
Neutrophil Extracellular Traps Promote NLRP3 Inflammasome Activation and Glomerular Endothelial Dysfunction in Diabetic Kidney Disease

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Mice
2.2. Determination of Albuminuria
2.3. ELISA
2.4. Human Renal Biopsies
2.5. Immunostaining
2.6. PAS Staining
2.7. Glomerular Fraction Isolation
2.8. Cell Culture
2.9. Neutrophil Isolation
2.10. Immunoblotting
2.11. Barrier Assay
2.12. Statistical Analysis
3. Results
3.1. NET Formation Is Associated with DKD
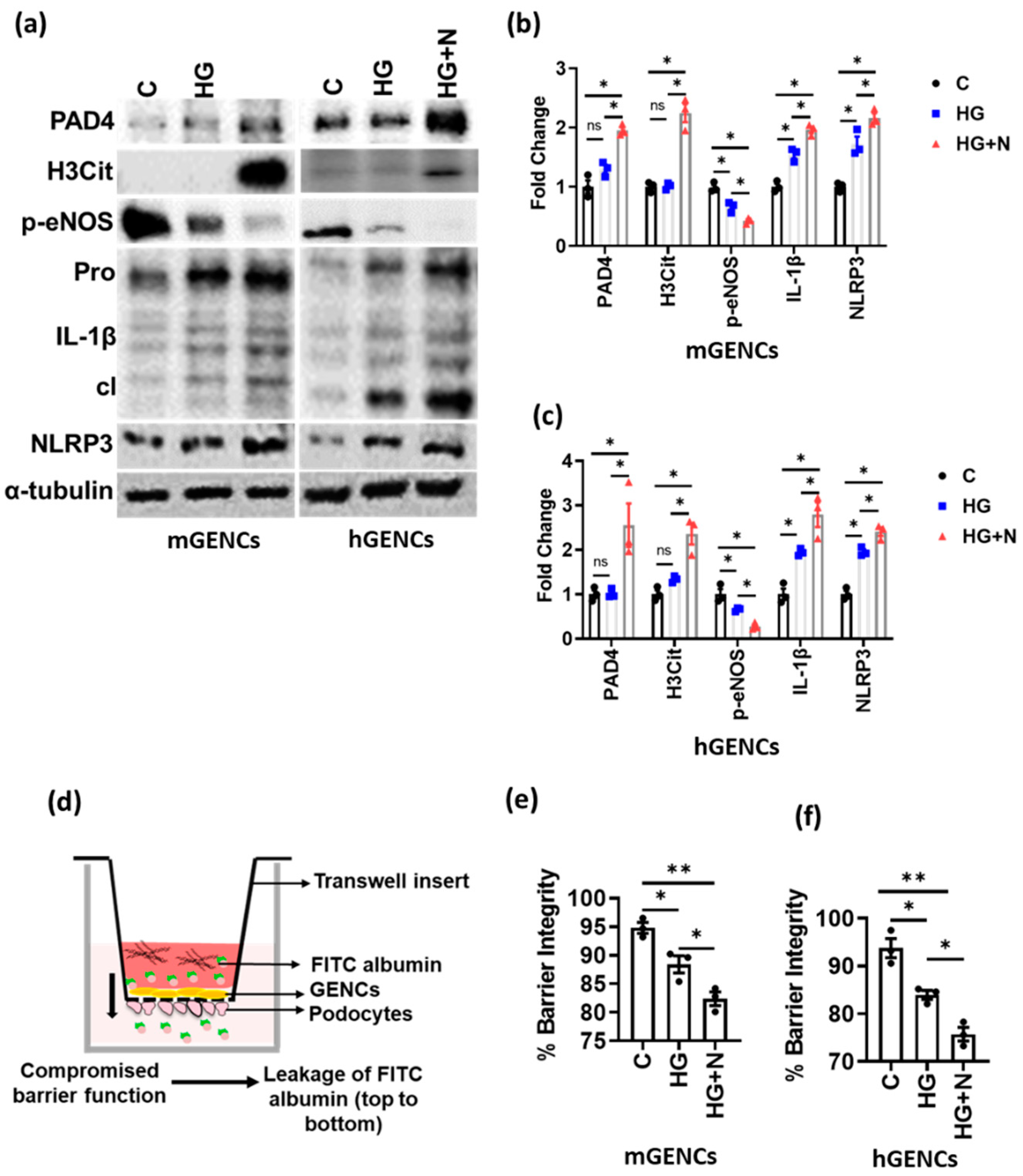
3.2. NETs Induce Inflammasome Activation and Endothelial Dysfunction in Glomerular Endothelial Cells
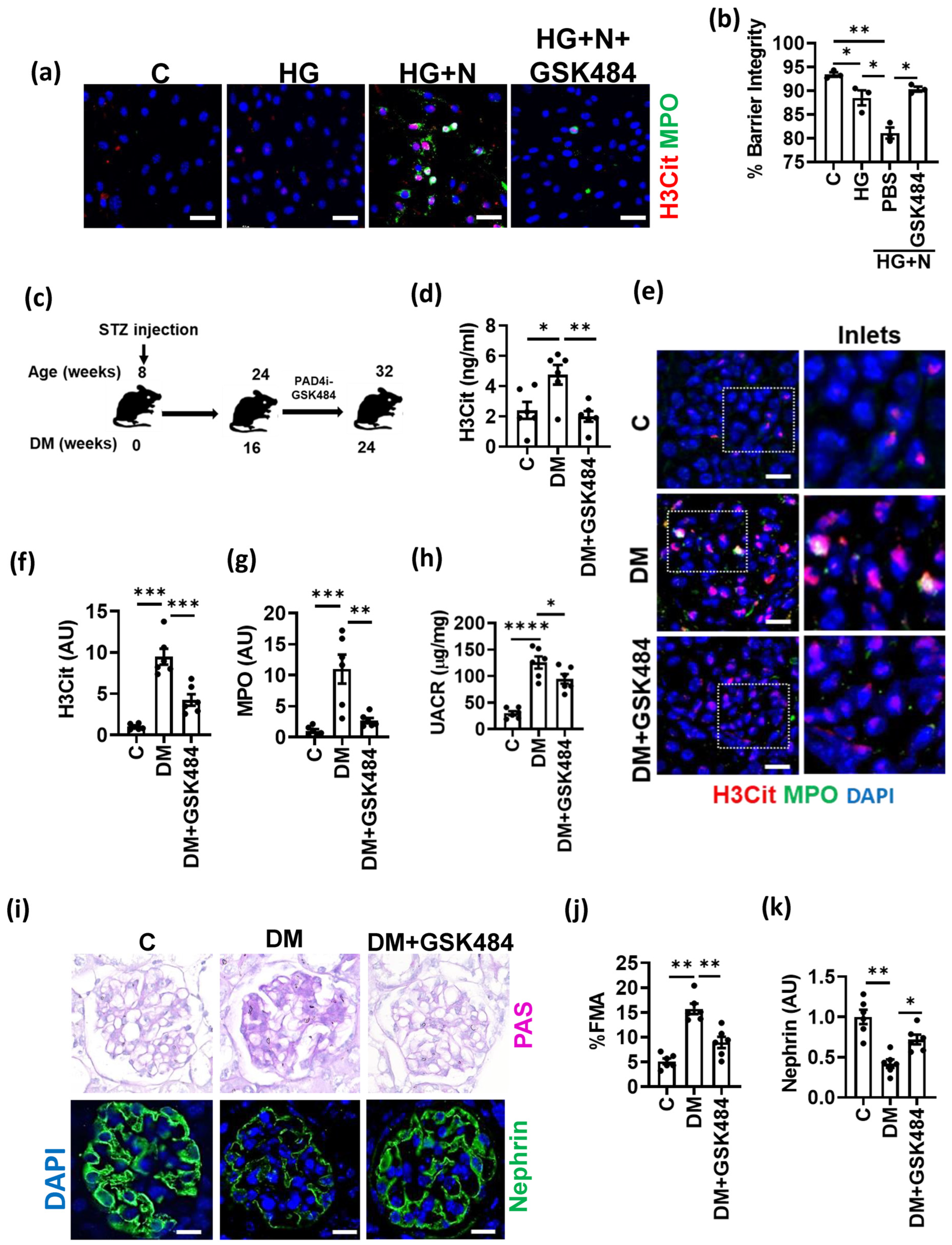
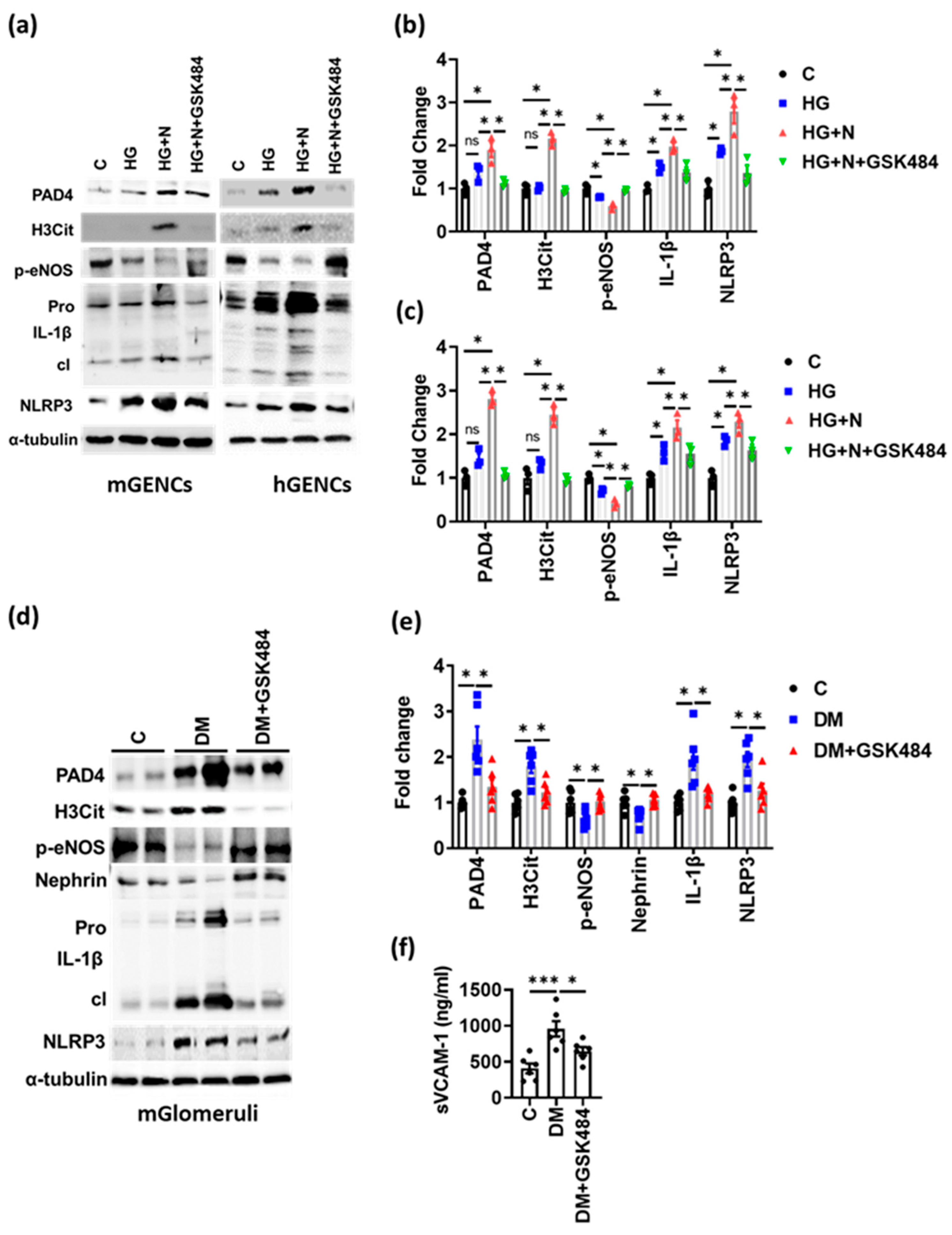
3.3. PAD4 Inhibition Ameliorates Experimental DKD
3.4. PAD4 Inhibition Inhibits NET-Induced Inflammasome Activation and Endothelial Dysfunction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Pantalone, K.M.; Hobbs, T.M.; Wells, B.J.; Kong, S.X.; Kattan, M.W.; Bouchard, J.; Yu, C.; Sakurada, B.; Milinovich, A.; Weng, W.; et al. Clinical characteristics, complications, comorbidities and treatment patterns among patients with type 2 diabetes mellitus in a large integrated health system. BMJ Open Diabetes Res. Care 2015, 3, e000093. [Google Scholar] [CrossRef] [PubMed]
- Domingueti, C.P.; Dusse, L.M.S.; Carvalho, M.D.G.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 3. Prevention or Delay of Type 2 Diabetes: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44, S34–S39. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef]
- Zheng, Y.; Gardner, S.E.; Clarke, M. Cell Death, Damage-Associated Molecular Patterns, and Sterile Inflammation in Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2011, 31, 2781–2786. [Google Scholar] [CrossRef]
- Herrero-Cervera, A.; Soehnlein, O.; Kenne, E. Neutrophils in chronic inflammatory diseases. Cell. Mol. Immunol. 2022, 19, 177–191. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.C.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J. Neutrophil Extracellular Traps Go Viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef]
- Jansen, M.P.; Emal, D.; Teske, G.J.; Dessing, M.C.; Florquin, S.; Roelofs, J.J. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. 2017, 91, 352–364. [Google Scholar] [CrossRef]
- Deparis, X.; Roche, C.; Murgue, B.; Chungue, E. Possible dengue sequential infection: Dengue spread in a neighbourhood during the 1996/97 dengue-2 epidemic in French Polynesia. Trop. Med. Int. Health 1998, 3, 866–871. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef] [PubMed]
- Westhorpe, C.L.; Bayard, J.E.; O’Sullivan, K.M.; Hall, P.; Cheng, Q.; Kitching, A.R.; Hickey, M.J. In Vivo Imaging of Inflamed Glomeruli Reveals Dynamics of Neutrophil Extracellular Trap Formation in Glomerular Capillaries. Am. J. Pathol. 2017, 187, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; Ponomaryov, T.; De Prendergast, A.; Whitworth, K.; Smith, C.W.; Khan, A.O.; Kavanagh, D.; Brill, A. Neutrophil extracellular traps and inflammasomes cooperatively promote venous thrombosis in mice. Blood Adv. 2021, 5, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R., Jr. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef]
- Madhusudhan, T.; Ghosh, S.; Wang, H.; Dong, W.; Gupta, D.; Elwakiel, A.; Stoyanov, S.; Al-Dabet, M.M.; Krishnan, S.; Biemann, R.; et al. Podocyte Integrin-β3 and Activated Protein C Coordinately Restrict RhoA Signaling and Ameliorate Diabetic Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 1762–1780. [Google Scholar] [CrossRef]
- Marquardt, A.; Al-Dabet, M.M.; Ghosh, S.; Kohli, S.; Manoharan, J.; Elwakiel, A.; Gadi, I.; Bock, F.; Nazir, S.; Wang, H.; et al. Farnesoid X Receptor Agonism Protects against Diabetic Tubulopathy: Potential Add-On Therapy for Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 3182–3189. [Google Scholar] [CrossRef]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [PubMed]
- Kohli, S.; Ranjan, S.; Hoffmann, J.; Kashif, M.; Daniel, E.A.; Al-Dabet, M.M.; Bock, F.; Nazir, S.; Huebner, H.; Mertens, P.R.; et al. Maternal extracellular vesicles and platelets promote preeclampsia via inflammasome activation in trophoblasts. Blood 2016, 128, 2153–2164. [Google Scholar] [CrossRef]
- Shahzad, K.; Fatima, S.; Al-Dabet, M.M.; Gadi, I.; Khawaja, H.; Ambreen, S.; Elwakiel, A.; Klöting, N.; Blüher, M.; Nawroth, P.P.; et al. CHOP-ASO Ameliorates Glomerular and Tubular Damage on Top of ACE Inhibition in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2021, 32, 3066–3079. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Kohli, S.; Nazir, S.; Ghosh, S.; Ranjan, S.; Wang, H.; Madhusudhan, T.; et al. Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sheng, J.; He, H.; Chen, X.; Li, J.; Tan, R.; Wang, L.; Lan, H.-Y. A simple and highly purified method for isolation of glomeruli from the mouse kidney. Am. J. Physiol. Ren. Physiol. 2019, 317, F1217–F1223. [Google Scholar] [CrossRef]
- Agak, G.W.; Mouton, A.; Teles, R.M.; Weston, T.A.; Morselli, M.; Andrade, P.R.; Pellegrini, M.; Modlin, R.L. Extracellular traps released by antimicrobial TH17 cells contribute to host defense. J. Clin. Investig. 2021, 131, e141594. [Google Scholar] [CrossRef]
- Gora, I.M.; Ciechanowska, A.; Ladyzynski, P. NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes. Cells 2021, 10, 314. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil extracellular traps participate in cardiovascular diseases: Recent experimental and clinical insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef]
- Carney, E. Acute kidney injury: Role of platelet activation and NETs in renal IRI. Nat. Rev. Nephrol. 2016, 12, 715. [Google Scholar] [CrossRef]
- Gupta, S.; Kaplan, S.G.M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced Formation and Disordered Regulation of NETs in Myeloperoxidase-ANCA–Associated Microscopic Polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2019, 9, 3076. [Google Scholar] [CrossRef] [PubMed]
- Njeim, R.; Azar, W.S.; Fares, A.H.; Azar, S.T.; Kassouf, H.K.; Eid, A.A. NETosis contributes to the pathogenesis of diabetes and its complications. J. Mol. Endocrinol. 2020, 65, R65–R76. [Google Scholar] [CrossRef]
- Diez-Delhoyo, F.; Gutierrez, E.; Sanz-Ruiz, R.; Vázquez-Álvarez, M.E.; Saldívar, H.G.; Juárez, A.R.; Sarnago, F.; Martínez-Sellés, M.; Bermejo, J.; Soriano, J.; et al. Prevalence of Microvascular and Endothelial Dysfunction in the Nonculprit Territory in Patients With Acute Myocardial Infarction. Circ. Cardiovasc. Interv. 2019, 12, e007257. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef]
- D’Abbondanza, M.; Martorelli, E.E.; Ricci, M.A.; De Vuono, S.; Migliola, E.N.; Godino, C.; Corradetti, S.; Siepi, D.; Paganelli, M.T.; Maugeri, N.; et al. Increased plasmatic NETs by-products in patients in severe obesity. Sci. Rep. 2019, 9, 14678. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cui, J.; Zhang, G.; Wu, C.; Abdel-Latif, A.; Smyth, S.S.; Shiroishi, T.; Mackman, N.; Wei, Y.; Tao, M.; et al. Inflammasome activation promotes venous thrombosis through pyroptosis. Blood Adv. 2021, 5, 2619–2623. [Google Scholar] [CrossRef] [PubMed]
- Bilal, J.; Berlinberg, A.; bin Riaz, I.; Faridi, W.; Bhattacharjee, S.; Ortega, G.; Murad, M.H.; Wang, Z.; Prokop, L.J.; Alhifany, A.A.; et al. Risk of Infections and Cancer in Patients With Rheumatologic Diseases Receiving Interleukin Inhibitors: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e1913102. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Nawroth, P.P.; Isermann, B. Mechanisms of Diabetic Nephropathy–Old Buddies and Newcomers Part 2. Exp. Clin. Endocrinol. Diabetes 2010, 118, 667–672. [Google Scholar] [CrossRef]
- Nawroth, P.P.; Isermann, B. Mechanisms of Diabetic Nephropathy–Old Buddies and Newcomers Part 1. Exp. Clin. Endocrinol. Diabetes 2010, 118, 571–576. [Google Scholar] [CrossRef]
- Refaeli, I.; Hughes, M.R.; Wong, A.K.-W.; Bissonnette, M.L.Z.; Roskelley, C.D.; Wayne Vogl, A.; Barbour, S.J.; Freedman, B.S.; McNagny, K.M. Distinct Functional Requirements for Podocalyxin in Immature and Mature Podocytes Reveal Mechanisms of Human Kidney Disease. Sci. Rep. 2020, 10, 9419. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, A.; Singh, K.; Fatima, S.; Ambreen, S.; Zimmermann, S.; Younis, R.; Krishnan, S.; Rana, R.; Gadi, I.; Schwab, C.; et al. Neutrophil Extracellular Traps Promote NLRP3 Inflammasome Activation and Glomerular Endothelial Dysfunction in Diabetic Kidney Disease. Nutrients 2022, 14, 2965. https://doi.org/10.3390/nu14142965
Gupta A, Singh K, Fatima S, Ambreen S, Zimmermann S, Younis R, Krishnan S, Rana R, Gadi I, Schwab C, et al. Neutrophil Extracellular Traps Promote NLRP3 Inflammasome Activation and Glomerular Endothelial Dysfunction in Diabetic Kidney Disease. Nutrients. 2022; 14(14):2965. https://doi.org/10.3390/nu14142965
Chicago/Turabian StyleGupta, Anubhuti, Kunal Singh, Sameen Fatima, Saira Ambreen, Silke Zimmermann, Ruaa Younis, Shruthi Krishnan, Rajiv Rana, Ihsan Gadi, Constantin Schwab, and et al. 2022. "Neutrophil Extracellular Traps Promote NLRP3 Inflammasome Activation and Glomerular Endothelial Dysfunction in Diabetic Kidney Disease" Nutrients 14, no. 14: 2965. https://doi.org/10.3390/nu14142965
APA StyleGupta, A., Singh, K., Fatima, S., Ambreen, S., Zimmermann, S., Younis, R., Krishnan, S., Rana, R., Gadi, I., Schwab, C., Biemann, R., Shahzad, K., Rani, V., Ali, S., Mertens, P. R., Kohli, S., & Isermann, B. (2022). Neutrophil Extracellular Traps Promote NLRP3 Inflammasome Activation and Glomerular Endothelial Dysfunction in Diabetic Kidney Disease. Nutrients, 14(14), 2965. https://doi.org/10.3390/nu14142965