
Activated Protein C Ameliorates Tubular Mitochondrial Reactive Oxygen Species and Inflammation in Diabetic Kidney Disease

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mice
2.3. Determination of Albuminuria
2.4. Cell Culture
2.5. RNA Expression Profiling
2.6. Flow Cytometry
2.7. Immunoblotting
2.8. Quantitative Polymerase Chain Reaction (qPCR)
2.9. Total DNA Isolation
2.10. ATP Level Quantification
2.11. Histochemistry
2.12. Immunofluorescence
2.13. Transmission Electron Microscopy
3. Results
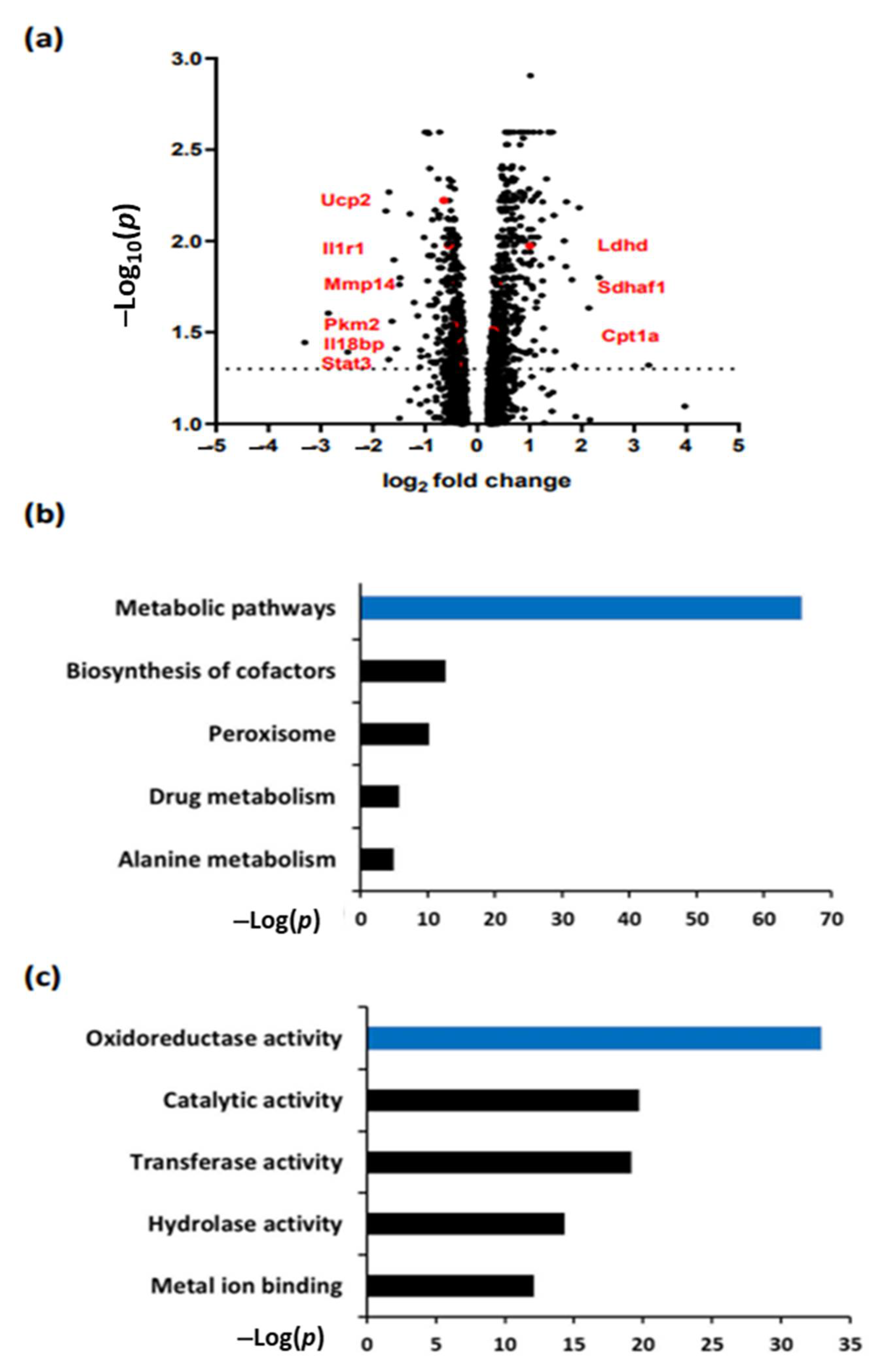
3.1. aPC Regulates Pathways Related to Metabolism and Oxidoreductase Activity in DKD
3.2. aPC Protects against Mitochondrial Dysfunction in DKD
3.3. aPC Reduces Tubular ROS in Diabetic Nephropathy
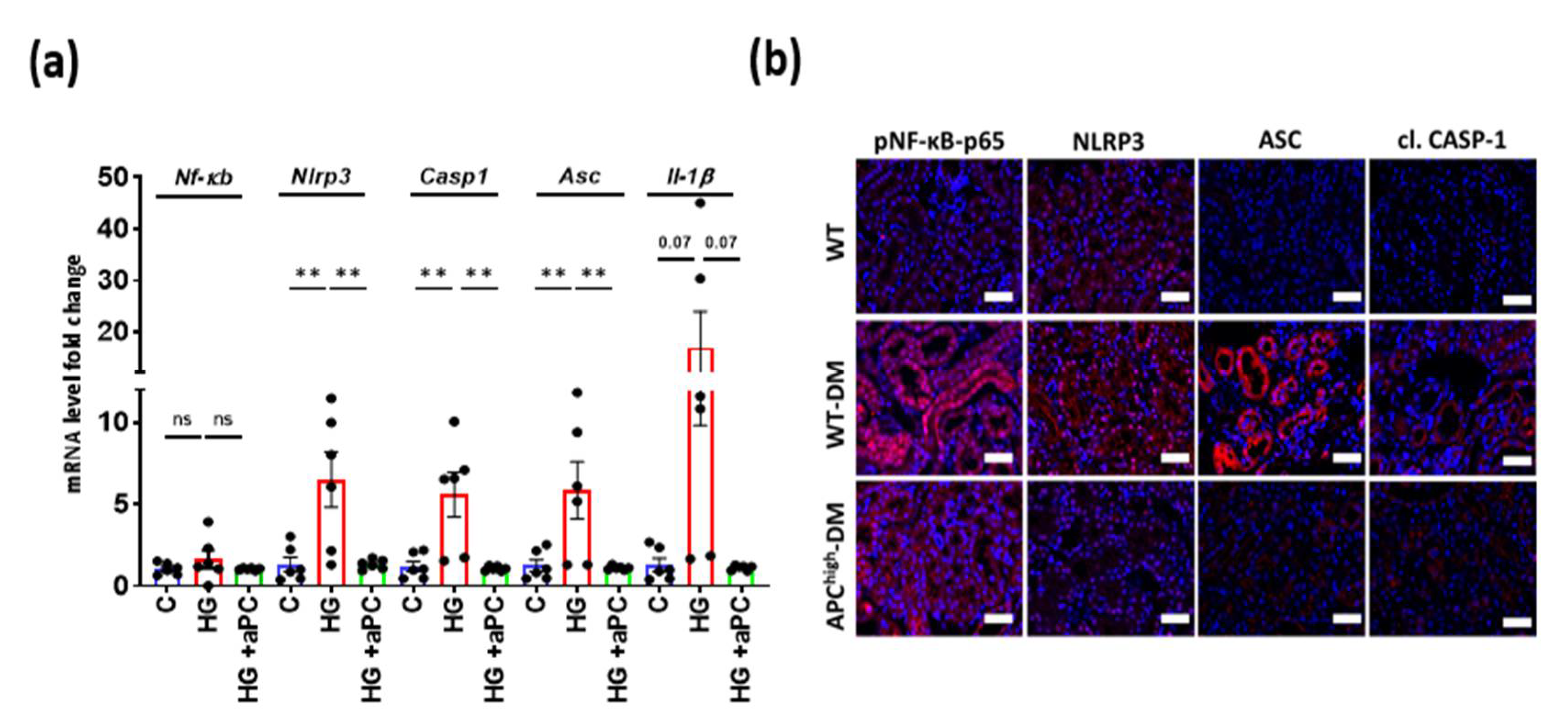
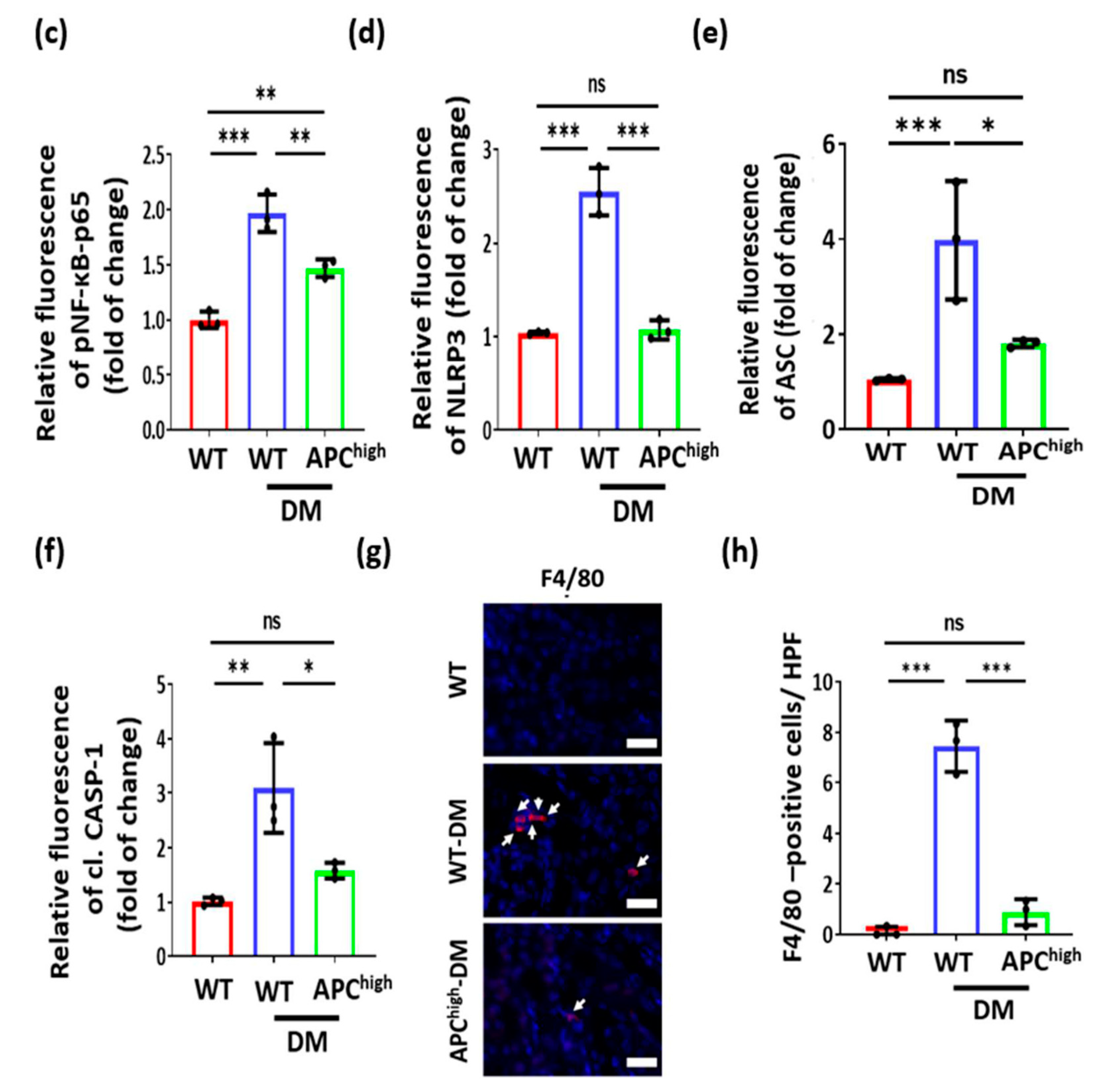
3.4. aPC Reduces Inflammasome Activation in Diabetic Nephropathy
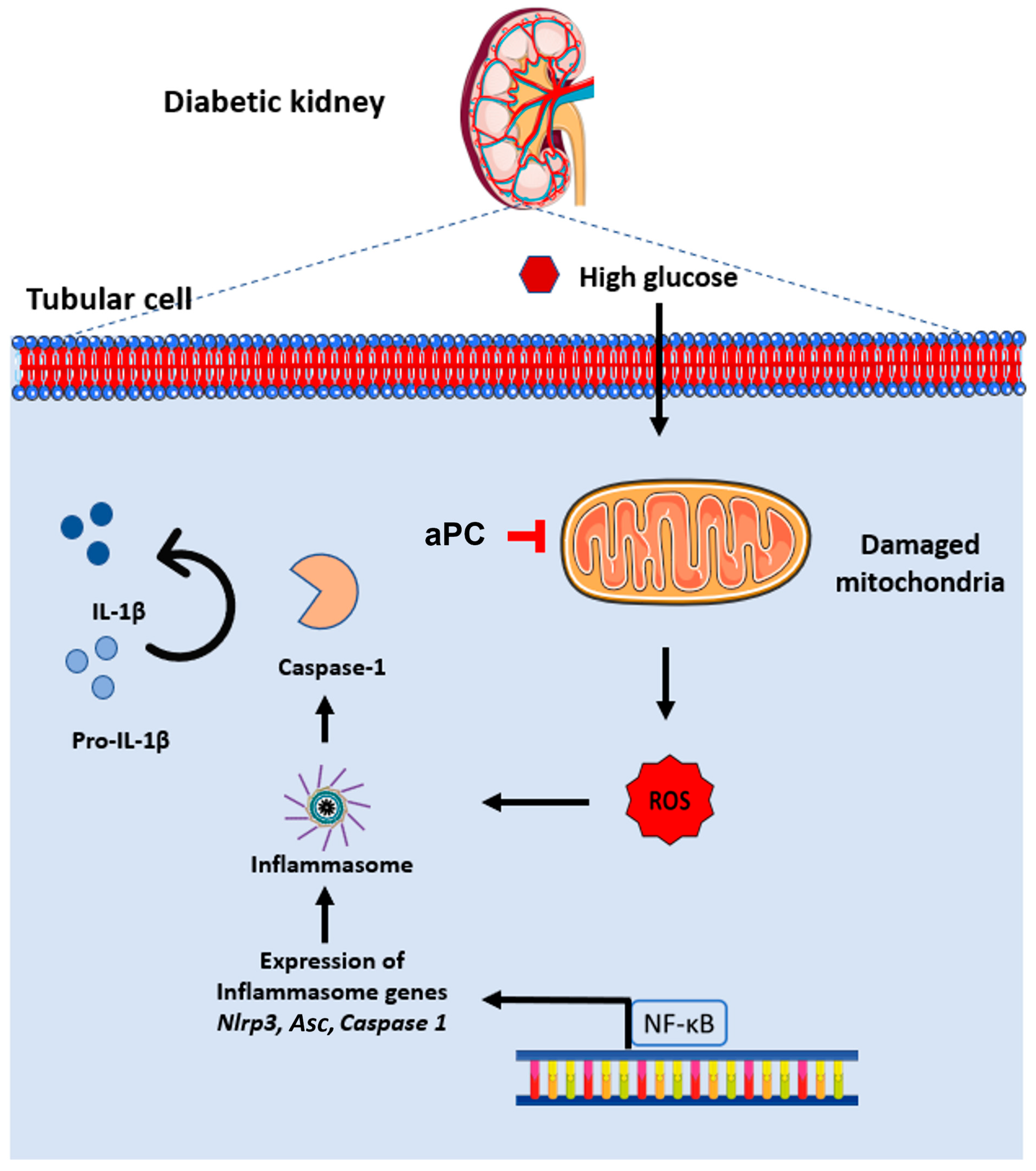
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 8-oxo-dG | 8-Oxo-2’-deoxyguanosine |
| aPC | Activated protein C |
| ANOVA | Analysis of variance |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| BUMPT | Boston university mouse proximal tubular cells |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| DKD | Diabetic kidney disease |
| DM | Diabetes mellitus |
| EPCR | Endothelial protein C receptor |
| FACS | Fluorescence-activated cell sorting |
| GBM | Glomerular basement membrane |
| APChigh | Hyperactivatable protein C |
| IL-1β | Interleukin 1 beta |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MFI | Mean Fluorescence Intensity |
| MTS | Masson’s trichrome stain |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NRLP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NO | Nitric oxide |
| PAS | Periodic acid–Schiff staining |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| qRT-PCR | Quantitative real time polymerase chain reaction |
| ROS | Reactive oxygen species |
| SEM | Standard error mean |
| STZ | Streptozotocin |
| UCAR | Urine albumin-creatinine ratio |
| VDAC | Voltage-dependent anion-selective channel |
References
- Anders, S.; Schroeter, C. Diabetes, Diet-health behavior, and obesity. Front. Endocrinol. 2015, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidell, J. Obesity, insulin resistance and diabetes—A worldwide epidemic. Br. J. Nutr. 2000, 83 (Suppl. S1), S5–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merker, L.; Bautsch, B.-W.; Ebert, T.; Guthoff, M.; Isermann, B. Nephropathy in diabetes. Exp. Clin. Endocrinol. Diabetes 2021, 129, S60–S63. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, A.; Knysak, M.; Nabrdalik, K.; Gumprecht, J.; Stompór, T. Inflammation and oxidative stress in diabetic kidney disease: The targets for SGLT2 inhibitors and GLP-1 receptor agonists. Int. J. Mol. Sci. 2021, 22, 10822. [Google Scholar] [CrossRef] [PubMed]
- Sawaf, H.; Thomas, G.; Taliercio, J.J.; Nakhoul, G.; Vachharajani, T.J.; Mehdi, A. Therapeutic advances in diabetic nephropathy. J. Clin. Med. 2022, 11, 378. [Google Scholar] [CrossRef]
- Isermann, B.; Vinnikov, I.A.; Madhusudhan, T.; Herzog, S.; Kashif, M.; Blautzik, J.; Corat, M.A.; Zeier, M.; Blessing, E.; Oh, J.; et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat. Med. 2007, 13, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Kanetsuna, Y.; Takahashi, K.; Nagata, M.; Gannon, M.A.; Breyer, M.D.; Harris, R.C.; Takahashi, T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am. J. Pathol. 2007, 170, 1473–1484. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Kohli, S.; Al-Dabet, M.M.; Isermann, B. Cell biology of activated protein C. Curr. Opin. Hematol. 2019, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Madhusudhan, T.; Kerlin, B.A.; Isermann, B. The emerging role of coagulation proteases in kidney disease. Nat. Rev. Nephrol. 2016, 12, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Bock, F.; Shahzad, K.; Wang, H.; Stoyanov, S.; Wolter, J.; Dong, W.; Pelicci, P.G.; Kashif, M.; Ranjan, S.; Schmidt, S.; et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc. Natl. Acad. Sci. USA 2013, 110, 648–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Activated protein C: Biased for translation. Blood 2015, 125, 2898–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Protein C anticoagulant and cytoprotective pathways. Int. J. Hematol. 2012, 95, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyden, P.; Pryor, K.E.; Coffey, C.S.; Cudkowicz, M.; Conwit, R.; Jadhav, A.; Sawyer, R.N., Jr.; Claassen, J.; Adeoye, O.; Song, S.; et al. Final Results of the RHAPSODY Trial: A Multi-Center, Phase 2 Trial Using a Continual Reassessment Method to Determine the Safety and Tolerability of 3K3A-APC, A Recombinant Variant of Human Activated Protein C, in Combination with Tissue Plasminogen Activator, Mechanical Thrombectomy or both in Moderate to Severe Acute Ischemic Stroke. Ann. Neurol. 2019, 85, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhusudhan, T.; Wang, H.; Ghosh, S.; Dong, W.; Kumar, V.; Al-Dabet, M.M.; Manoharan, J.; Nazir, S.; Elwakiel, A.; Bock, F.; et al. Signal integration at the PI3K-p85-XBP1 hub endows coagulation protease activated protein C with insulin-like function. Blood 2017, 130, 1445. [Google Scholar] [CrossRef]
- Gil-Bernabe, P.; D’Alessandro-Gabazza, C.N.; Toda, M.; Boveda Ruiz, D.; Miyake, Y.; Suzuki, T.; Onishi, Y.; Morser, J.; Gabazza, E.C.; Takei, Y.; et al. Exogenous activated protein C inhibits the progression of diabetic nephropathy. J. Thromb. Haemost. 2012, 10, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauta, F.L.; Boertien, W.E.; Bakker, S.J.; van Goor, H.; van Oeveren, W.; de Jong, P.E.; Bilo, H.; Gansevoort, R.T. Glomerular and tubular damage markers are elevated in patients with diabetes. Diabetes Care 2011, 34, 975–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zylka, A.; Dumnicka, P.; Kusnierz-Cabala, B.; Gala-Bladzinska, A.; Ceranowicz, P.; Kucharz, J.; Zabek-Adamska, A.; Maziarz, B.; Drozdz, R.; Kuzniewski, M. Markers of Glomerular and Tubular Damage in the Early Stage of Kidney Disease in Type 2 Diabetic Patients. Mediat. Inflamm. 2018, 2018, 7659243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, A.; Al-Dabet, M.M.; Ghosh, S.; Kohli, S.; Manoharan, J.; ElWakiel, A.; Gadi, I.; Bock, F.; Nazir, S.; Wang, H.; et al. Farnesoid X Receptor Agonism Protects against Diabetic Tubulopathy: Potential Add-On Therapy for Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 3182–3189. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Fatima, S.; Al-Dabet, M.M.; Gadi, I.; Khawaja, H.; Ambreen, S.; Elwakiel, A.; Kloting, N.; Bluher, M.; Nawroth, P.P.; et al. CHOP-ASO Ameliorates Glomerular and Tubular Damage on Top of ACE Inhibition in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2021, 10, 3066–3079. [Google Scholar] [CrossRef]
- Madhusudhan, T.; Ghosh, S.; Wang, H.; Dong, W.; Gupta, D.; Elwakiel, A.; Stoyanov, S.; Al-Dabet, M.M.; Krishnan, S.; Biemann, R.; et al. Podocyte Integrin-beta 3 and Activated Protein C Coordinately Restrict RhoA Signaling and Ameliorate Diabetic Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 1762–1780. [Google Scholar] [CrossRef]
- Sheridan, A.M.; Schwartz, J.H.; Kroshian, V.M.; Tercyak, A.M.; Laraia, J.; Masino, S.; Lieberthal, W. Renal mouse proximal tubular cells are more susceptible than MDCK cells to chemical anoxia. Am. J. Physiol. Physiol. 1993, 265, F342–F350. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhusudhan, T.; Wang, H.; Straub, B.K.; Grone, E.; Zhou, Q.; Shahzad, K.; Muller-Krebs, S.; Schwenger, V.; Gerlitz, B.; Grinnell, B.W.; et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood 2012, 119, 874–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, A.; Prefontaine, K.E. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 752–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashihara, N.; Haruna, Y.; Kondeti, V.K.; Kanwar, Y.S. Oxidative stress in diabetic nephropathy. Curr. Med. Chem. 2010, 17, 4256–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Nazir, S.; Wang, H.; Kohli, S.; Ranjan, S.; Mertens, P.R.; Nawroth, P.P.; et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci. Rep. 2016, 6, 34228. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, G.R.; Vincent, J.L.; Laterre, P.F.; LaRosa, S.P.; Dhainaut, J.F.; Lopez-Rodriguez, A.; Steingrub, J.S.; Garber, G.E.; Helterbrand, J.D.; Ely, E.W.; et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N. Engl. J. Med. 2001, 344, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti-Carvajal, A.J.; Sola, I.; Lathyris, D.; Cardona, A.F. Human recombinant activated protein C for severe sepsis. Cochrane Database Syst. Rev. 2012, 1–68. [Google Scholar] [CrossRef]
- Shahzad, K.; Gadi, I.; Nazir, S.; Al-Dabet, M.M.; Kohli, S.; Bock, F.; Breitenstein, L.; Ranjan, S.; Fuchs, T.; Halloul, Z.; et al. Activated protein C reverses epigenetically sustained p66(Shc) expression in plaque-associated macrophages in diabetes. Commun. Biol. 2018, 1, 104. [Google Scholar] [CrossRef]
- Yamaji, K.; Wang, Y.; Liu, Y.; Abeyama, K.; Hashiguchi, T.; Uchimura, T.; Krishna Biswas, K.; Iwamoto, H.; Maruyama, I. Activated protein C, a natural anticoagulant protein, has antioxidant properties and inhibits lipid peroxidation and advanced glycation end products formation. Thromb. Res. 2005, 115, 319–325. [Google Scholar] [CrossRef]
- Toltl, L.J.; Austin, R.C.; Liaw, P.C. Activated protein C modulates inflammation, apoptosis and tissue factor procoagulant activity by regulating endoplasmic reticulum calcium depletion in blood monocytes. J. Thromb. Haemost. 2011, 9, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.; Schild, L.; Bock, F.; Hellwig, A.; Gadi, I.; Al-Dabet, M.M.; Ranjan, S.; Ronicke, R.; Nawroth, P.P.; Petersen, K.U.; et al. Thrombomodulin-dependent protein C activation is required for mitochondrial function and myelination in the central nervous system. J. Thromb. Haemost. 2016, 14, 2212–2226. [Google Scholar] [CrossRef]
- Yang, S.M.; Ka, S.M.; Wu, H.L.; Yeh, Y.C.; Kuo, C.H.; Hua, K.F.; Shi, G.Y.; Hung, Y.J.; Hsiao, F.C.; Yang, S.S.; et al. Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice via anti-NF-kappaB/NLRP3 inflammasome-mediated inflammation, enhancement of NRF2 antioxidant activity and inhibition of apoptosis. Diabetologia 2014, 57, 424–434. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Kohli, S.; Nazir, S.; Ghosh, S.; Ranjan, S.; Wang, H.; Madhusudhan, T.; et al. Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- Dockendorff, C.; Aisiku, O.; Verplank, L.; Dilks, J.R.; Smith, D.A.; Gunnink, S.F.; Dowal, L.; Negri, J.; Palmer, M.; Macpherson, L.; et al. Discovery of 1,3-Diaminobenzenes as Selective Inhibitors of Platelet Activation at the PAR1 Receptor. ACS Med. Chem. Lett. 2012, 3, 232–237. [Google Scholar] [CrossRef]
- Gadi, I.; Fatima, S.; Elwakiel, A.; Nazir, S.; Mohanad Al-Dabet, M.; Rana, R.; Bock, F.; Manoharan, J.; Gupta, D.; Biemann, R.; et al. Different DOACs Control Inflammation in Cardiac Ischemia-Reperfusion Differently. Circ. Res. 2021, 128, 513–529. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, R.; Manoharan, J.; Gupta, A.; Gupta, D.; Elwakiel, A.; Khawaja, H.; Fatima, S.; Zimmermann, S.; Singh, K.; Ambreen, S.; et al. Activated Protein C Ameliorates Tubular Mitochondrial Reactive Oxygen Species and Inflammation in Diabetic Kidney Disease. Nutrients 2022, 14, 3138. https://doi.org/10.3390/nu14153138
Rana R, Manoharan J, Gupta A, Gupta D, Elwakiel A, Khawaja H, Fatima S, Zimmermann S, Singh K, Ambreen S, et al. Activated Protein C Ameliorates Tubular Mitochondrial Reactive Oxygen Species and Inflammation in Diabetic Kidney Disease. Nutrients. 2022; 14(15):3138. https://doi.org/10.3390/nu14153138
Chicago/Turabian StyleRana, Rajiv, Jayakumar Manoharan, Anubhuti Gupta, Dheerendra Gupta, Ahmed Elwakiel, Hamzah Khawaja, Sameen Fatima, Silke Zimmermann, Kunal Singh, Saira Ambreen, and et al. 2022. "Activated Protein C Ameliorates Tubular Mitochondrial Reactive Oxygen Species and Inflammation in Diabetic Kidney Disease" Nutrients 14, no. 15: 3138. https://doi.org/10.3390/nu14153138
APA StyleRana, R., Manoharan, J., Gupta, A., Gupta, D., Elwakiel, A., Khawaja, H., Fatima, S., Zimmermann, S., Singh, K., Ambreen, S., Gadi, I., Biemann, R., Jiang, S., Shahzad, K., Kohli, S., & Isermann, B. (2022). Activated Protein C Ameliorates Tubular Mitochondrial Reactive Oxygen Species and Inflammation in Diabetic Kidney Disease. Nutrients, 14(15), 3138. https://doi.org/10.3390/nu14153138