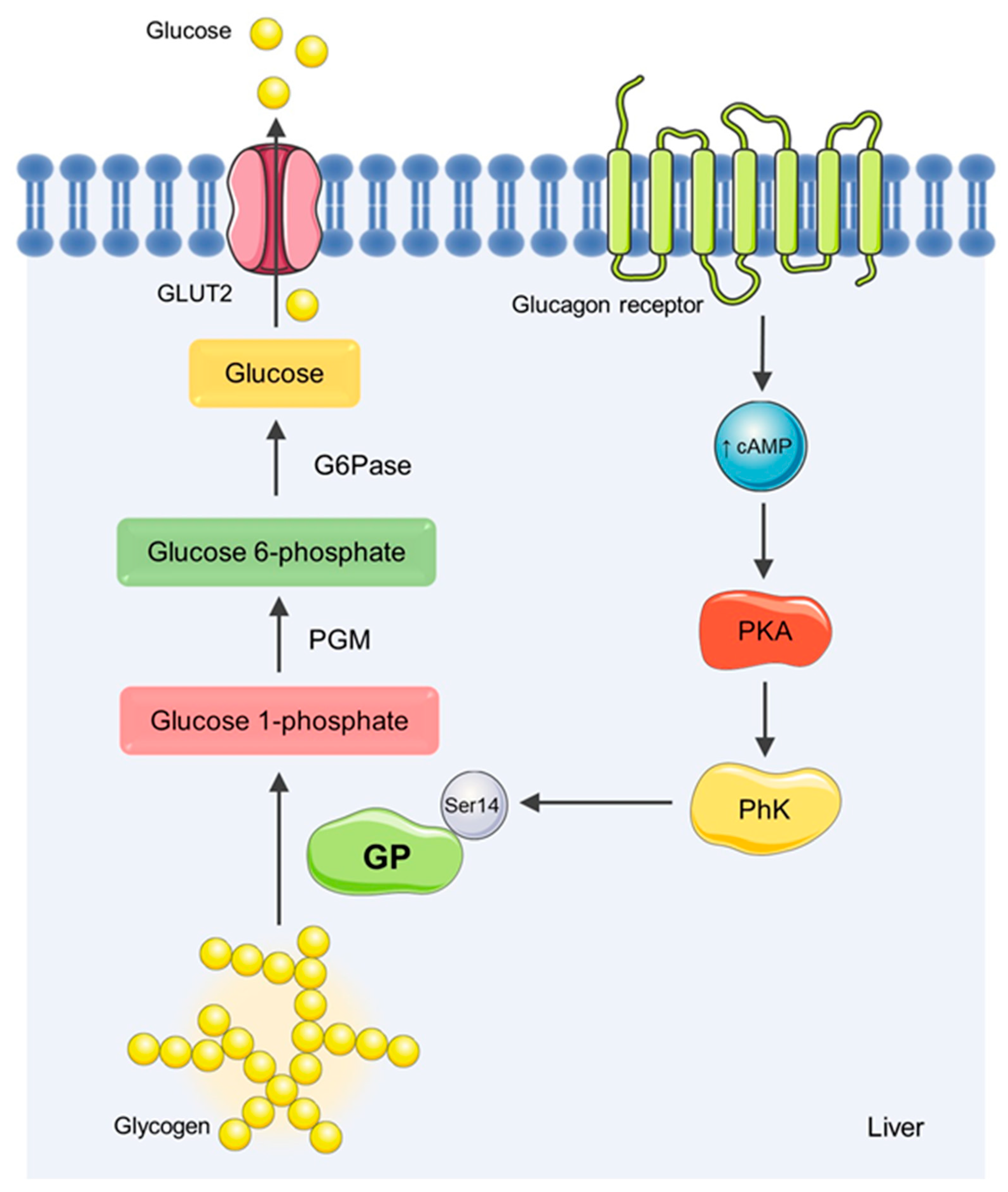
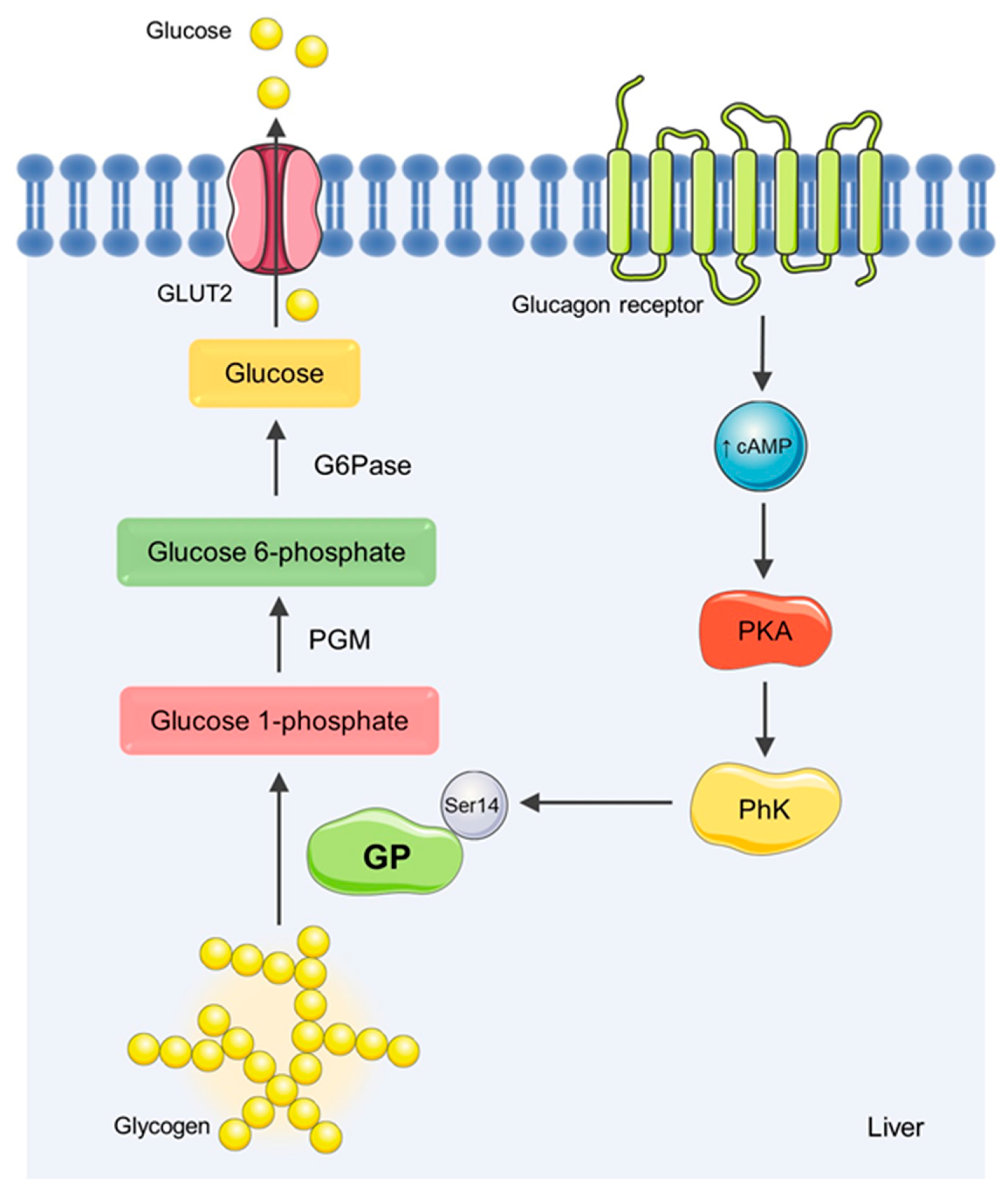
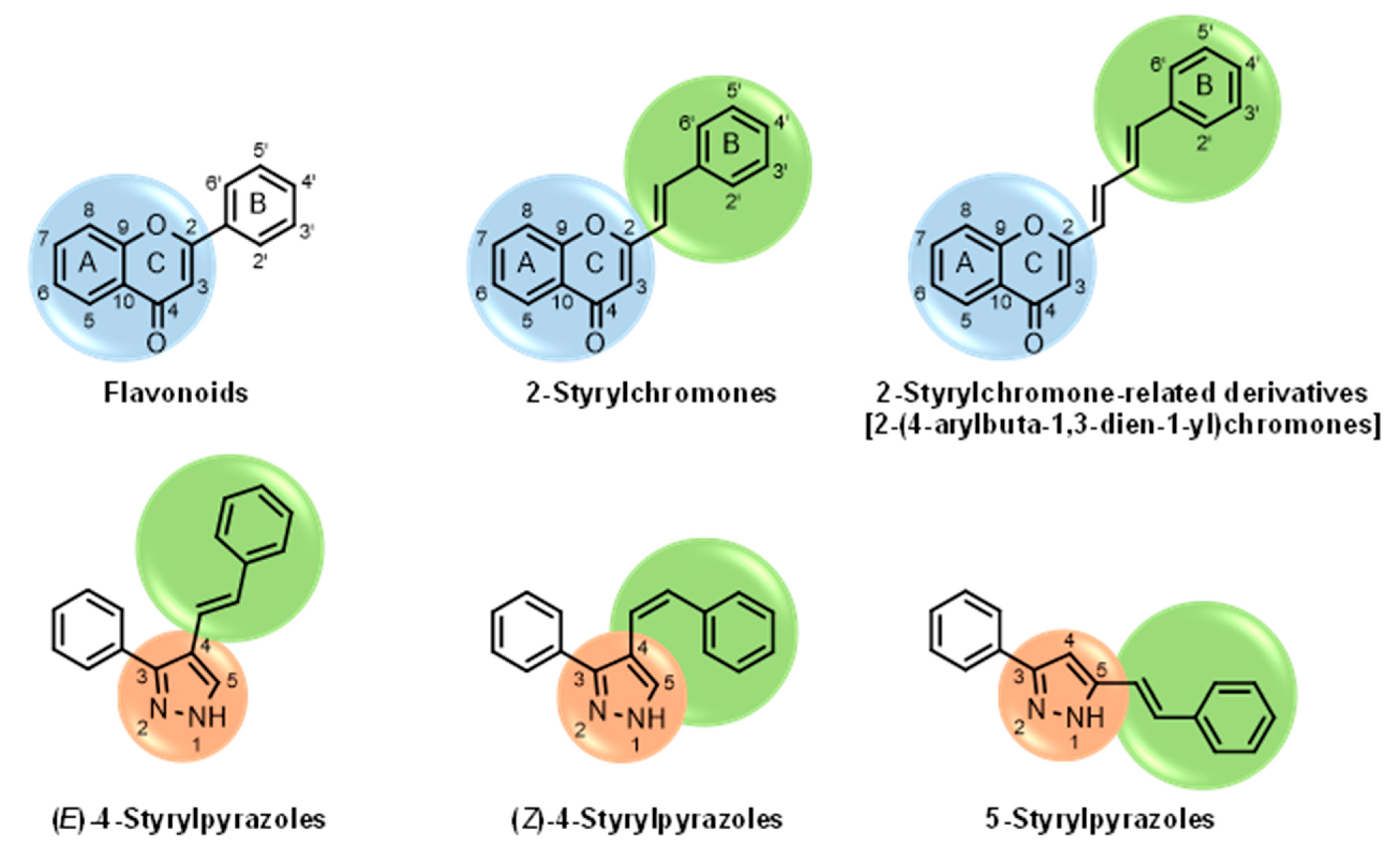
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. The In Vitro Glycogen Phosphorylase Inhibition Assay
2.3. The In Vitro Effect of Glucose on Inhibitor Potency
2.4. Molecular Docking Calculations and Data Analyses
2.5. Statistical Analyses
3. Results
3.1. The In Vitro Glycogen Phosphorylase Inhibition
3.2. In Vitro Effects of Glucose on Inhibitor Potency
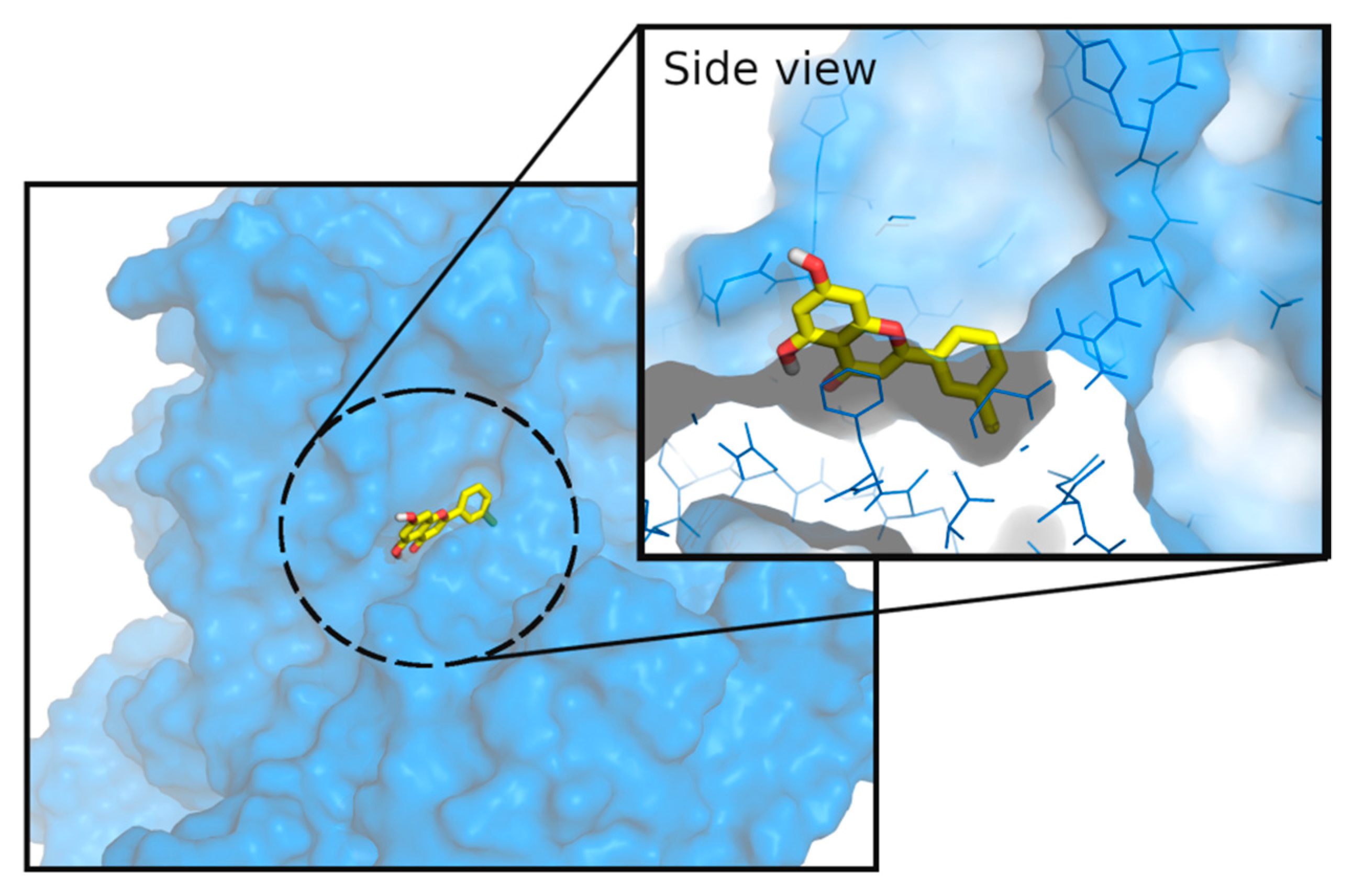
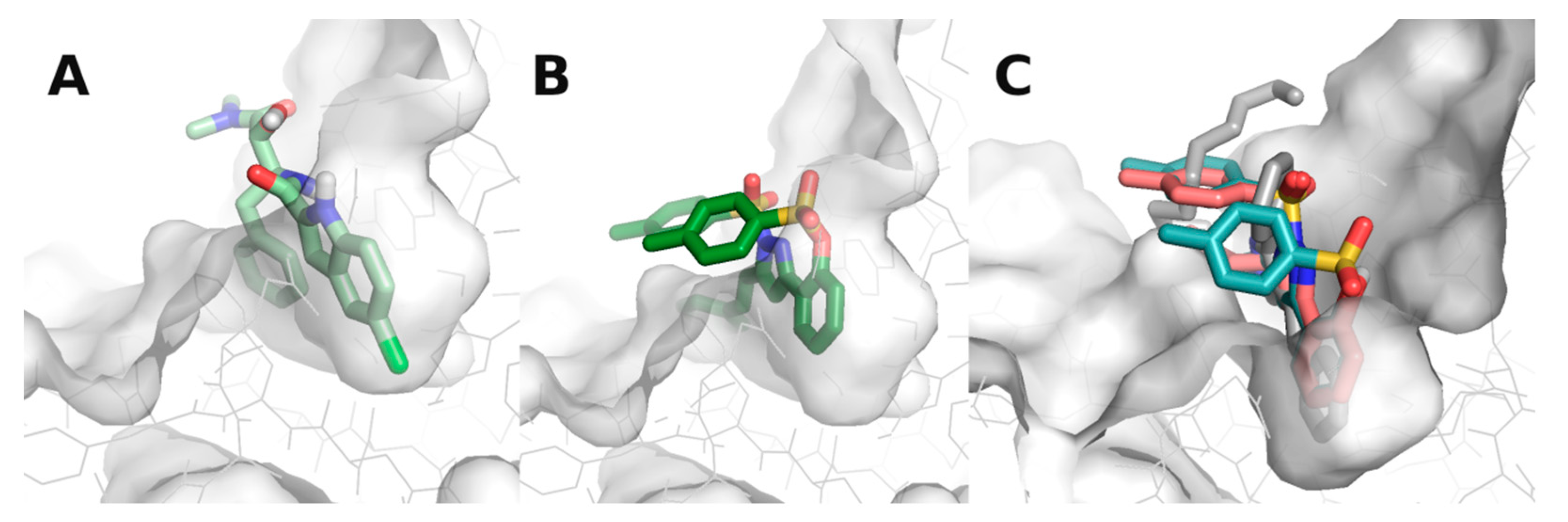
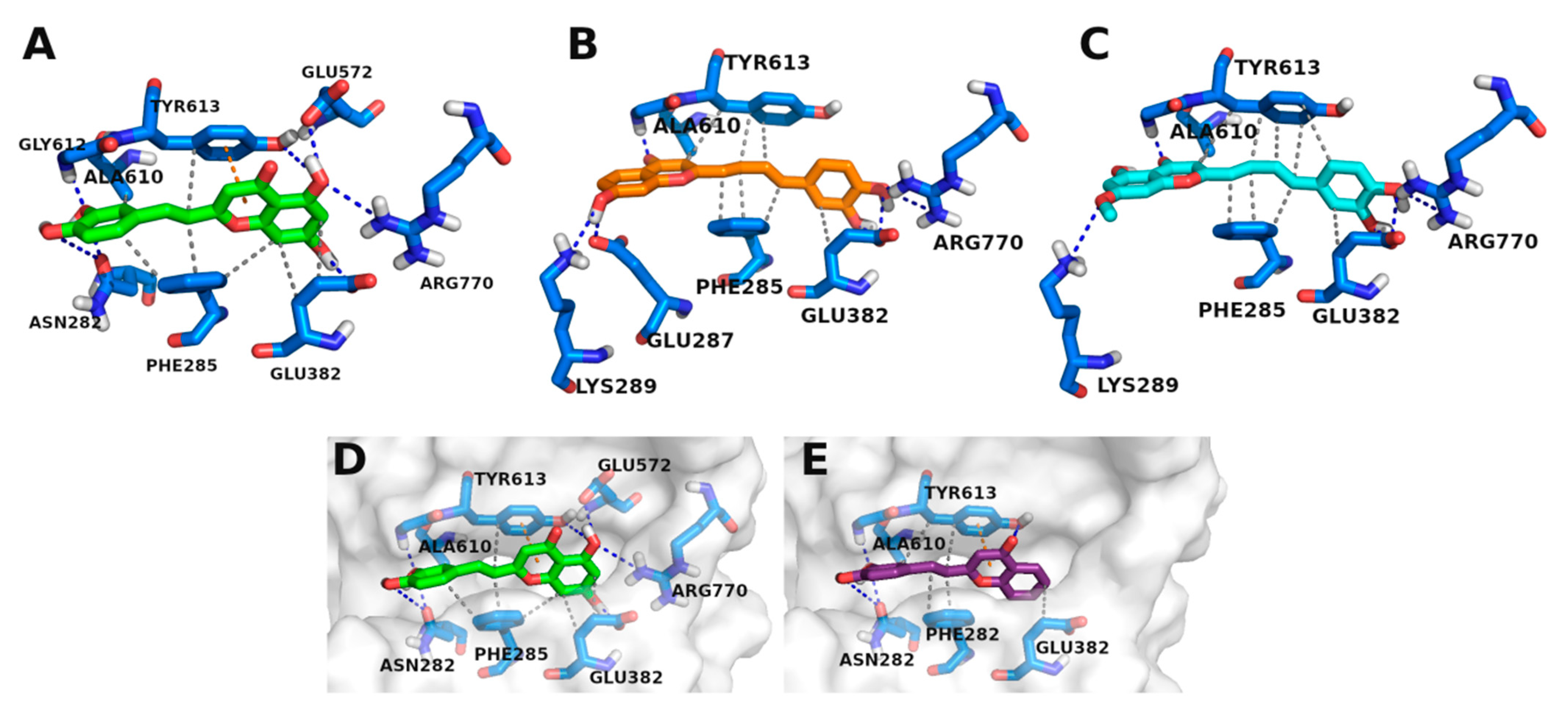
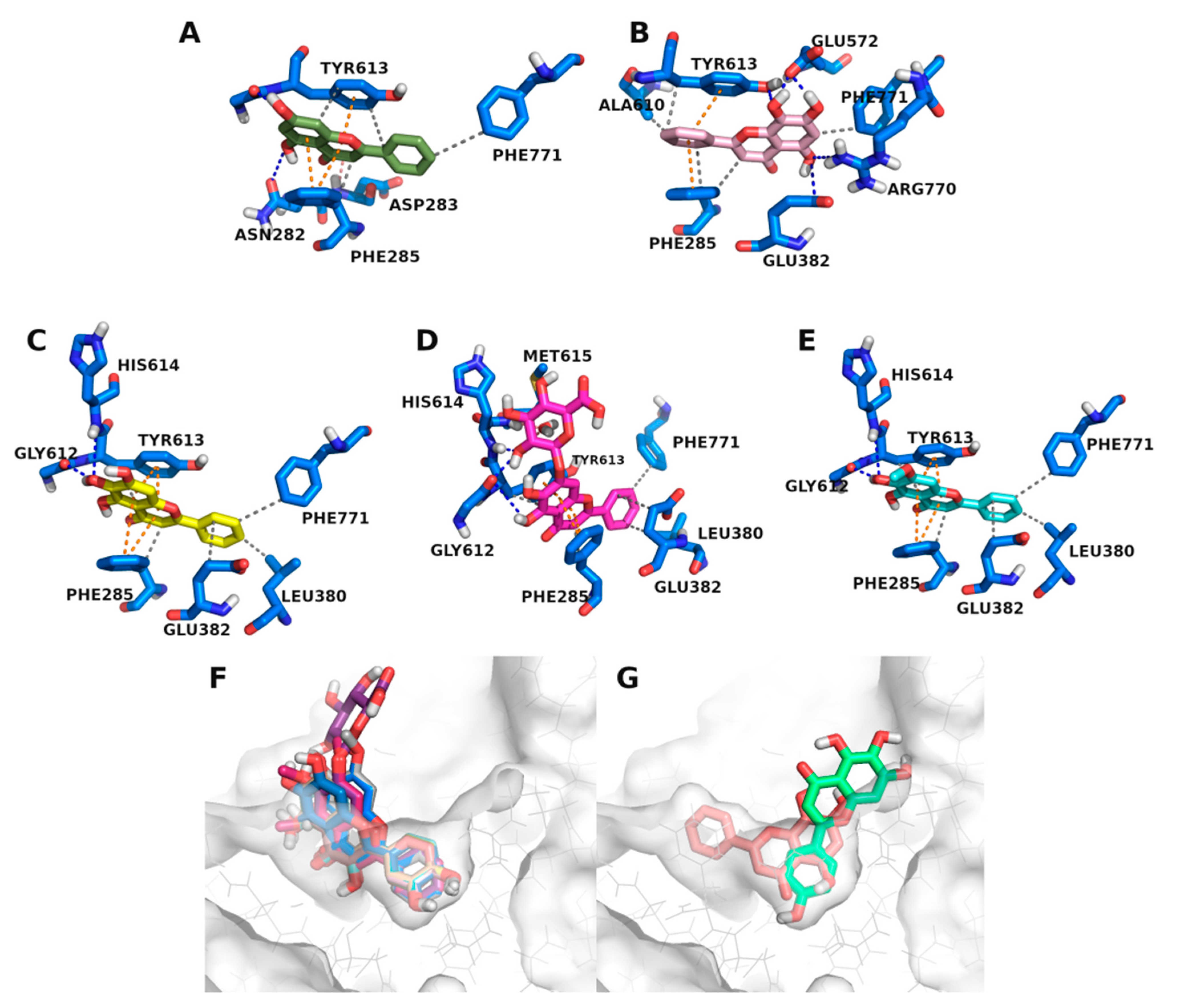
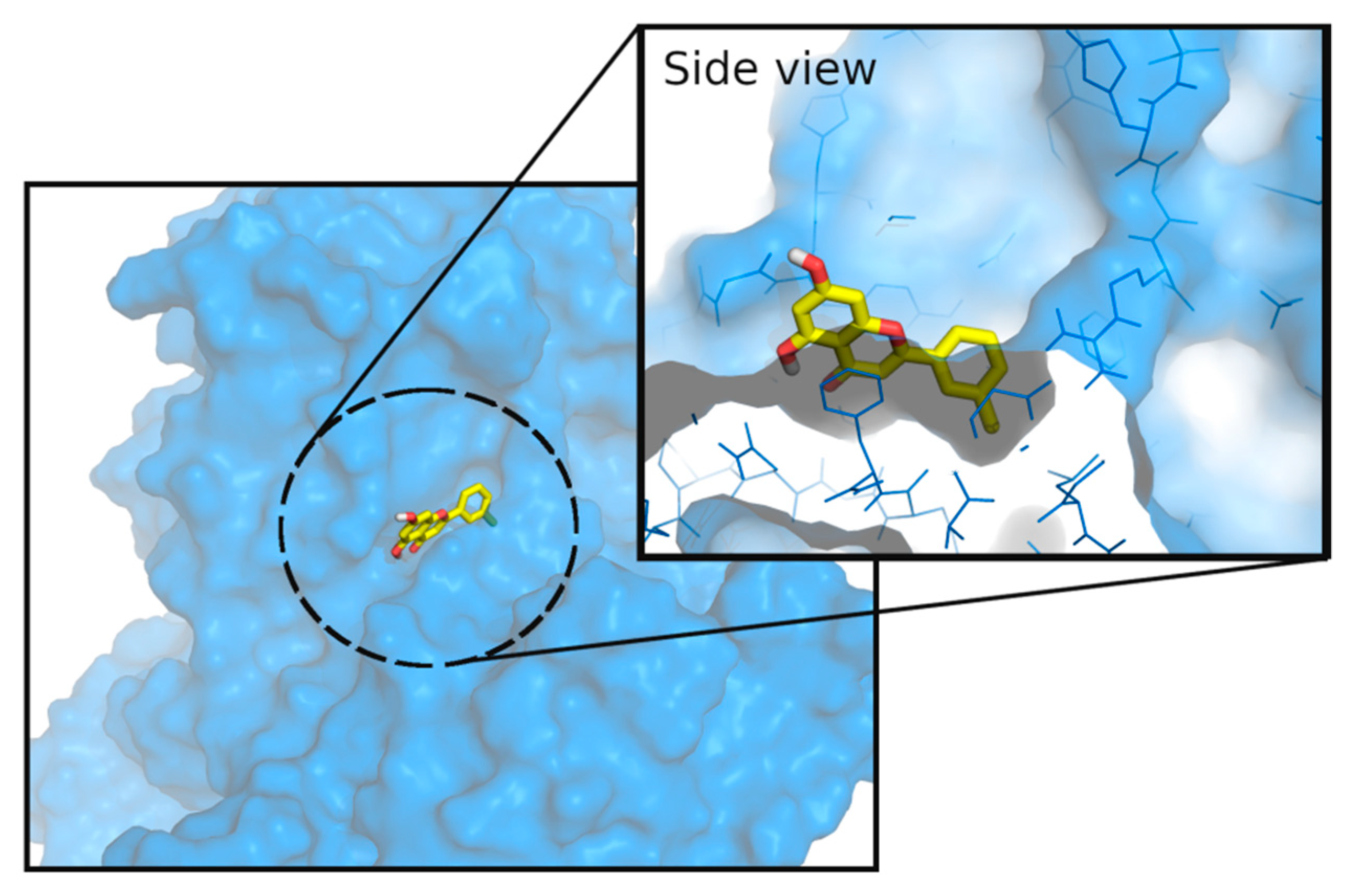
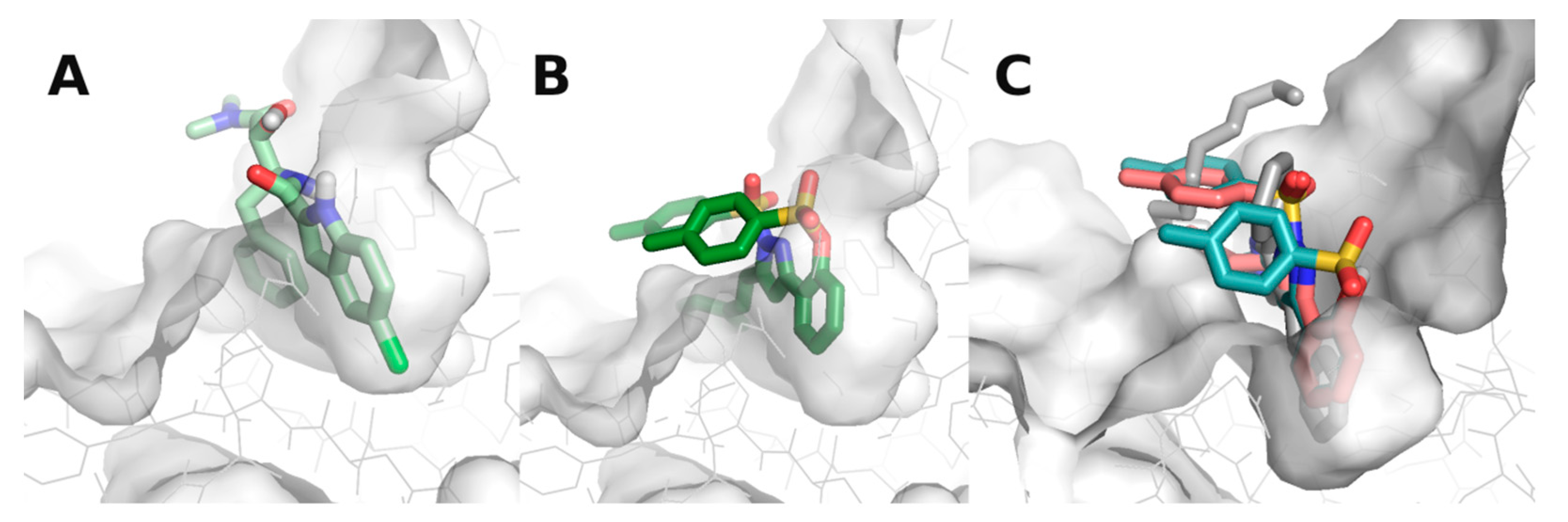
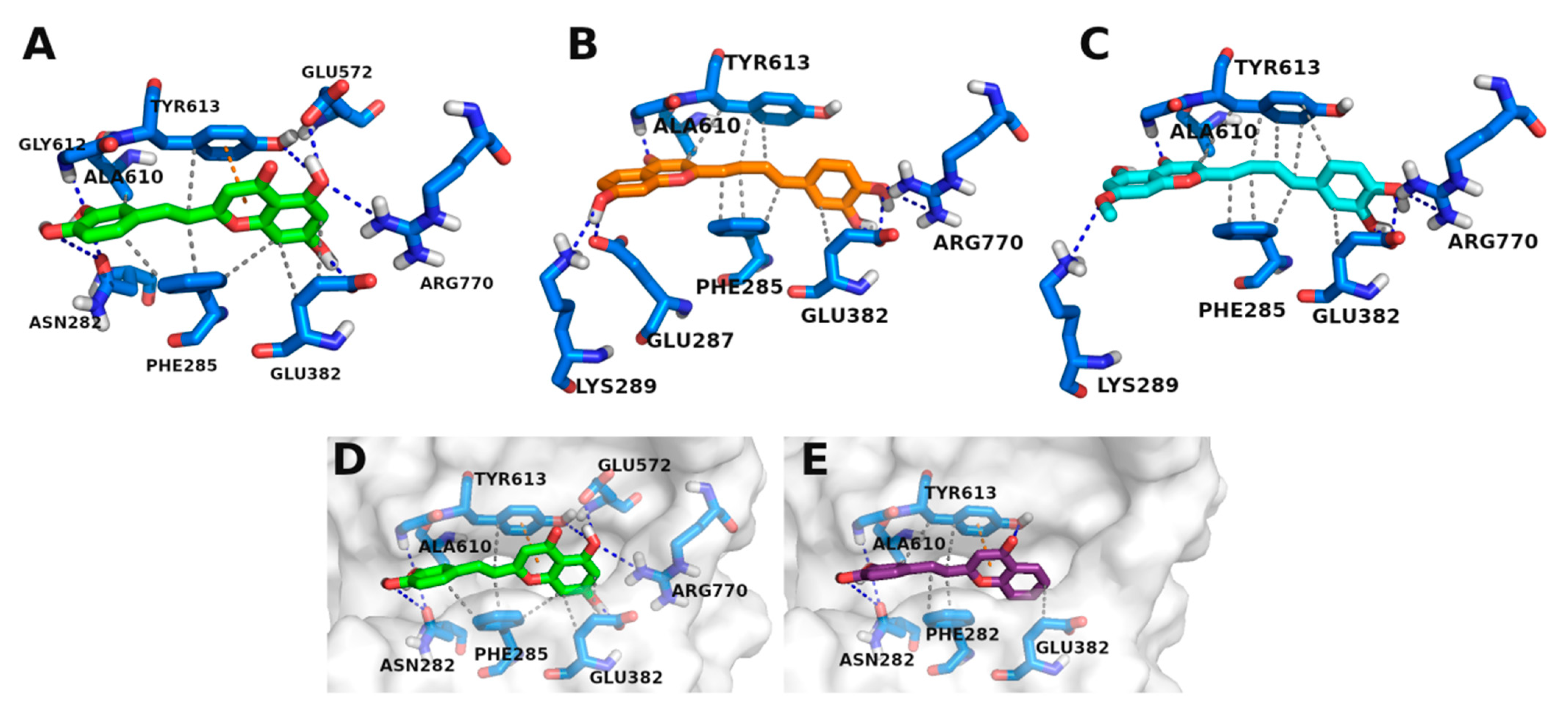
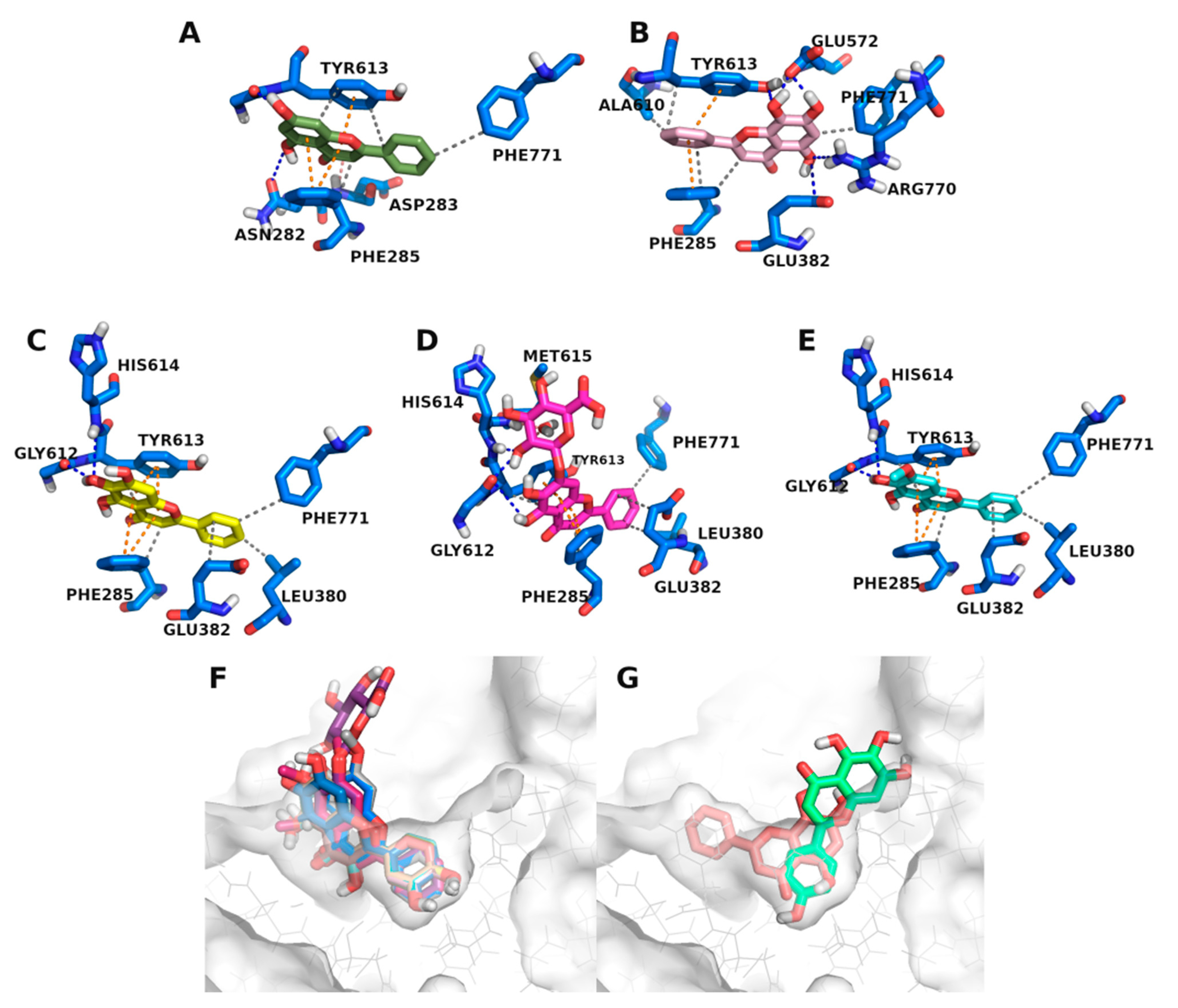
3.3. Molecular Docking Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.-F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artasensi, A.; Pedretti, A.; Vistoli, G.; Fumagalli, L. Type 2 diabetes mellitus: A review of multi-target drugs. Molecules 2020, 25, 1987. [Google Scholar] [CrossRef]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and molecular mechanisms of metformin action. Endocr. Rev. 2020, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Rines, A.K.; Sharabi, K.; Tavares, C.D.; Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.M.; Kantsadi, A.L.; Leonidas, D.D. Natural products and their derivatives as inhibitors of glycogen phosphorylase: Potential treatment for type 2 diabetes. Phytochem. Rev. 2014, 13, 471–498. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; González-Lucán, M.; Donapetry-García, C.; Fernández-Fernández, C.; Ameneiros-Rodríguez, E. Glycogen metabolism in humans. BBA Clin. 2016, 5, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brás, N.F.; Fernandes, P.A.; Ramos, M.J. Understanding the rate-limiting step of glycogenolysis by using QM/MM calculations on human glycogen phosphorylase. ChemMedChem 2018, 13, 1608–1616. [Google Scholar] [CrossRef]
- Gaboriaud-Kolar, N.; Skaltsounis, A.L. Glycogen phosphorylase inhibitors: A patent review (2008–2012). Expert Opin. Ther. Pat. 2013, 23, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Song, C.; Miao, G.; Zhao, L.; Yan, Z.; Li, J.; Wang, Y. Novel liver-targeted conjugates of glycogen phosphorylase inhibitor PSN-357 for the treatment of diabetes: Design, synthesis, pharmacokinetic and pharmacological evaluations. Sci. Rep. 2017, 7, 42251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, L.; Márton, J.; Vida, A.; Kis, G.; Bokor, É.; Kun, S.; Gönczi, M.; Docsa, T.; Tóth, A.; Antal, M.; et al. Glycogen phosphorylase inhibition improves beta cell function. Br. J. Pharmacol. 2018, 175, 301–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnier-Maréchal, M.; Vidal, S. Glycogen phosphorylase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Pat. 2016, 26, 199–212. [Google Scholar] [CrossRef]
- Servier Medical ART: SMART. Available online: http://smart.servier.com/ (accessed on 3 December 2021).
- Baker, D.J.; Greenhaff, P.L.; Timmons, J.A. Glycogen phosphorylase inhibition as a therapeutic target: A review of the recent patent literature. Expert Opin. Ther. Pat. 2006, 16, 459–466. [Google Scholar] [CrossRef]
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A valid scaffold in medicinal chemistry. Chem. Rev. 2014, 114, 4960–4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a privileged scaffold in drug discovery: Recent advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef]
- Sharma, S.K.; Kumar, S.; Chand, K.; Kathuria, A.; Gupta, A.; Jain, R. An update on natural occurrence and biological activity of chromones. Curr. Med. Chem. 2011, 18, 3825–3852. [Google Scholar] [CrossRef] [PubMed]
- Proença, C.; Ribeiro, D.; Freitas, M.; Carvalho, F.; Fernandes, E. A comprehensive review on the antidiabetic activity of flavonoids targeting PTP1B and DPP-4: A structure-activity relationship analysis. Crit. Rev. Food Sci. Nutr. 2021, 1–57. [Google Scholar] [CrossRef]
- Proença, C.; Ribeiro, D.; Freitas, M.; Fernandes, E. Flavonoids as potential agents in the management of type 2 diabetes through the modulation of α-amylase and α-glucosidase activity: A review. Crit. Rev. Food Sci. Nutr. 2021, 1–71. [Google Scholar] [CrossRef] [PubMed]
- Al-Ishaq, R.K.; Abotaleb, M.; Kubatka, P.; Kajo, K.; Büsselberg, D. Flavonoids and their anti-diabetic effects: Cellular mechanisms and effects to improve blood sugar levels. Biomolecules 2019, 9, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, S.; Lucas, M.; Ribeiro, D.; Corvo, M.L.; Fernandes, E.; Freitas, M. Nano-based drug delivery systems used as vehicles to enhance polyphenols therapeutic effect for diabetes mellitus treatment. Pharmacol. Res. 2021, 169, 105604. [Google Scholar] [CrossRef]
- Santos, C.M.M.; Silva, A.M.S. An overview of 2-styrylchromones: Natural occurrence, synthesis, reactivity and biological properties. Eur. J. Org. Chem. 2017, 2017, 3115–3133. [Google Scholar] [CrossRef]
- Proença, C.; Albuquerque, H.M.T.; Ribeiro, D.; Freitas, M.; Santos, C.M.M.; Silva, A.M.S.; Fernandes, E. Novel chromone and xanthone derivatives: Synthesis and ROS/RNS scavenging activities. Eur. J. Med. Chem. 2016, 115, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Faisal, M.; Saeed, A.; Hussain, S.; Dar, P.; Larik, F.A. Recent developments in synthetic chemistry and biological activities of pyrazole derivatives. J. Chem. Sci. 2019, 131, 70. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, I.C.S.; Silva, V.L.M.; Silva, A.M.S. Exploring the reactivity of (E)-3(5)-(2-hydroxyphenyl)-5(3)-styryl-1H-pyrazoles as dienes in the Diels–Alder reaction: A new synthesis of 1H-indazoles. Synlett 2015, 26, 945–949. [Google Scholar] [CrossRef]
- Gomes, A.; Neuwirth, O.; Freitas, M.; Couto, D.; Ribeiro, D.; Figueiredo, A.G.P.R.; Silva, A.M.; Seixas, R.S.; Pinto, D.C.; Tomé, A.C.; et al. Synthesis and antioxidant properties of new chromone derivatives. Bioorg. Med. Chem. 2009, 17, 7218–7226. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.M.M.; Silva, A.M.S.; Cavaleiro José, A.S. Synthesis of new hydroxy-2-styrylchromones. Eur. J. Org. Chem. 2003, 2003, 4575–4585. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Elguero, J. 3(5)-(2-Hydroxyphenyl)-5(3)-styrylpyrazoles: Synthesis and Diels−Alder transformations. Eur. J. Org. Chem. 2004, 2004, 4348–4356. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Elguero, J. Novel (E)- and (Z)-3(5)-(2-hydroxyphenyl)-4-styrylpyrazoles from (E)- and (Z)-3-styrylchromones: The unexpected case of (E)-3(5)-(2-hydroxyphenyl)-4-(4-nitrostyryl)pyrazoles. Tetrahedron Lett. 2007, 48, 3859–3862. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Elguero, J. Synthesis of (E)- and (Z)-3(5)-(2-hydroxyphenyl)-4-styrylpyrazoles. Monatsh. Chem. 2009, 140, 87–95. [Google Scholar] [CrossRef]
- Sousa, J.L.C.; Proença, C.; Freitas, M.; Fernandes, E.; Silva, A.M.S. New polyhydroxylated flavon-3-ols and 3-hydroxy-2-styrylchromones: Synthesis and ROS/RNS scavenging activities. Eur. J. Med. Chem. 2016, 119, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Lucas, M.; Araújo, A.N.; Corvo, M.L.; Fernandes, E.; Freitas, M. Optimization and validation of an in vitro standardized glycogen phosphorylase activity assay. Molecules 2021, 26, 4635. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE), 2020; Chemical Computing Group ULC: Montreal, QC, Canada, 2021. [Google Scholar]
- LeDock. LePhar 2021. Available online: http://www.lephar.com/download.htm (accessed on 18 April 2021).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Chetter, B.A.; Kyriakis, E.; Barr, D.; Karra, A.G.; Katsidou, E.; Koulas, S.M.; Skamnaki, V.T.; Snape, T.J.; Psarra, A.-M.G.; Leonidas, D.D.; et al. Synthetic flavonoid derivatives targeting the glycogen phosphorylase inhibitor site: QM/MM-PBSA motivated synthesis of substituted 5,7-dihydroxyflavones, crystallography, in vitro kinetics and ex-vivo cellular experiments reveal novel potent inhibitors. Bioorg. Chem. 2020, 102, 104003. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 1.8.4.0. Available online: http://www.pymol.org (accessed on 4 January 2022).
- Henke, B.R. Chapter 12: Inhibition of glycogen phosphorylase as a strategy for the treatment of type 2 diabetes. In New Therapeutic Strategies for Type 2 Diabetes: Small Molecule Approaches; The Royal Society of Chemistry: Bengaluru, India, 2012; pp. 324–365. [Google Scholar]
- Szabó, K.; Kandra, L.; Gyémánt, G. Studies on the reversible enzyme reaction of rabbit muscle glycogen phosphorylase b using isothermal titration calorimetry. Carbohydr. Res. 2019, 477, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Kattimani, P.P.; Somagond, S.M.; Bayannavar, P.K.; Kamble, R.R.; Bijjaragi, S.C.; Hunnur, R.K.; Joshi, S.D. Novel 5-(1-aryl-1H-pyrazol-3-yl)-1H-tetrazoles as glycogen phosphorylase inhibitors: An in vivo antihyperglycemic activity study. Drug Dev. Res. 2020, 81, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Brás, N.F.; Neves, R.P.P.; Lopes, F.A.A.; Correia, M.A.S.; Palma, A.S.; Sousa, S.F.; Ramos, M.J. Combined in silico and in vitro studies to identify novel antidiabetic flavonoids targeting glycogen phosphorylase. Bioorg. Chem. 2021, 108, 104552. [Google Scholar] [CrossRef]
- Jakobs, S.; Fridrich, D.; Hofem, S.; Pahlke, G.; Eisenbrand, G. Natural flavonoids are potent inhibitors of glycogen phosphorylase. Mol. Nutr. Food Res. 2006, 50, 52–57. [Google Scholar] [CrossRef]
- Kato, A.; Nasu, N.; Takebayashi, K.; Adachi, I.; Minami, Y.; Sanae, F.; Asano, N.; Watson, A.A.; Nash, R.J. Structure-activity relationships of flavonoids as potential inhibitors of glycogen phosphorylase. J. Agric Food Chem. 2008, 56, 4469–4473. [Google Scholar] [CrossRef]
- Oikonomakos, N.G.; Schnier, J.B.; Zographos, S.E.; Skamnaki, V.T.; Tsitsanou, K.E.; Johnson, L.N. Flavopiridol inhibits glycogen phosphorylase by binding at the inhibitor site. J. Biol. Chem. 2000, 275, 34566–34573. [Google Scholar] [CrossRef] [Green Version]
- Tsitsanou, K.E.; Hayes, J.M.; Keramioti, M.; Mamais, M.; Oikonomakos, N.G.; Kato, A.; Leonidas, D.; Zographos, S.E. Sourcing the affinity of flavonoids for the glycogen phosphorylase inhibitor site via crystallography, kinetics and QM/MM-PBSA binding studies: Comparison of chrysin and flavopiridol. Food Chem. Toxicol. 2013, 61, 14–27. [Google Scholar] [CrossRef]
- Johnson-Rabbett, B.; Seaquist, E.R. Hypoglycemia in diabetes: The dark side of diabetes treatment. A patient-centered review. J. Diabetes 2019, 11, 711–718. [Google Scholar] [CrossRef]
- Martin, W.H.; Hoover, D.J.; Armento, S.J.; Stock, I.A.; McPherson, R.K.; Danley, D.E.; Stevenson, R.W.; Barrett, E.J.; Treadway, J.L. Discovery of a human liver glycogen phosphorylase inhibitor that lowers blood glucose in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 1776–1781. [Google Scholar] [CrossRef] [Green Version]
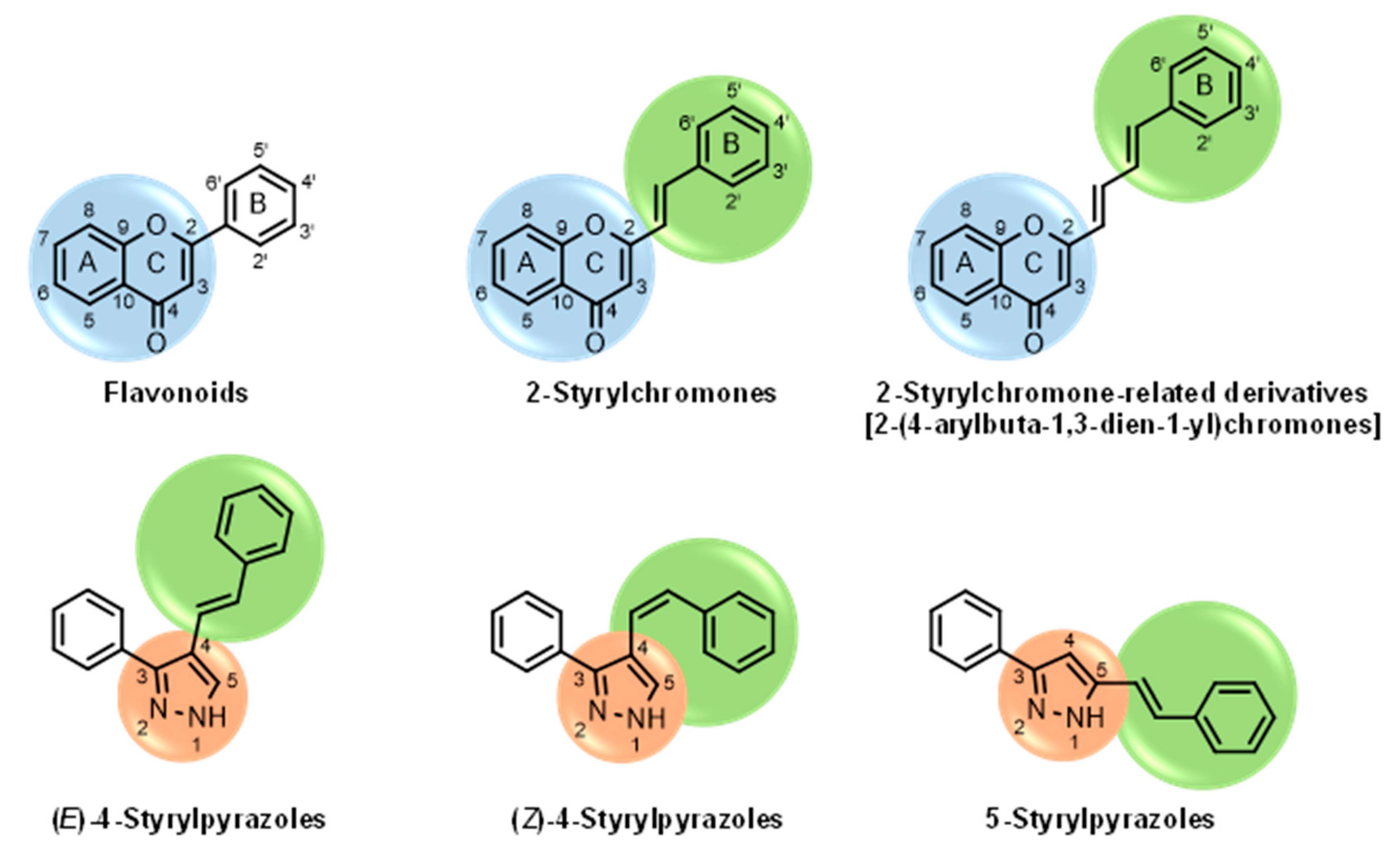

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4-Styrylpyrazoles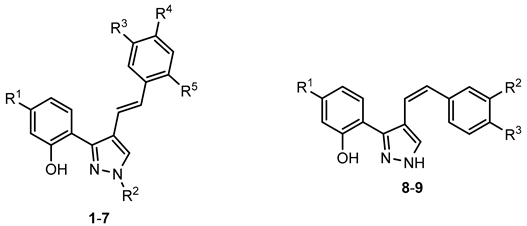 (1–9) | ||||||||
| Compounds | R1 | R2 | R3 | R4 | R5 | GPa inhibitory activity (%) or IC50 value | ||
| 1 | - | - | - | - | - | <30% 50 μM | ||
| 2 | - | - | - | Cl | - | <30% 50 μM | ||
| 3 | - | - | - | OCH3 | - | <30% 50 μM | ||
| 4 | - | - | - | - | CF3 | <30% 50 μM | ||
| 5 | - | (CH2)9CH3 | - | - | - | <30% 50 μM | ||
| 6 | - | (CH2)11CH3 | - | - | - | <30% 50 μM | ||
| 7 | - | (CH2)11CH3 | - | Cl | - | <30% 50 μM | ||
| 8 | - | NO2 | - | - | - | <30% 50 μM | ||
| 9 | OCH3 | - | NO2 | - | - | <30% 50 μM | ||
5-Styrylpyrazoles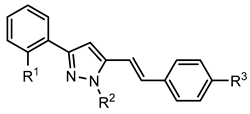 (10–15) | ||||||||
| Compounds | R1 | R2 | R3 | GPa inhibitory activity (%) or IC50 value | ||||
| 10 | OH | - | - | <30% 50 μM | ||||
| 11 | OH | - | Cl | <30% 50 μM | ||||
| 12 | OH |  | OCH3 | <30% 50 μM | ||||
| 13 | OH |  | OCH3 | <30% 50 μM | ||||
| 14 | 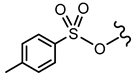 | 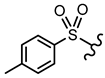 | - | <30% 50 μM | ||||
| 15 | 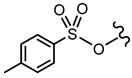 | 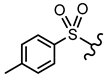 | OCH3 | <30% 50 μM | ||||
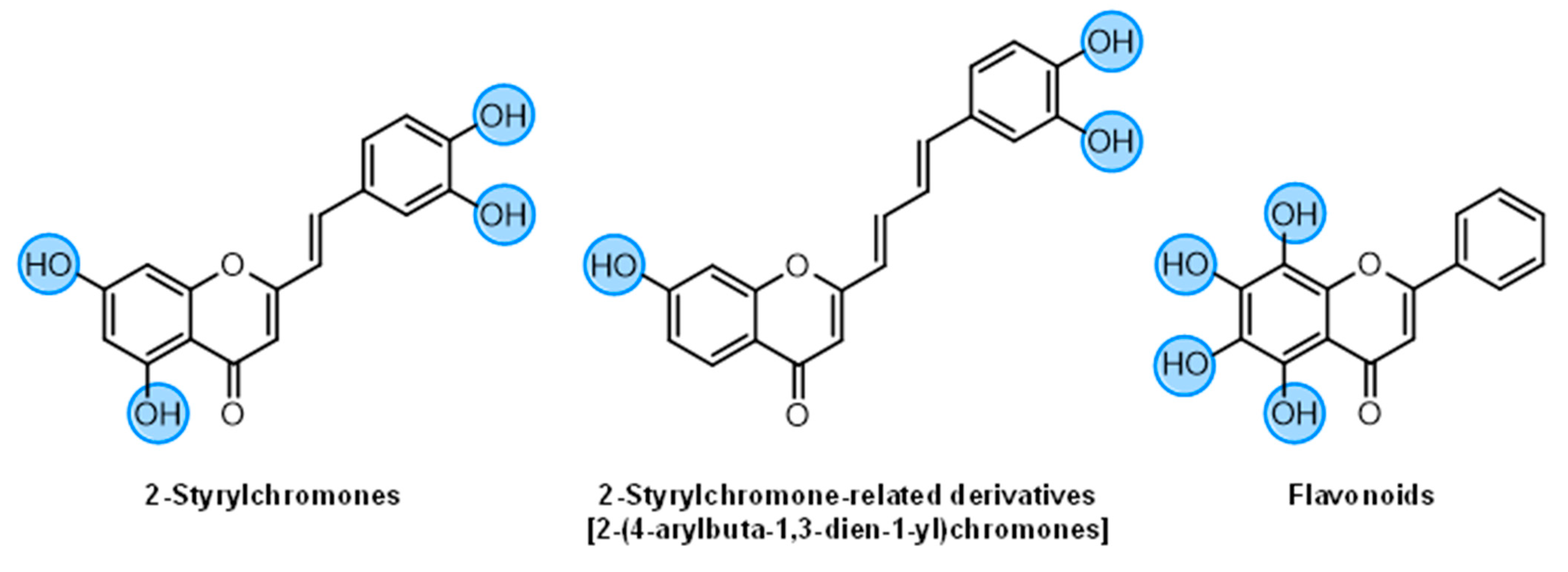
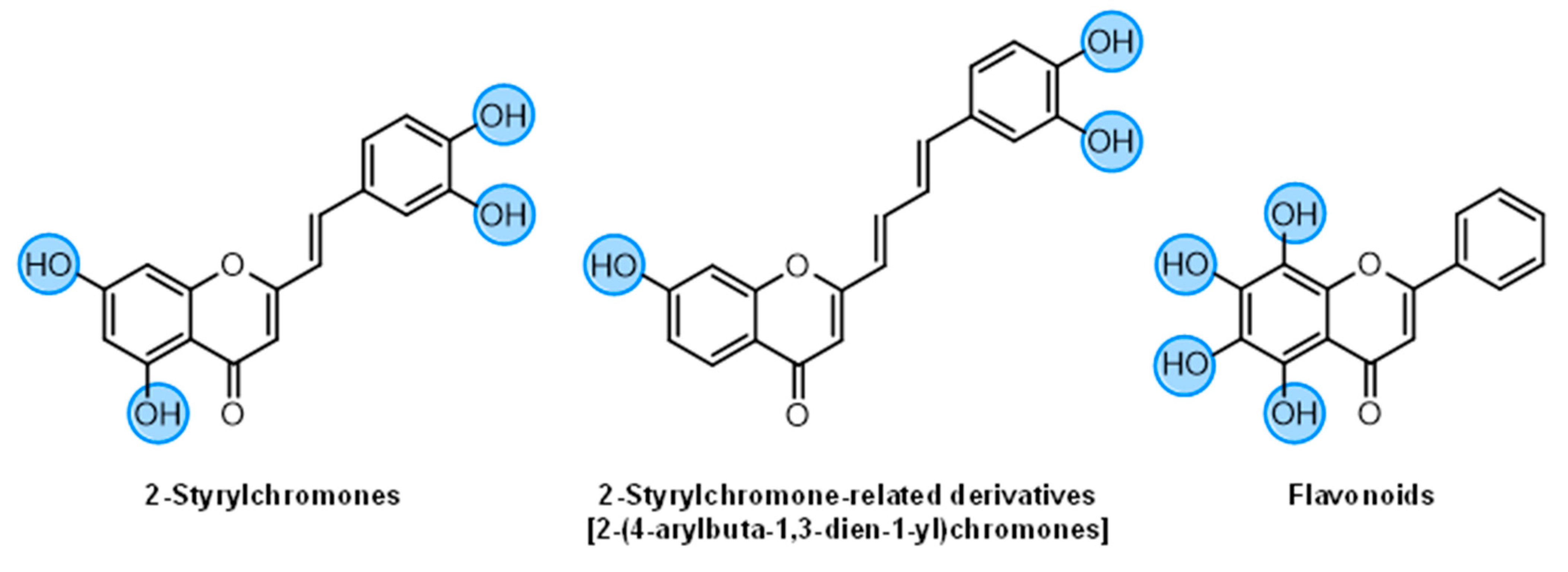
2-Styrylchromones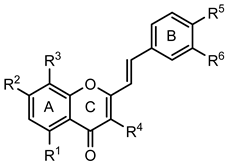 (16–29) | ||||||||
| Compounds | R1 | R2 | R3 | R4 | R5 | R6 | GPa inhibitory activity (%) or IC50 value | |
| 16 | - | OH | - | - | - | - | <30% 50 μM | |
| 17 | - | OH | - | - | OH | - | <30% 50 μM | |
| 18 | - | - | - | - | OH | OH | <30% 50 μM | |
| 19 | - | OH | - | - | OH | OH | <30% 50 μM | |
| 20 | OH | - | - | - | OH | OH | <30% 50 μM | |
| 21 | OH | OH | - | - | - | - | 30.1 ± 6.1 % 50 μM | |
| 22 | OH | OH | - | - | OH | - | 43.2 ± 5.1 % 50 μM | |
| 23 | OH | OH | - | - | OH | OH | 31.7 ± 2.4 µM (b) | |
| 24 | - | OH | OH | OH | OH | OH | 33.7 ± 6.5% 25 μM | |
| 25 | OCH3 | OCH3 | - | - | OH | OH | <30% 50 μM | |
| 26 | OCH3 | OCH3 | - | - | OCH3 | OCH3 | 42.2 ± 5.6% 50 μM | |
| 27 | - | OCH3 | OCH3 | - | OCH3 | OCH3 | <30% 50 μM | |
| 28 | - | OCH3 | - | - | OCH3 | OCH3 | <30% 50 μM | |
| 29 | OCH3 | - | - | - | OCH3 | OCH3 | <30% 50 μM | |
| 2-Styrylchromone-related derivatives [2-(4-arylbuta-1,3-dien-1-yl)chromones] 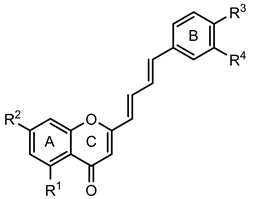 (30–35) | ||||||||
| Compounds | R1 | R2 | R3 | R4 | GPa inhibitory activity (%) or IC50 value | |||
| 30 | - | - | OH | OH | <30% 50 μM | |||
| 31 | OH | - | OH | OH | <30% 50 μM | |||
| 32 | - | OH | OH | OH | 16.7 ± 1.5 µM (c) | |||
| 33 | OH | OCH3 | OH | OH | 15.9 ± 1.1 µM (c) | |||
| 34 | - | OCH3 | OH | OH | 51.9 ± 1.8% 50 μM | |||
| 35 | - | OCH3 | OCH3 | OCH3 | 35.8 ± 4.6% 50 μM | |||
Flavonoids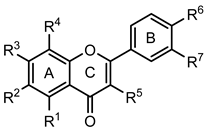 (36–52) | ||||||||
| Compounds | R1 | R2 | R3 | R4 | R5 | R6 | R7 | GPa inhibitory activity (%) or IC50 value |
| 36 | - | - | OH | - | - | - | - | <30% 50 μM |
| 37 | - | - | OH | - | - | OH | - | <30% 50 μM |
| 38 Chrysin | OH | - | OH | - | - | - | - | 19.3 ± 1.5 µM (c) |
| 39 | - | - | - | - | OH | OH | <30% 50 μM | |
| 40 | - | - | OH | - | - | OH | OH | <30% 50 μM |
| 41 | OH | - | - | - | OH | OH | <30% 50 μM | |
| 42 Apigenin | OH | - | OH | - | - | OH | - | 35.7 ± 2.1% 50 μM |
| 43 Luteolin | OH | - | OH | - | - | OH | OH | <30% 50 μM |
| 44 Galangin | OH | - | OH | - | OH | - | - | <30% 50 μM |
| 45 Quercetin | OH | - | OH | - | OH | OH | OH | <30% 50 μM |
| 46 Norwogonin | OH | - | OH | OH | - | - | - | 13.2 ± 1.4 µM (c) |
| 47 Baicalein | OH | OH | OH | - | - | - | - | 23.5 ± 2.9 µM (b, c) |
| 48 Scutellarein | OH | OH | OH | - | - | OH | - | <30% 50 μM |
| 49 Baicalin | OH | OH | O-Glu curoni de | - | - | - | - | 20.5 ± 2.5 µM (c) |
| 50 Quercetagetin | OH | OH | OH | - | OH | OH | OH | <30% 50 μM |
| 51 Baicalein-7-methylether | OH | OH | OCH3 | - | - | - | - | 22.6 ± 0.4 µM (b, c) |
| 52 Baicalein-5,6,7-trimethylether | OCH3 | OCH3 | OCH3 | - | - | - | - | <30% 50 μM |
| Positive control CP-91149 | 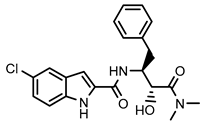 | 0.58 ± 0.09 μM (a) | ||||||
| 0 mM of Glucose | 5 mM of Glucose | 10 mM of Glucose | |
|---|---|---|---|
| GPa Inhibitory Activity (%) or IC50 Value | |||
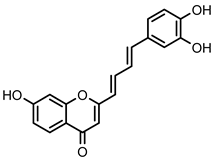 2-Styrylchromone-related derivative 32 | 16.7 ± 1.5 (b,c) | 13.4 ± 1.6 (b) | 10.8 ± 1.2 (c) |
 2-Styrylchromone-related derivative 33 | 15.9 ± 1.1 (b,c) | 53.9 ± 2.5% 40 μM | <30% 40 μM |
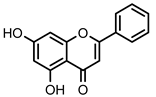 Flavonoid 38 Chrysin | 19.3 ± 1.5 (b) | 8.8 ± 0.8 (b) | 5.2 ± 0.3 (b) |
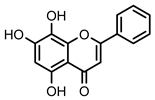 Flavonoid 46 Norwogonin | 13.2 ± 1.4 (c) | 8.9 ± 1.0 (b) | 3.7 ± 0.5 (a; b) |
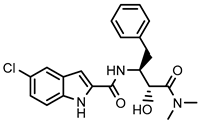 Positive control CP-91149 | 0.58 ± 0.09 (a) | 0.39 ± 0.05 (a) | 0.22 ± 0.04 (a) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, S.; Aniceto, N.; Guedes, R.C.; Albuquerque, H.M.T.; Silva, V.L.M.; Silva, A.M.S.; Corvo, M.L.; Fernandes, E.; Freitas, M. An In Silico and an In Vitro Inhibition Analysis of Glycogen Phosphorylase by Flavonoids, Styrylchromones, and Pyrazoles. Nutrients 2022, 14, 306. https://doi.org/10.3390/nu14020306
Rocha S, Aniceto N, Guedes RC, Albuquerque HMT, Silva VLM, Silva AMS, Corvo ML, Fernandes E, Freitas M. An In Silico and an In Vitro Inhibition Analysis of Glycogen Phosphorylase by Flavonoids, Styrylchromones, and Pyrazoles. Nutrients. 2022; 14(2):306. https://doi.org/10.3390/nu14020306
Chicago/Turabian StyleRocha, Sónia, Natália Aniceto, Rita C. Guedes, Hélio M. T. Albuquerque, Vera L. M. Silva, Artur M. S. Silva, Maria Luísa Corvo, Eduarda Fernandes, and Marisa Freitas. 2022. "An In Silico and an In Vitro Inhibition Analysis of Glycogen Phosphorylase by Flavonoids, Styrylchromones, and Pyrazoles" Nutrients 14, no. 2: 306. https://doi.org/10.3390/nu14020306
APA StyleRocha, S., Aniceto, N., Guedes, R. C., Albuquerque, H. M. T., Silva, V. L. M., Silva, A. M. S., Corvo, M. L., Fernandes, E., & Freitas, M. (2022). An In Silico and an In Vitro Inhibition Analysis of Glycogen Phosphorylase by Flavonoids, Styrylchromones, and Pyrazoles. Nutrients, 14(2), 306. https://doi.org/10.3390/nu14020306