
Clostridium butyricum Potentially Improves Immunity and Nutrition through Alteration of the Microbiota and Metabolism of Elderly People with Malnutrition in Long-Term Care


Abstract
:1. Introduction
2. Materials and Methods
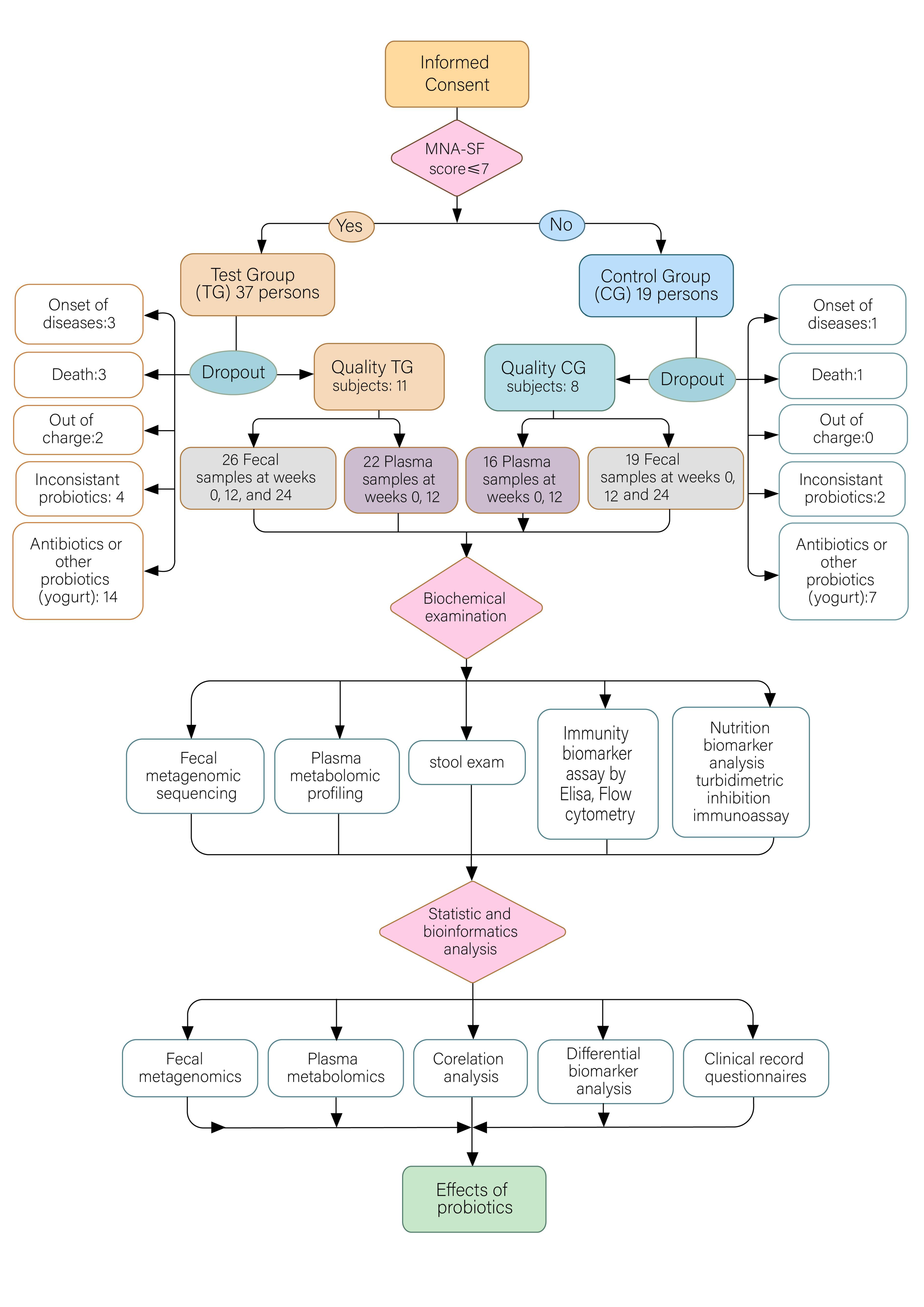
2.1. Study Design and Enrolled Subjects
2.2. Metagenomic Sequencing, Assembly, and Functional Annotation
2.3. Phylogenetic Analysis, Species, and KEGG Ortholog (KO) Abundance Profiling
2.4. Plasma Metabolome Quantification
2.5. Differential Metagenomic Analysis
2.6. Cytokines and Biomarker Analysis
3. Results
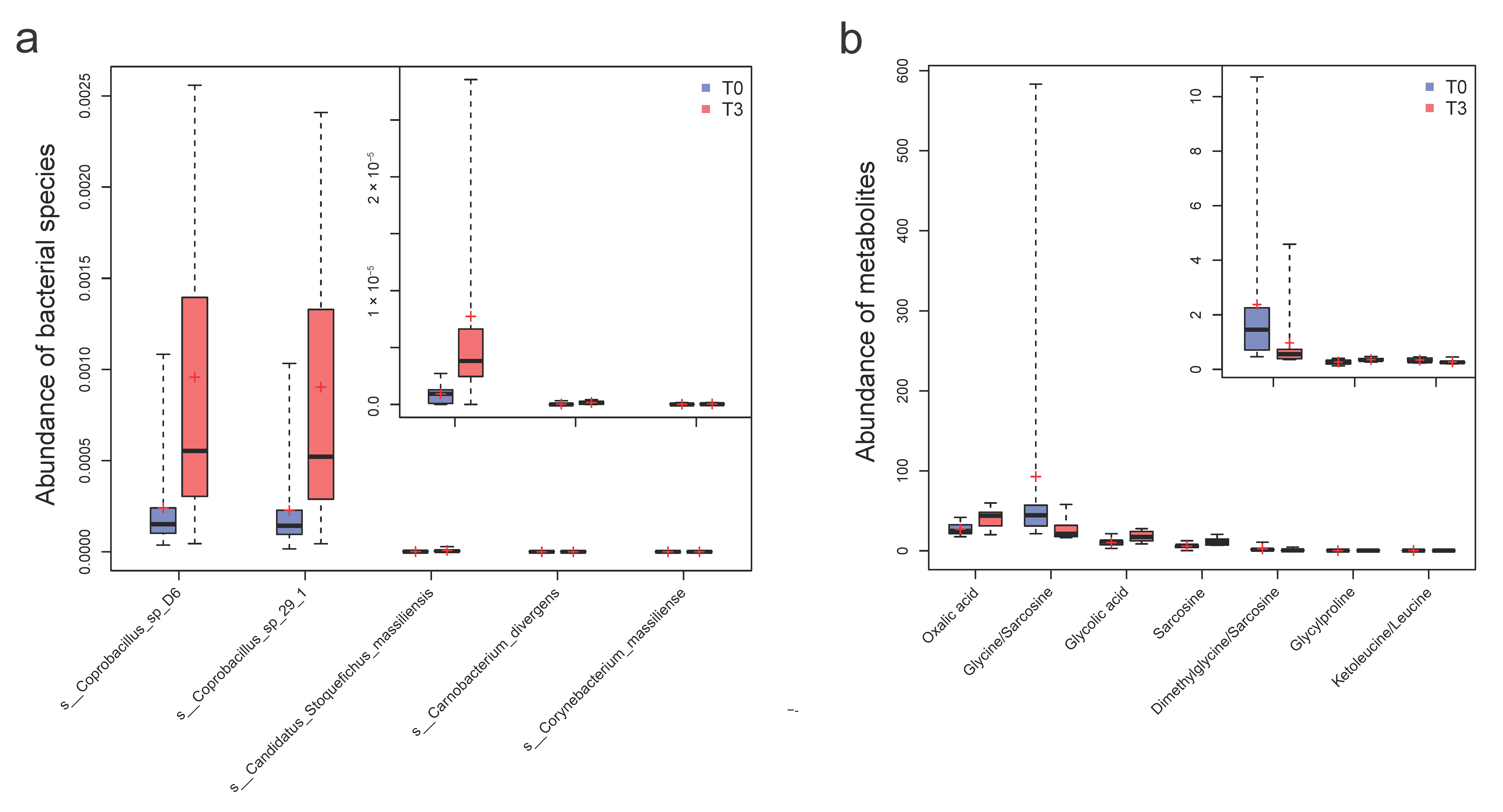
3.1. Metagenomic Analysis Reveals an Altered Human Intestinal Microbiome and Augmented Beneficial Microbes with Probiotic Intervention
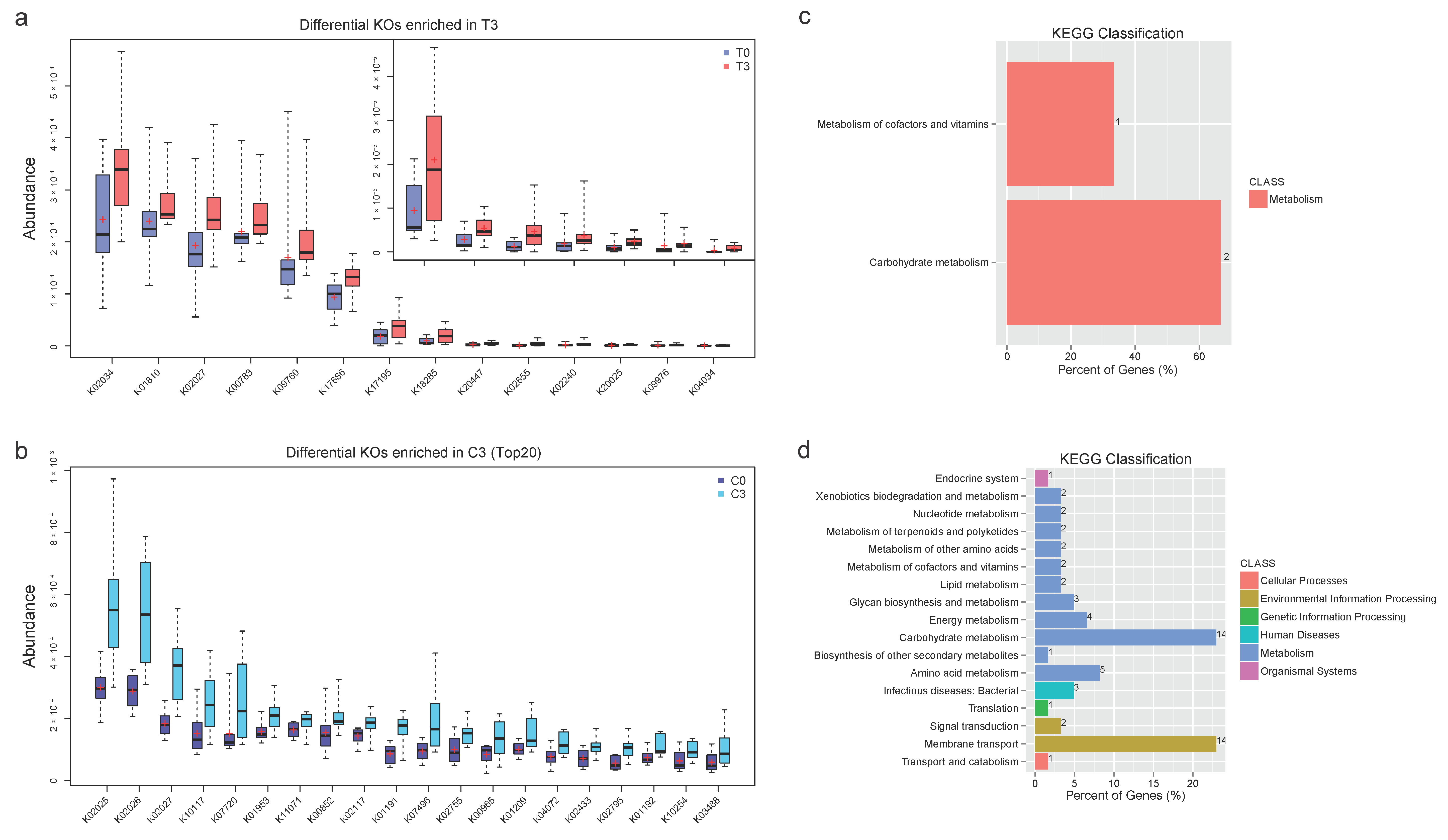
3.2. Functional Annotation and Analysis Revealed Concentrated Differential KOs and Active Microbial Activities with Probiotics Compared with Scattered Changes in Control
3.3. Quantitative Plasma Metabolomic Changes Involved in the Glycine and One-Carbon Metabolism with Probiotics, Compared with Bile Acid and Lipid Metabolism in the Control
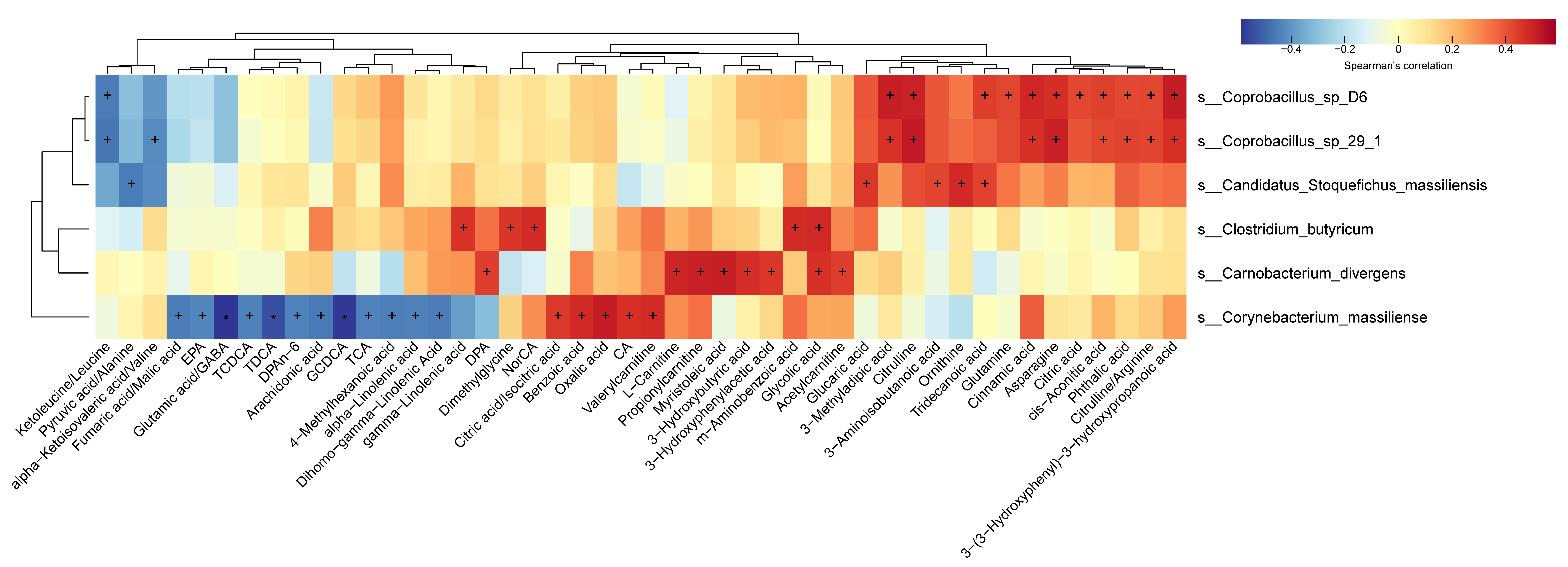
3.4. Correlation Analysis between Intestinal Differential Microbes and the Plasma Metabolome Revealed Coordinated Changes in Generating SCFAs, MUFA, LUFA, and Efficient Energy Production in the TG
3.5. Significant KOs Were Correlated with Multiple Plasma Metabolites and Species
3.6. Biomarker Assessment Indicated Improvement in Immunity and Nutrition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Xie, G.; Wang, L.; Chen, T.; Zhou, K.; Zhang, Z.; Li, J.; Sun, B.; Guo, Y.; Wang, X.; Wang, Y.; et al. A Metabolite Array Technology for Precision Medicine. Anal. Chem. 2021, 93, 5709–5717. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Gingrich, A.; Volkert, D.; Kiesswetter, E.; Thomanek, M.; Bach, S.; Sieber, C.C.; Zopf, Y. Prevalence and overlap of sarcopenia, frailty, cachexia and malnutrition in older medical inpatients. BMC Geriatr. 2019, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Garay, J.A.R.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Kumar, M.; Babaei, P.; Ji, B.; Nielsen, J. Human gut microbiota and healthy aging: Recent developments and future prospective. Nutr. Healthy Aging 2016, 4, 3–16. [Google Scholar] [CrossRef]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, R. Immunosenescence and nutrition: Reviewing clinical evidence on pre-, pro- and synbiotics in aging. J. Allergy Immunol. 2017, 1, 1–7. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; MacPherson, A.J. Interactions Between the Microbiota and the Immune System. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Malaguarnera, G.; Leggio, F.; Vacante, M.; Motta, M.; Giordano, M.; Biondi, A.; Basile, F.; Mastrojeni, S.; Mistretta, A.; Malaguarnera, M.; et al. Probiotics in the gastrointestinal diseases of the elderly. J. Nutr. Health Aging 2012, 16, 402–410. [Google Scholar] [CrossRef]
- Tomita, Y.; Ikeda, T.; Sakata, S.; Saruwatari, K.; Sato, R.; Iyama, S.; Jodai, T.; Akaike, K.; Ishizuka, S.; Saeki, S.; et al. Association of Probiotic Clostridium butyricum Therapy with Survival and Response to Immune Checkpoint Blockade in Patients with Lung Cancer. Cancer Immunol. Res. 2020, 8, 1236–1242. [Google Scholar] [CrossRef]
- Dizman, N.; Meza, L.; Bergerot, P.; Alcantara, M.; Dorff, T.; Lyou, Y.; Frankel, P.; Cui, Y.; Mira, V.; Llamas, M.; et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carci-noma: A randomized phase 1 trial. Nat. Med. 2022, 28, 704–712. [Google Scholar] [CrossRef]
- Mekonnen, S.A.; Merenstein, D.; Fraser, C.M.; Marco, M.L. Molecular mechanisms of probiotic prevention of antibiotic-associated diarrhea. Curr. Opin. Biotechnol. 2020, 61, 226–234. [Google Scholar] [CrossRef]
- Gao, R.; Zhang, X.; Huang, L.; Shen, R.; Qin, H. Gut Microbiota Alteration After Long-Term Consumption of Probiotics in the Elderly. Probiotics Antimicrob. Proteins 2019, 11, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Gareau, M.G.; Sherman, P.M.; Walker, W.A. Probiotics and the gut microbiota in intestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Butel, M.J. Probiotics, gut microbiota and health. Med. Mal. Infect. 2014, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; Zhou, J.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; De Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef]
- Ariyoshi, T.; Hagihara, M.; Eguchi, S.; Fukuda, A.; Iwasaki, K.; Oka, K.; Takahashi, M.; Yamagishi, Y.; Mikamo, H. Clostridium butyricum MIYAIRI 588-Induced Protectin D1 Has an Anti-inflammatory Effect on Antibiotic-Induced Intestinal Disorder. Front. Microbiol. 2020, 11, 587725. [Google Scholar] [CrossRef]
- Bolte, L.A.; Vich Vila, A.; Imhann, F.; Collij, V.; Gacesa, R.; Peters, V.; Wijmenga, C.; Kurilshikov, A.; Campmans-Kuijpers, M.J.E.; Fu, J.; et al. Long-term dietary patterns are associated with pro-inflammatory and anti-inflammatory features of the gut microbiome. Gut 2021, 70, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wu, C.; Zhu, Q.; Gao, R.; Lu, J.; Valles-Colomer, M.; Zhu, J.; Yin, F.; Huang, L.; Ding, L.; et al. Metagenomic and metabolomic remodeling in nonagenarians and centenarians and its association with genetic and socioeconomic factors. Nat. Aging 2022, 2, 438–452. [Google Scholar] [CrossRef]
- Leisner, J.J.; Laursen, B.G.; Prévost, H.; Drider, D.; Dalgaard, P. Carnobacterium: Positive and negative effects in the environment and in foods. FEMS Microbiol. Rev. 2007, 31, 592–613. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Zhu, X.; Qin, J.; Tan, C.; Ning, K. The seasonal changes of the gut microbiome of the population living in traditional lifestyles are represented by characteristic species-level and functional-level SNP enrichment patterns. BMC Genom. 2021, 22, 83. [Google Scholar] [CrossRef]
- Regan, M.D.; Chiang, E.; Liu, Y.; Tonelli, M.; Verdoorn, K.M.; Gugel, S.R.; Suen, G.; Carey, H.V.; Assadi-Porter, F.M. Nitrogen recycling via gut symbionts increases in ground squirrels over the hibernation season. Science 2022, 375, 460–463. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Abratt, V.R.; Reid, S.J. Oxalate-Degrading Bacteria of the Human Gut as Probiotics in the Management of Kidney Stone Disease. Adv. Appl. Microbiol. 2010, 72, 63–87. [Google Scholar] [PubMed]
- Robertson, D.S. The function of oxalic acid in the human metabolism. Clin. Chem. Lab. Med. 2011, 49, 1405–1412. [Google Scholar]
- Fu, Z.; Wu, Q.; Guo, W.; Gu, J.; Zheng, X.; Gong, Y.; Lu, C.; Ye, J.; Ye, X.; Jiang, W.; et al. Impaired Insulin Clearance as the Initial Regulator of Obesity-Associated Hyperinsulinemia: Novel Insight Into the Underlying Mechanism Based on Serum Bile Acid Profiles. Diabetes Care 2022, 45, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Nho, K.; Kueider-Paisley, A.; MahmoudianDehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; Kastenmüller, G.; et al. Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: Relationship to neuroimaging and CSF biomarkers. Alzheimers Dement. 2019, 15, 232–244. [Google Scholar] [CrossRef]
- Chen, T.; Zhou, K.; Sun, T.; Sang, C.; Jia, W.; Xie, G. Altered bile acid glycine: Taurine ratio in the progression of chronic liver disease. J. Gastroenterol. Hepatol. 2022, 37, 208–215. [Google Scholar] [CrossRef]
- O’Sullivan, J.F.; Morningstar, J.E.; Yang, Q.; Zheng, B.; Gao, Y.; Jeanfavre, S.; Scott, J.; Fernandez, C.; Zheng, H.; O’Connor, S.; et al. Dimethylguanidino valeric acid is a marker of liver fat and predicts diabetes. J. Clin. Investig. 2017, 127, 4394–4402. [Google Scholar] [CrossRef]
- Grajeda-Iglesias, C.; Durand, S.; Daillère, R.; Iribarren, K.; Lemaitre, F.; Derosa, L.; Aprahamian, F.; Bossut, N.; Nirmalathasan, N.; Madeo, F.; et al. Oral administration of Akkermansia muciniphila elevates systemic antiaging and anticancer metab-olites. Aging 2021, 13, 6375–6405. [Google Scholar] [CrossRef]
- Bárcena, C.; Valdés-Mas, R.; Mayoral, P.; Garabaya, C.; Durand, S.; Rodríguez, F.; Fernández-García, M.T.; Salazar, N.; Nogacka, A.M.; Garatachea, N.; et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 2019, 25, 1234–1242. [Google Scholar] [CrossRef]
- Ren, L.; Peng, C.; Hu, X.; Han, Y.; Huang, H. Microbial production of vitamin K2: Current status and future prospects. Biotechnol. Adv. 2020, 39, 107453. [Google Scholar] [CrossRef]
- Ligthart, K.; Belzer, C.; de Vos, W.M.; Tytgat, H.L.P. Bridging Bacteria and the Gut: Functional Aspects of Type IV Pili. Trends Microbiol. 2020, 28, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lin, J.-X.; Leonard, W.J. Interleukin-2 at the Crossroads of Effector Responses, Tolerance, and Immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Jackson, T.; Sapey, E.; Lord, J.M. Frailty and sarcopenia: The potential role of an aged immune system. Ageing Res. Rev. 2017, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Silvester, J.; Chen, X.; Xu, H.; Sawhney, V.; Rangan, V.; Iturrino, J.; Nee, J.; Duerksen, D.R.; Lembo, A. Serum zonulin is elevated in IBS and correlates with stool frequency in IBS-D. United Eur. Gastroenterol. J. 2019, 7, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Wen, C.; Zheng, Z.; Shao, T.; Liu, L.; Xie, Z.; le Chatelier, E.; He, Z.; Zhong, W.; Fan, Y.; Zhang, L.; et al. Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 2017, 18, 142. [Google Scholar] [CrossRef]
- Li, S.S.; Zhu, A.; Benes, V.; Costea, P.I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojärvi, J.; Voigt, A.Y.; et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016, 352, 586–589. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Hu-man Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Probiotic Group (n = 11) | Control Group (n = 8) |
|---|---|---|
| Age (year), mean ± SD | 81.64 ± 5.01 | 85.38 ± 4.92 |
| Gender (n), female/male | 8/3 | 5/3 |
| BMI (kg/m2), mean ± SD | 19.64 ± 5.29 | 21.18 ± 1.64 |
| AB (g/L), mean ± SD | 37.14 ± 2.53 (0 weeks)/36.29 ± 3.61(12 weeks) | 37.50 ± 1.80 (0 weeks)/34.50 ± 2.63 (12 weeks) |
| PA (mg/L), mean ± SD | 221.57 ± 50.51 (0 weeks)/231.57 ± 45.86 (12 weeks) | 222.83 ± 51.67 (0 weeks)/215.33 ± 52.33 (12 weeks) |
| Hb (g/L), mean ± SD | 107.71 ± 11.57 (0 weeks)/102.14 ± 21.56 (12 weeks) | 126.33 ± 21.08 (0 weeks)/115.33 ± 24.57 (12 weeks) |
| CI disease, n (%) | 9 (81.82) | 8 (100.00) |
| HBP disease, n (%) | 10 (90.91) | 7 (87.50) |
| CAD disease, n (%) | 9 (81.82) | 6 (75.00) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Chen, X.; Liu, L.; Qin, H. Clostridium butyricum Potentially Improves Immunity and Nutrition through Alteration of the Microbiota and Metabolism of Elderly People with Malnutrition in Long-Term Care. Nutrients 2022, 14, 3546. https://doi.org/10.3390/nu14173546
Liu L, Chen X, Liu L, Qin H. Clostridium butyricum Potentially Improves Immunity and Nutrition through Alteration of the Microbiota and Metabolism of Elderly People with Malnutrition in Long-Term Care. Nutrients. 2022; 14(17):3546. https://doi.org/10.3390/nu14173546
Chicago/Turabian StyleLiu, Lin, Xiang Chen, Lu Liu, and Huanlong Qin. 2022. "Clostridium butyricum Potentially Improves Immunity and Nutrition through Alteration of the Microbiota and Metabolism of Elderly People with Malnutrition in Long-Term Care" Nutrients 14, no. 17: 3546. https://doi.org/10.3390/nu14173546
APA StyleLiu, L., Chen, X., Liu, L., & Qin, H. (2022). Clostridium butyricum Potentially Improves Immunity and Nutrition through Alteration of the Microbiota and Metabolism of Elderly People with Malnutrition in Long-Term Care. Nutrients, 14(17), 3546. https://doi.org/10.3390/nu14173546