
Retinoic Acid Signaling Is Compromised in DSS-Induced Dysbiosis



 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Colitis Model
2.2. Gene Expression
2.3. Quantification of Bacterial DNA
2.4. 16S rRNA Gene Pyrosequencing
2.5. Statistical Analysis
3. Results
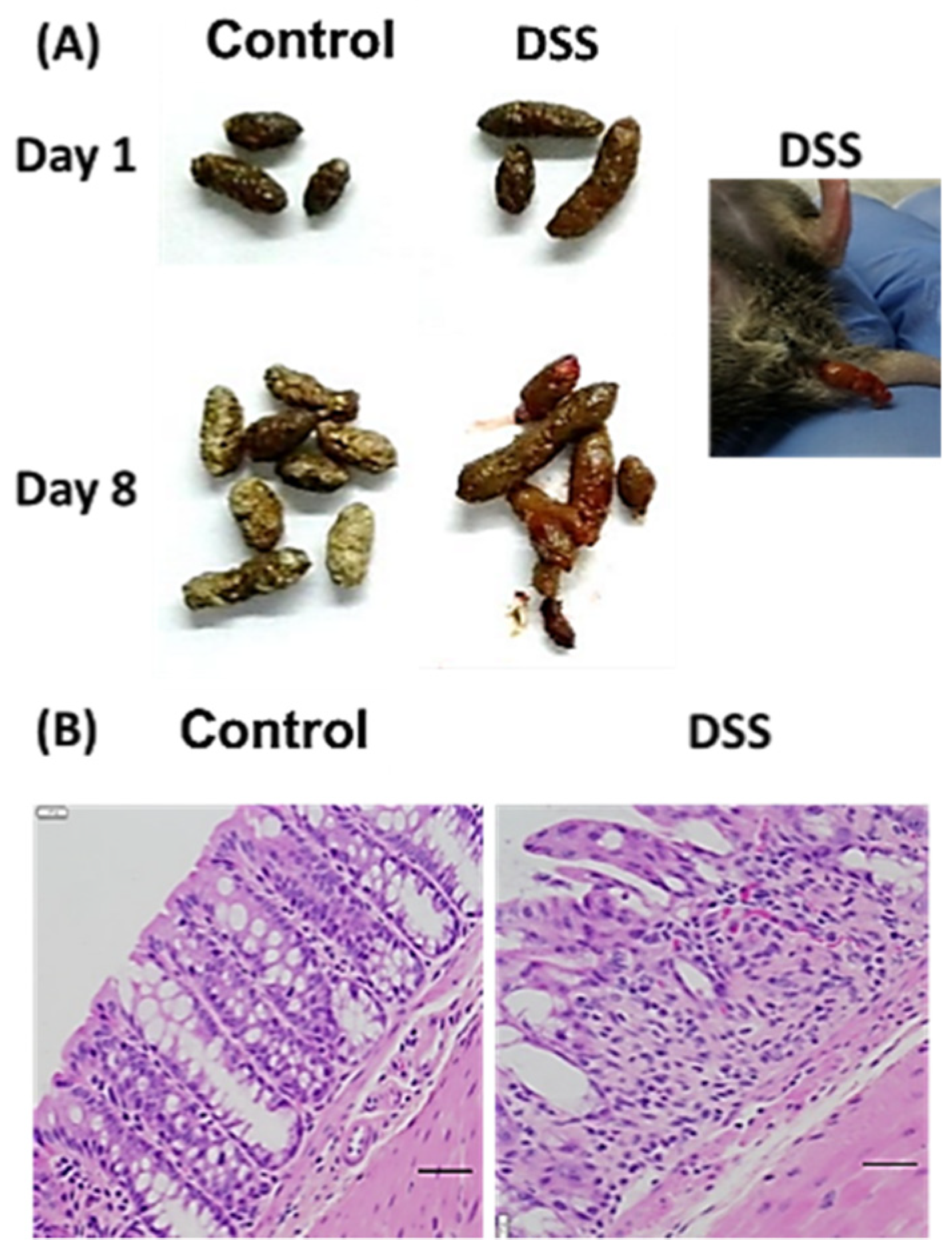
3.1. Mouse Colitis Model
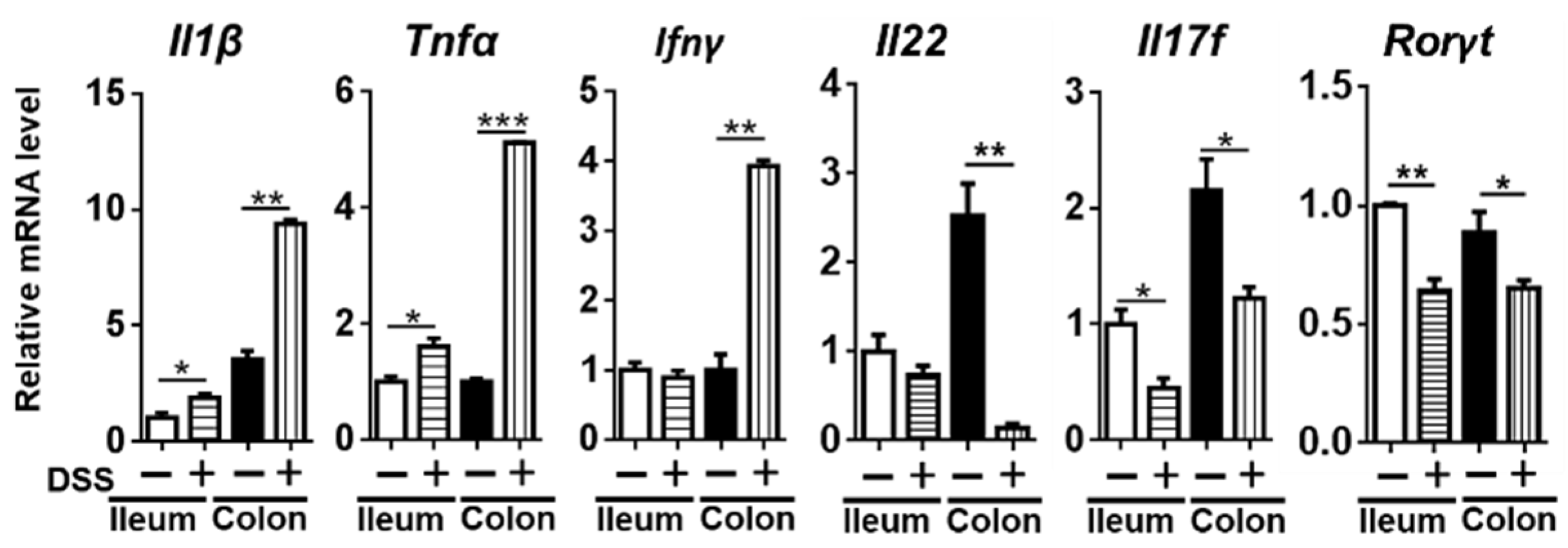
3.2. Changes in Inflammatory Cytokines
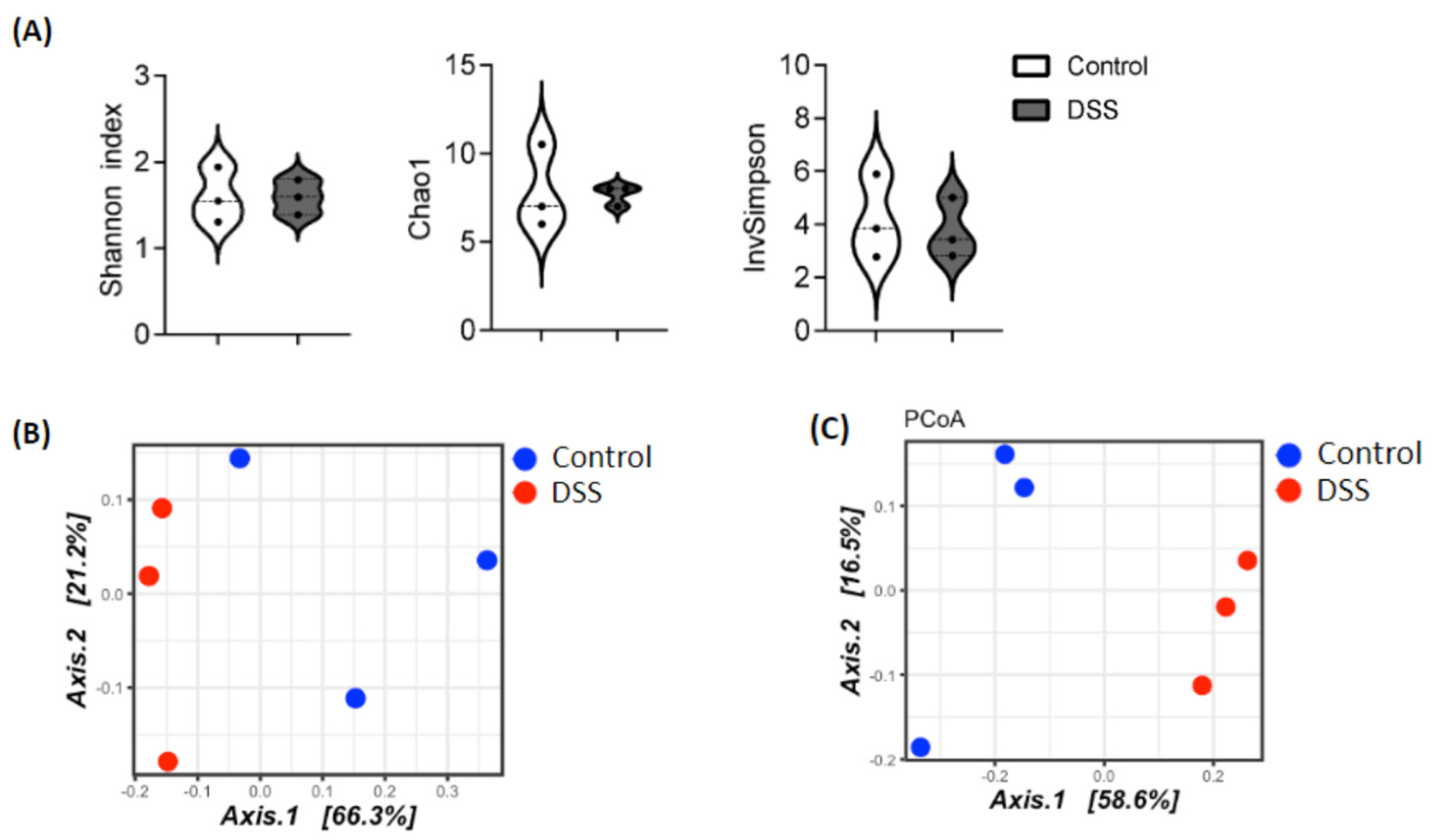
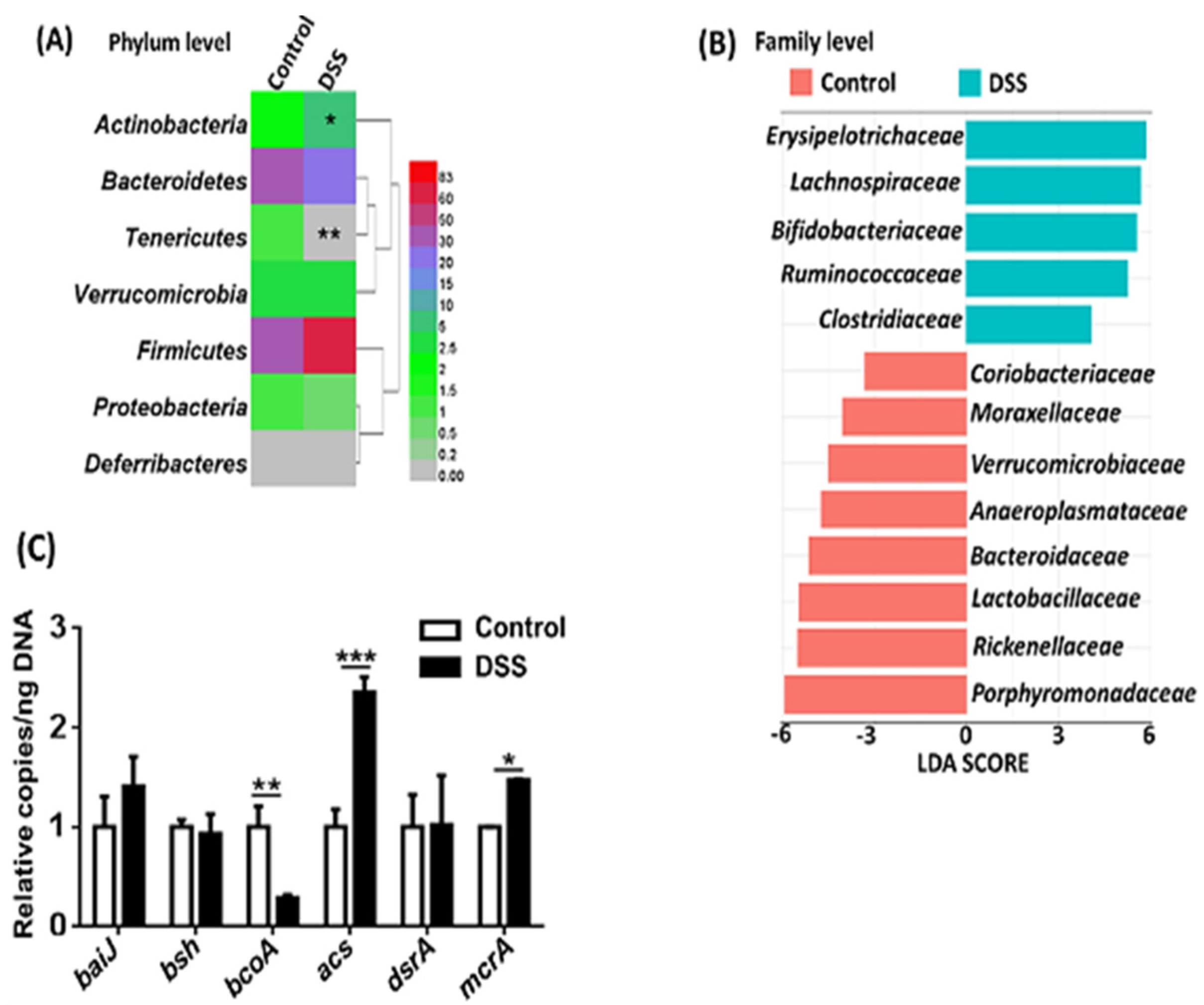
3.3. Dysbiosis Occurs in Colitis Mice
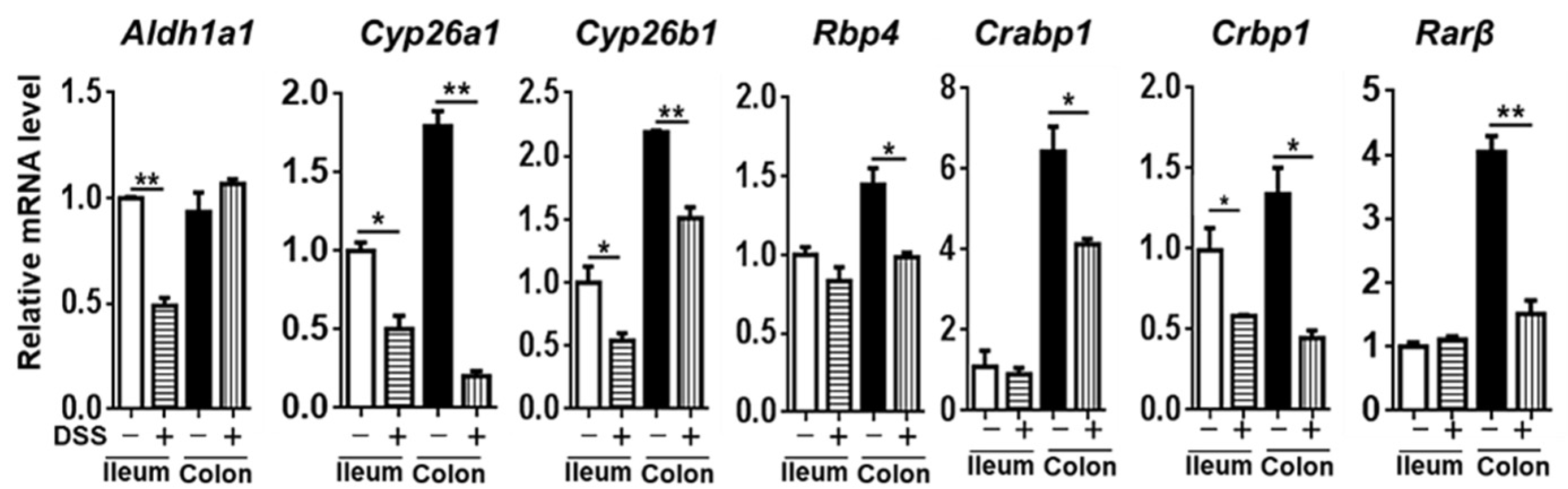
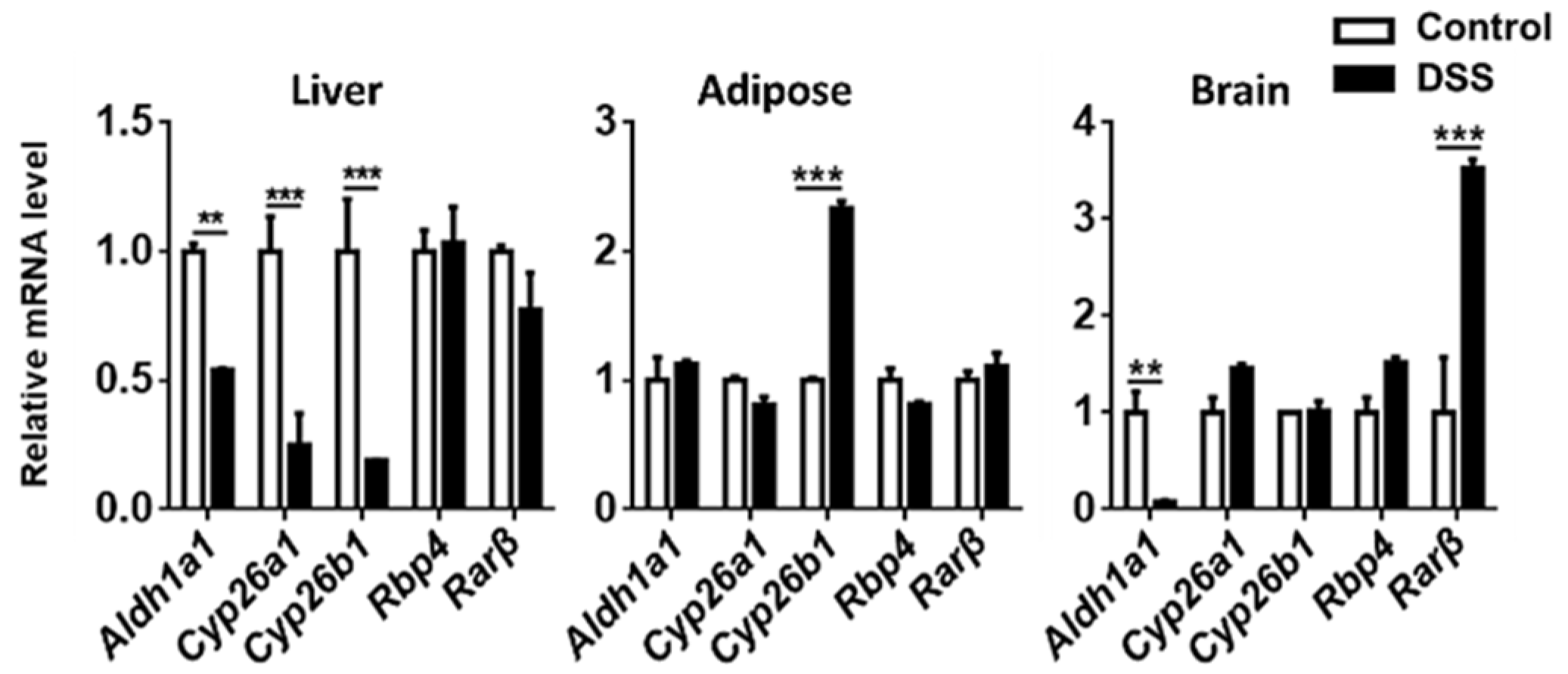
3.4. The Expression of Genes Implicated in RA Signaling
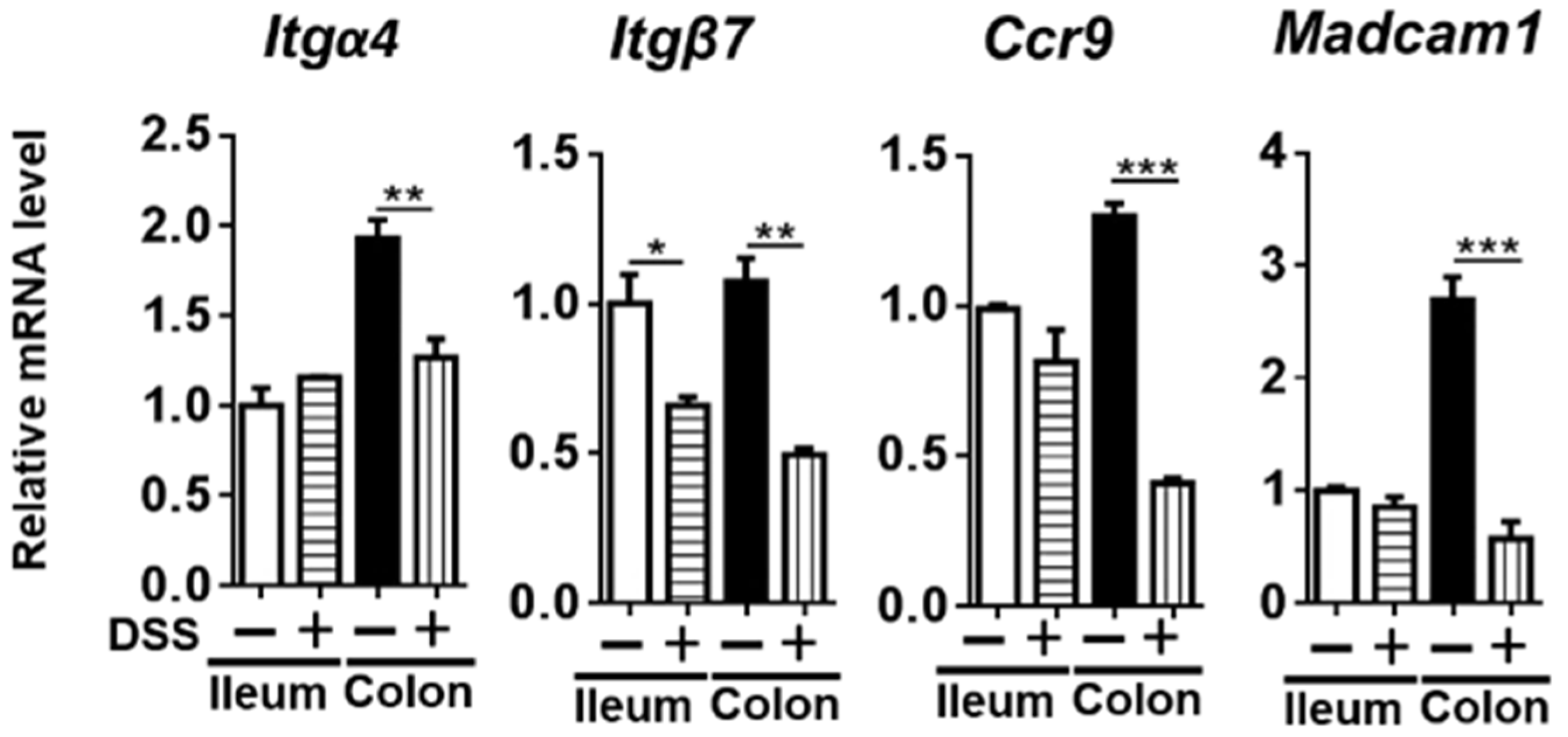
3.5. The Expression of RA-Regulated Gut-Homing Genes
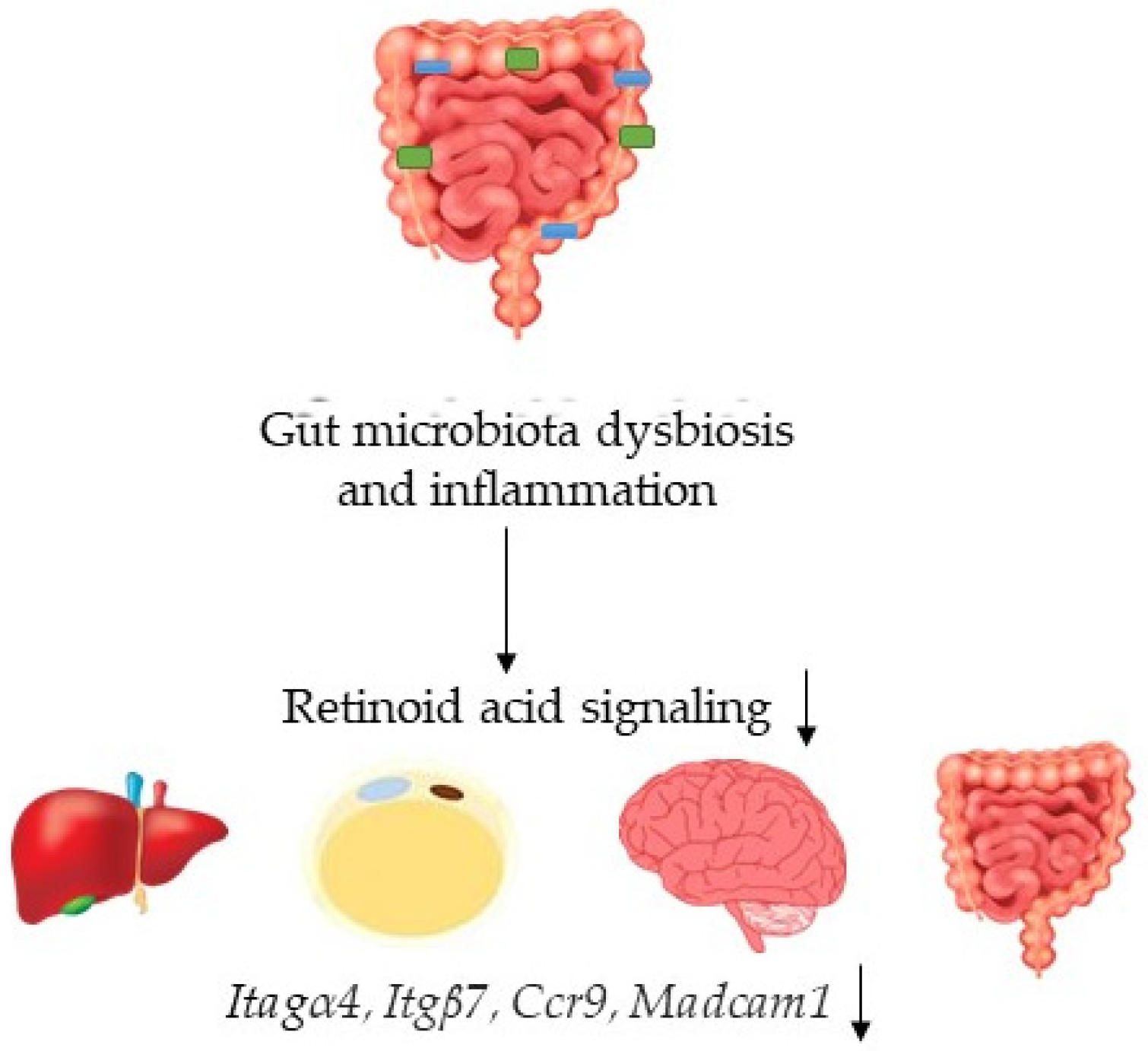
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burisch, J.; Munkholm, P. The epidemiology of inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Cosnes, J.; Gower-Rousseau, C.; Seksik, P.; Cortot, A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011, 140, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Yuille, S.; Reichardt, N.; Panda, S.; Dunbar, H.; Mulder, I.E. Human gut bacteria as potent class I histone deacetylase inhibitors in vitro through production of butyric acid and valeric acid. PLoS ONE 2018, 13, e0201073. [Google Scholar] [CrossRef] [Green Version]
- Grabiec, A.M.; Potempa, J. Epigenetic regulation in bacterial infections: Targeting histone deacetylases. Crit. Rev. Microbiol. 2018, 44, 336–350. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.J.; An, D.; Cai, Y.; Repa, J.J.; Hung-Po Chen, T.; Flores, M.; Postic, C.; Magnuson, M.A.; Chen, J.; Chien, K.R.; et al. Hepatocyte-specific mutation establishes retinoid X receptor alpha as a heterodimeric integrator of multiple physiological processes in the liver. Mol. Cell Biol. 2000, 20, 4436–4444. [Google Scholar] [CrossRef]
- He, Y.; Gong, L.; Fang, Y.; Zhan, Q.; Liu, H.X.; Lu, Y.; Guo, G.L.; Lehman-McKeeman, L.; Fang, J.; Wan, Y.J. The role of retinoic acid in hepatic lipid homeostasis defined by genomic binding and transcriptome profiling. BMC Genom. 2013, 14, 575. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Fu, X.; Liang, X.; Deavila, J.M.; Wang, Z.; Zhao, L.; Tian, Q.; Zhao, J.; Gomez, N.A.; Trombetta, S.C.; et al. Retinoic acid induces white adipose tissue browning by increasing adipose vascularity and inducing beige adipogenesis of PDGFRalpha(+) adipose progenitors. Cell Discov. 2017, 3, 17036. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; He, Y.; Liu, H.X.; Tsuei, J.; Jiang, X.; Yang, L.; Wang, Z.T.; Wan, Y.J. All-trans retinoic acid regulates hepatic bile acid homeostasis. Biochem. Pharm. 2014, 91, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trasino, S.E.; Tang, X.H.; Jessurun, J.; Gudas, L.J. Obesity Leads to Tissue, but not Serum Vitamin A Deficiency. Sci. Rep. 2015, 5, 15893. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Sheng, L.; Di Lucente, J.; Jin, L.W.; Maezawa, I.; Wan, Y.Y. Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J. 2018, 32, 2866–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waszkielewicz, A.M.; Gunia, A.; Szkaradek, N.; Sloczynska, K.; Krupinska, S.; Marona, H. Ion Channels as Drug Targets in Central Nervous System Disorders. Curr. Med. Chem. 2013, 20, 1241–1285. [Google Scholar] [CrossRef] [Green Version]
- Das, B.C.; Dasgupta, S.; Ray, S.K. Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer’s disease. Neural. Regen Res. 2019, 14, 1880–1892. [Google Scholar] [CrossRef]
- Xu, J.; Drew, P.D. 9-Cis-retinoic acid suppresses inflammatory responses of microglia and astrocytes. J. Neuroimmunol. 2006, 171, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Erkelens, M.N.; Mebius, R.E. Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends Immunol. 2017, 38, 168–180. [Google Scholar] [CrossRef]
- Bakdash, G.; Vogelpoel, L.T.; van Capel, T.M.; Kapsenberg, M.L.; de Jong, E.C. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal. Immunol. 2015, 8, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Taparowsky, E.J.; Kim, C.H. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 2015, 43, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.A.; Cannons, J.L.; Grainger, J.R.; Dos Santos, L.M.; Hand, T.W.; Naik, S.; Wohlfert, E.A.; Chou, D.B.; Oldenhove, G.; Robinson, M.; et al. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 2011, 34, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Mucida, D.; Pino-Lagos, K.; Kim, G.; Nowak, E.; Benson, M.J.; Kronenberg, M.; Noelle, R.J.; Cheroutre, H. Retinoic Acid Can Directly Promote TGF-beta-Mediated Foxp3(+) Treg Cell Conversion of Naive T Cells. Immunity 2009, 30, 471–472. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.; Zhang, Y.; Guo, Y.; Xie, J.; Wang, J.; He, X.; Lu, N.; Bai, A. All-trans retinoic acid attenuates experimental colitis through inhibition of NF-kappaB signaling. Immunol. Lett. 2014, 162, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Hana, K.; Nemoto, N.; Yoshida, T.; Saegusa, M.; Yokota-Nakatsuma, A.; Song, S.Y.; Iwata, M. Vitamin A Inhibits Development of Dextran Sulfate Sodium-Induced Colitis and Colon Cancer in a Mouse Model. Biomed. Res. Int. 2016, 2016, 4874809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, T.J.; McCarthy, N.E.; Giles, E.M.; Davidson, K.L.; Haltalli, M.L.; Hazell, S.; Lindsay, J.O.; Stagg, A.J. Increased production of retinoic acid by intestinal macrophages contributes to their inflammatory phenotype in patients with Crohn’s disease. Gastroenterology 2014, 146, 1278–1288.e1271-1272. [Google Scholar] [CrossRef] [PubMed]
- Oehlers, S.H.; Flores, M.V.; Hall, C.J.; Crosier, K.E.; Crosier, P.S. Retinoic acid suppresses intestinal mucus production and exacerbates experimental enterocolitis. Dis. Model. Mech. 2012, 5, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.A.; Grainger, J.R.; Spencer, S.P.; Belkaid, Y. The role of retinoic acid in tolerance and immunity. Immunity 2011, 35, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15–25. [Google Scholar] [CrossRef]
- Bell, C.J.; Gall, D.G.; Wallace, J.L. Disruption of colonic electrolyte transport in experimental colitis. Am. J. Physiol 1995, 268, G622–G630. [Google Scholar] [CrossRef]
- Neurath, M.F.; Fuss, I.; Kelsall, B.L.; Stuber, E.; Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 1995, 182, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- National Research Council Committee for the Update of the Guide for the Care and Use of Laboratory Animals. The National Academies Collection: Reports funded by National Institutes of Health. In Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Bushue, N.; Wan, Y.J. Retinoid pathway and cancer therapeutics. Adv. Drug Deliv. Rev. 2010, 62, 1285–1298. [Google Scholar] [CrossRef] [Green Version]
- Eksteen, B.; Liaskou, E.; Adams, D.H. Lymphocyte homing and its role in the pathogenesis of IBD. Inflamm. Bowel. Dis. 2008, 14, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim. Biophys. Acta 1999, 1440, 139–162. [Google Scholar] [CrossRef]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Odorico, A.; Bortolan, S.; Cardin, R.; D’Inca, R.; Martines, D.; Ferronato, A.; Sturniolo, G.C. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand. J. Gastroenterol. 2001, 36, 1289–1294. [Google Scholar] [PubMed]
- Hengstermann, S.; Valentini, L.; Schaper, L.; Buning, C.; Koernicke, T.; Maritschnegg, M.; Buhner, S.; Tillinger, W.; Regano, N.; Guglielmi, F.; et al. Altered status of antioxidant vitamins and fatty acids in patients with inactive inflammatory bowel disease. Clin. Nutr. 2008, 27, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Schippers, A.; Muschaweck, M.; Clahsen, T.; Tautorat, S.; Grieb, L.; Tenbrock, K.; Gassler, N.; Wagner, N. beta7-Integrin exacerbates experimental DSS-induced colitis in mice by directing inflammatory monocytes into the colon. Mucosal. Immunol. 2016, 9, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.; Colombel, J.F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.J.; Danese, S.; et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2013, 369, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Soler, D.; Chapman, T.; Yang, L.L.; Wyant, T.; Egan, R.; Fedyk, E.R. The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J. Pharm. Exp. 2009, 330, 864–875. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.L.; Zheng, Y.J.; Pan, Y.D.; Xie, C.; Sun, H.; Zhang, Y.H.; Yuan, M.Y.; Song, B.L.; Chen, J.F. Regulatory T-cell depletion in the gut caused by integrin beta7 deficiency exacerbates DSS colitis by evoking aberrant innate immunity. Mucosal. Immunol. 2016, 9, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Li, L.J.; Gong, C.; Zhao, M.H.; Feng, B.S. Role of interleukin-22 in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 18177–18188. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielke, L.A.; Jones, S.A.; Raverdeau, M.; Higgs, R.; Stefanska, A.; Groom, J.R.; Misiak, A.; Dungan, L.S.; Sutton, C.E.; Streubel, G.; et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J. Exp. Med. 2013, 210, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. 2013, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Galvez, J. Role of Th17 Cells in the Pathogenesis of Human IBD. ISRN Inflamm 2014, 2014, 928461. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [Green Version]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [Green Version]
- Seiderer, J.; Elben, I.; Diegelmann, J.; Glas, J.; Stallhofer, J.; Tillack, C.; Pfennig, S.; Jurgens, M.; Schmechel, S.; Konrad, A.; et al. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): Upregulated colonic IL-17F expression in active Crohn’s disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm. Bowel. Dis. 2008, 14, 437–445. [Google Scholar] [CrossRef]
- Yang, X.O.; Chang, S.H.; Park, H.; Nurieva, R.; Shah, B.; Acero, L.; Wang, Y.H.; Schluns, K.S.; Broaddus, R.R.; Zhu, Z.; et al. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 2008, 205, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Gianotti, R.J.; Moss, A.C. Fecal Microbiota Transplantation: From Clostridium difficile to Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2017, 13, 209–213. [Google Scholar]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, L.M.; Kawamoto, S.; Maruya, M.; Fagarasan, S. The role of the adaptive immune system in regulation of gut microbiota. Immunol. Rev. 2014, 260, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef]
- Zhu, Q.; Jin, Z.; Wu, W.; Gao, R.; Guo, B.; Gao, Z.; Yang, Y.; Qin, H. Analysis of the intestinal lumen microbiota in an animal model of colorectal cancer. PLoS ONE 2014, 9, e90849. [Google Scholar] [CrossRef]
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal microbiota, microbial translocation, and systemic inflammmation in chronic HIV infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liao, X.; Sparks, J.B.; Luo, X.M. Dynamics of gut microbiota in autoimmune lupus. Appl. Environ. Microbiol. 2014, 80, 7551–7560. [Google Scholar] [CrossRef] [Green Version]
- Navaneethan, U.; Venkatesh, P.G.; Shen, B. Clostridium difficile infection and inflammatory bowel disease: Understanding the evolving relationship. World J. Gastroenterol. 2010, 16, 4892–4904. [Google Scholar] [CrossRef]
- Chen, Y.; Furuya-Kanamori, L.; Doi, S.A.; Ananthakrishnan, A.N.; Kirk, M. Clostridium difficile Infection and Risk of Colectomy in Patients with Inflammatory Bowel Disease: A Bias-adjusted Meta-analysis. Inflamm. Bowel Dis. 2017, 23, 200–207. [Google Scholar] [CrossRef]
- Sheng, L.; Jena, P.K.; Liu, H.X.; Hu, Y.; Nagar, N.; Bronner, D.N.; Settles, M.L.; Baumler, A.J.; Wan, Y.Y. Obesity treatment by epigallocatechin-3-gallate-regulated bile acid signaling and its enriched Akkermansia muciniphila. FASEB J. 2018, 32, 6371–6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol 2011, 17, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Duncan, S.H.; McCrae, S.I.; Millar, J.; Jackson, M.S.; Flint, H.J. Restricted distribution of the butyrate kinase pathway among butyrate-producing bacteria from the human colon. J. Bacteriol. 2004, 186, 2099–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharm. 2008, 27, 104–119. [Google Scholar] [CrossRef]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 2004, 91, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Pimental, M.G.R.; Rao, S.S.; Zhang, H. Methanogens in human health and disease. Am. J. Gastroenterol. 2012, 6, 28–33. [Google Scholar] [CrossRef]
- Pique, J.M.; Pallares, M.; Cuso, E.; Vilarbonet, J.; Gassull, M.A. Methane Production and Colon Cancer. Gastroenterology 1984, 87, 601–605. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphology of the Colon and Ileum | Score |
|---|---|
| No ulcers, no inflammation | 0 |
| No ulcers, local mucosal congestion and edema | 1 |
| Single ulceration, but no hyperemia of mucosa | 2 |
| Single ulceration, mucosal inflammation | 3 |
| Multiple ulcers and mucosal inflammation | 4 |
| Ulcer area >2 cm, severe erosion, mucosal hyperemia and edema | 5 |
| Histopathological Manifestations of the Ileum and Colon | Score |
|---|---|
| No obvious inflammation reaction | 0 |
| Low-level inflammatory cell infiltration, no damage to intestinal villi structure | 1 |
| Moderate inflammatory cell infiltration, deepening of the intestinal crypts, thickening of the intestinal wall but not invading the muscle layer | 2 |
| High levels of inflammatory cell infiltration, vascular proliferation, thickening of the intestinal wall and invasion of the muscle layer | 3 |
| Infiltration of inflammatory cells, reduction in goblet cells, vascular proliferation, thickening of the intestinal wall and invasion of the muscle layer | 4 |
| Gene | Primers (5′–3′) | Tm | Amplicon Size | NCBI Accession Number |
|---|---|---|---|---|
| Aldh1a1 | F:AGGCCCTCAGATTGACAAGGAACA | 63.89 | 135 | NM_013467 |
| R:AACACTGTGGGCTGCACAAAGAAG | 64.25 | |||
| Ccl25 | F:CCAAGGTGCCTTTGAAGACT | 58.01 | 400 | NM_009138 |
| R:TCCTCCAGCTGGTGGTTACT | 60.18 | |||
| Ccr9 | F:CTTGCCACTCTTCCCTTCTG | 58.18 | 170 | NM_001166625 |
| R:GCCTTCATGGCCTGTACAAT | 58.23 | |||
| Crabp1 | F:GCTGGCCAACGATGAGCTAA | 60.74 | 74 | NM_013496 |
| R:ACTCCCGGACATAAATTCT GTG | 58.74 | |||
| Crbp1 | F:AGCAGGTGAGAAGGGATAAAG GT | 61.08 | 75 | NM_011254 |
| R:CGTGTTCCGTGGCTTCTGAT | 60.67 | |||
| Cyp26a1 | F:GCACAAGCAGCGAAGAAGGTGAT | 64.38 | 104 | NM_007811 |
| R:ACTGCTCCAGACAACTGCTGACTT | 64.31 | |||
| Cyp26b1 | F:GGCGGCTACCGCACTGT | 62.54 | 88 | NM_175475 |
| R:TGTCTCGGATGCTAT CATGACACT | 61.44 | |||
| Il1β | F:AAGATGAAGGGCTGCTTCCA | 59.30 | 78 | NM_008361 |
| R:GTGCTGCTGCGAGATTTGAA | 59.48 | |||
| Il-17a | F:TCCAGAAGGCCCTCAGACTA | 59.29 | 248 | NM_010552 |
| R:ACACCCACCAGCATCTTCTC | 59.67 | |||
| Il22 | F:TTGAGGTGTCCAACTTCCAGCA | 62.08 | 97 | NM_016971 |
| R:AGCCGGACGTCTGTGTTGTTA | 61.96 | |||
| Il-6 | F:GTTGCCTTCTTGGGACTGATG | 59.18 | 90 | NM_001314054 |
| R:GGGAGTGGTATCCTCTGTGAAGTCT | 63.10 | |||
| Infγ | F:AGCTCTTCCTCATGGCTGTT | 59.01 | 148 | NM_008337 |
| R:TCCTTTTGCCAGTTCCTCCA | 59.15 | |||
| Itgα4 | F:GCCTGGAGGAGAGGGATAAC | 58.95 | 159 | NM_010576 |
| R:CAGAAGGCATGACGTAGCAA | 58.27 | |||
| Itgβ7 | F:CTACGACTCTGGGCTCTTGG | 59.54 | 193 | NM_013566 |
| R:ACAGGTCAGCCTCAGAGCAT | 60.91 | |||
| Madcam1 | F:GCATGGTGACCTGGCAGT GAAG | 63.96 | 397 | NM_001358785 |
| R:GGCAGCAGTATCCTCTCTGTAC | 59.96 | |||
| Rarβ | F:GCACTGACGCCATAGTGGTA | 59.83 | 88 | NM_001289762 |
| R:CACCATCTCCACTTCCTCCT | 58.42 | |||
| Rbp4 | F:TCTGTGGACGAGAAGGGTCAT | 60.55 | 72 | NM_001159487 |
| R:CACTTCCCAGTGCTCAGAAGA | 60.22 | |||
| Rorγt | F:ACCTCCATGCCAGCTGTGTGCTGTC | 70.66 | 725 | NM_011281 |
| R:CAAGTTCAGGATGCCTGGTTTCCTC | 63.79 | |||
| Tnfα | F:TCGAGTGACAAGCCTGTAG | 56.51 | 121 | NM_013693 |
| R:GTTGGTTGTCTTTGAGATCC | 54.22 |
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| acs | CTYTGYCAGTCMTTYGCBCC | CCCATAAABCCYGGDGTYTG |
| baij | TCAGGACGTGGAGGCGATCCA | TACRTGATACTGGTAGCTCCA |
| bcoa | GCIGAICATTTCACITGGAAYWSITGGCAYATG | CCTGCCTTTGCAATRTCIACRAANGC |
| bsh | ATGGGCGGACTAGGATTACC | TGCCACTCTCTGTCTGCATC |
| dsra | GCCGTTACTGTGACCAGCC | GGTGGAGCCGTGCATGTT |
| mcra | TTCGGTGGATCDCARAGRGC | GBARGTCGWAWCCGTAGAATCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Sheng, L.; Jena, P.K.; Gilbert, M.C.; Wan, Y.-J.Y.; Mao, H. Retinoic Acid Signaling Is Compromised in DSS-Induced Dysbiosis. Nutrients 2022, 14, 2788. https://doi.org/10.3390/nu14142788
Li Y, Sheng L, Jena PK, Gilbert MC, Wan Y-JY, Mao H. Retinoic Acid Signaling Is Compromised in DSS-Induced Dysbiosis. Nutrients. 2022; 14(14):2788. https://doi.org/10.3390/nu14142788
Chicago/Turabian StyleLi, Yongchun, Lili Sheng, Prasant Kumar Jena, Miranda Claire Gilbert, Yu-Jui Yvonne Wan, and Hua Mao. 2022. "Retinoic Acid Signaling Is Compromised in DSS-Induced Dysbiosis" Nutrients 14, no. 14: 2788. https://doi.org/10.3390/nu14142788
APA StyleLi, Y., Sheng, L., Jena, P. K., Gilbert, M. C., Wan, Y.-J. Y., & Mao, H. (2022). Retinoic Acid Signaling Is Compromised in DSS-Induced Dysbiosis. Nutrients, 14(14), 2788. https://doi.org/10.3390/nu14142788