
The Comparative Analysis of Genomic Diversity and Genes Involved in Carbohydrate Metabolism of Eighty-Eight Bifidobacterium pseudocatenulatum Isolates from Different Niches of China

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Probiotic Strains and Culture
2.2. Genome Assembly, Genome Annotation and Pangenome Analysis
2.3. Animals and Experimental Design
2.4. Biochemical Parameters
2.5. Gut Microbiota Sequencing and Short Chain Fatty Acids (SCFA) Determination
2.6. Statistical Analysis
3. Results
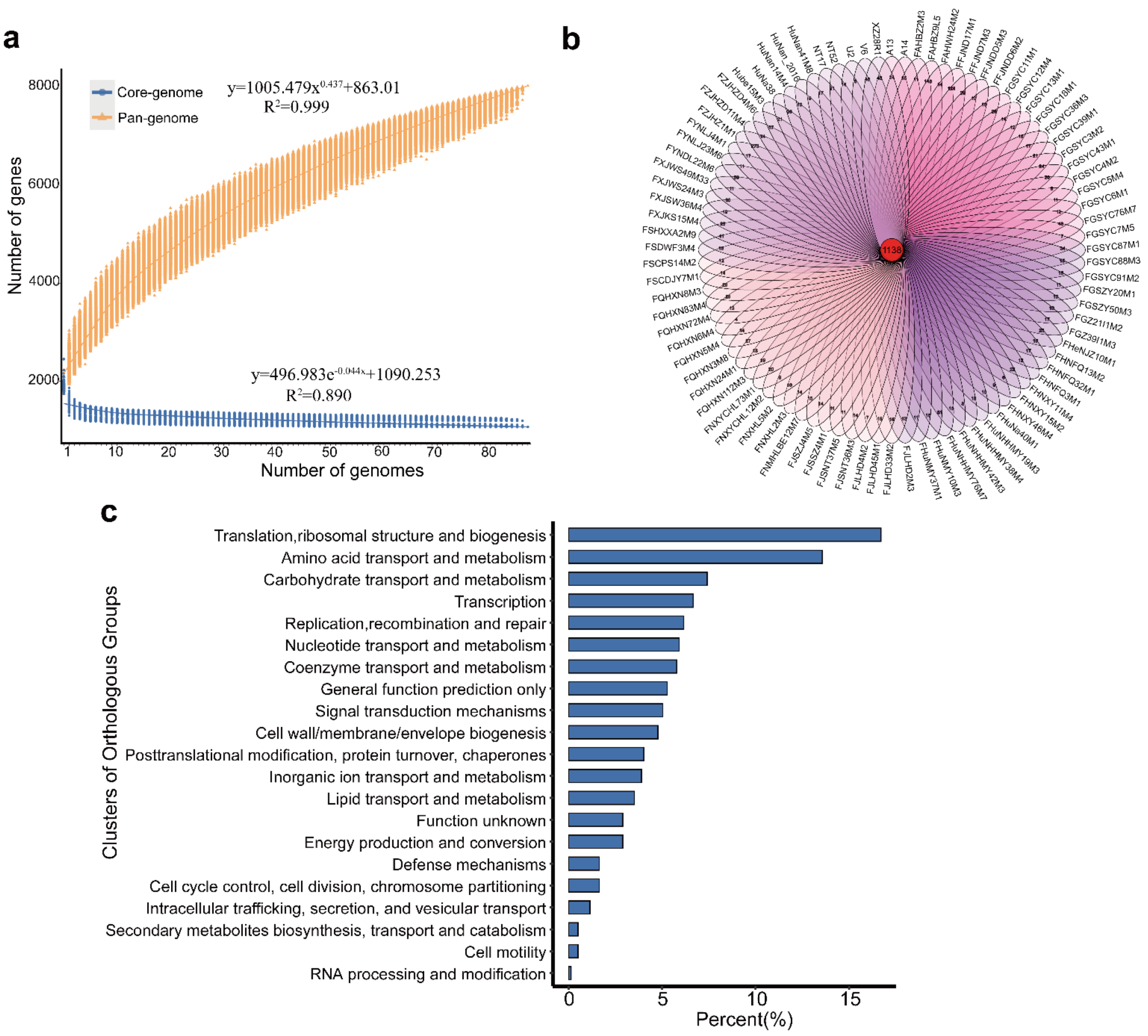
3.1. General Features of the Genomes of B. pseudocatenulatum Strains
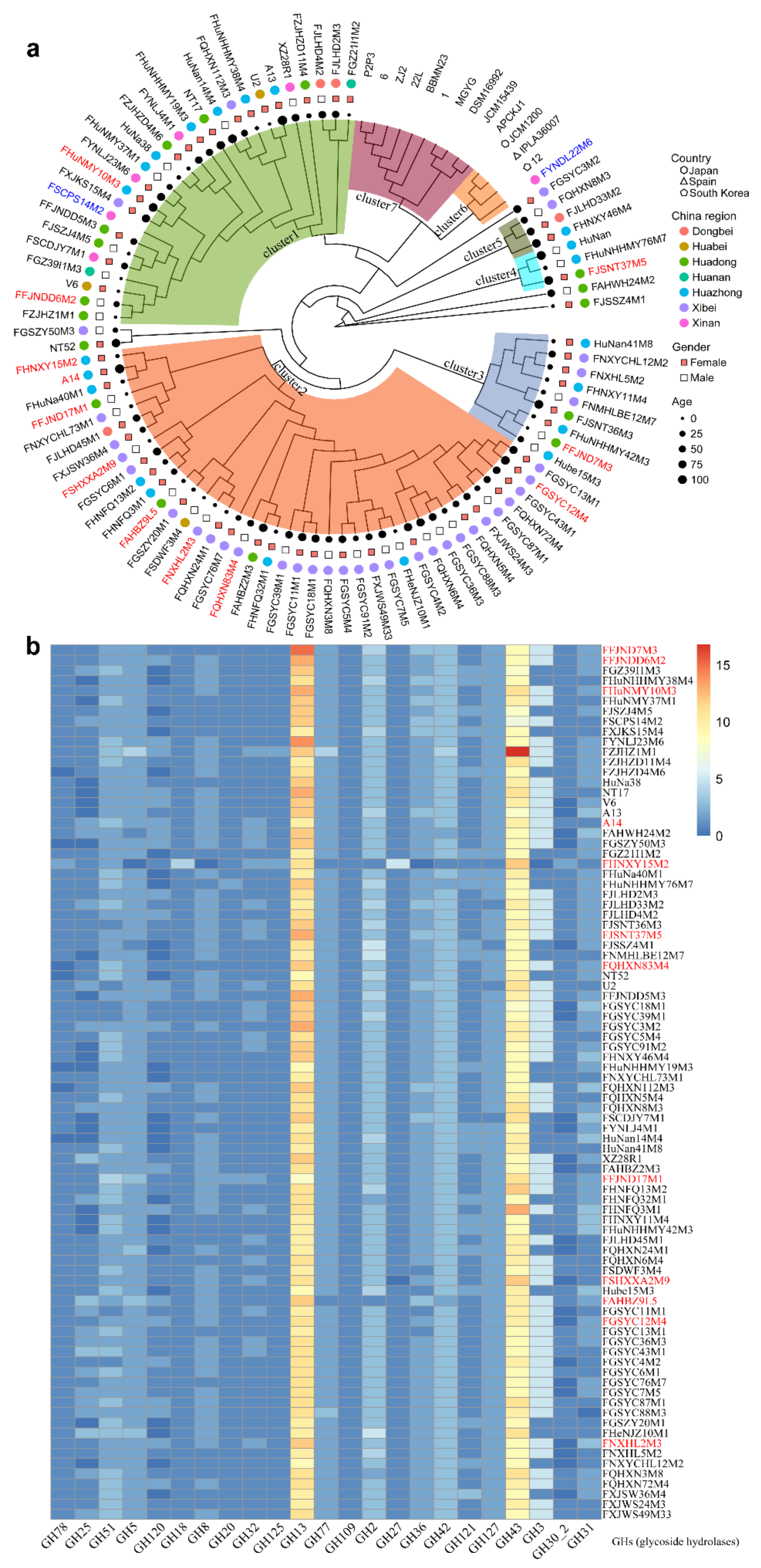
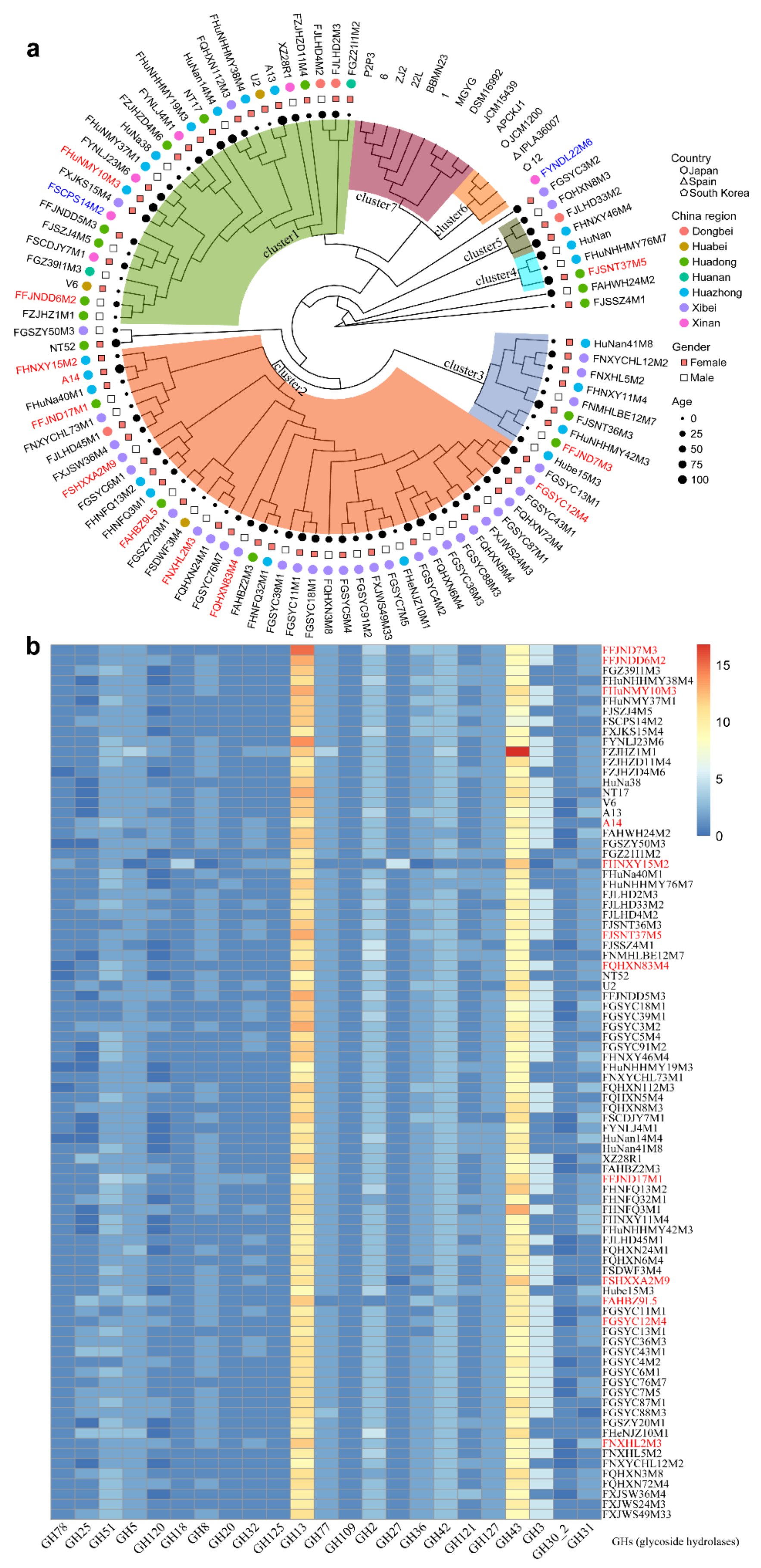
3.2. B. pseudocatenulatum Genomic Diversity
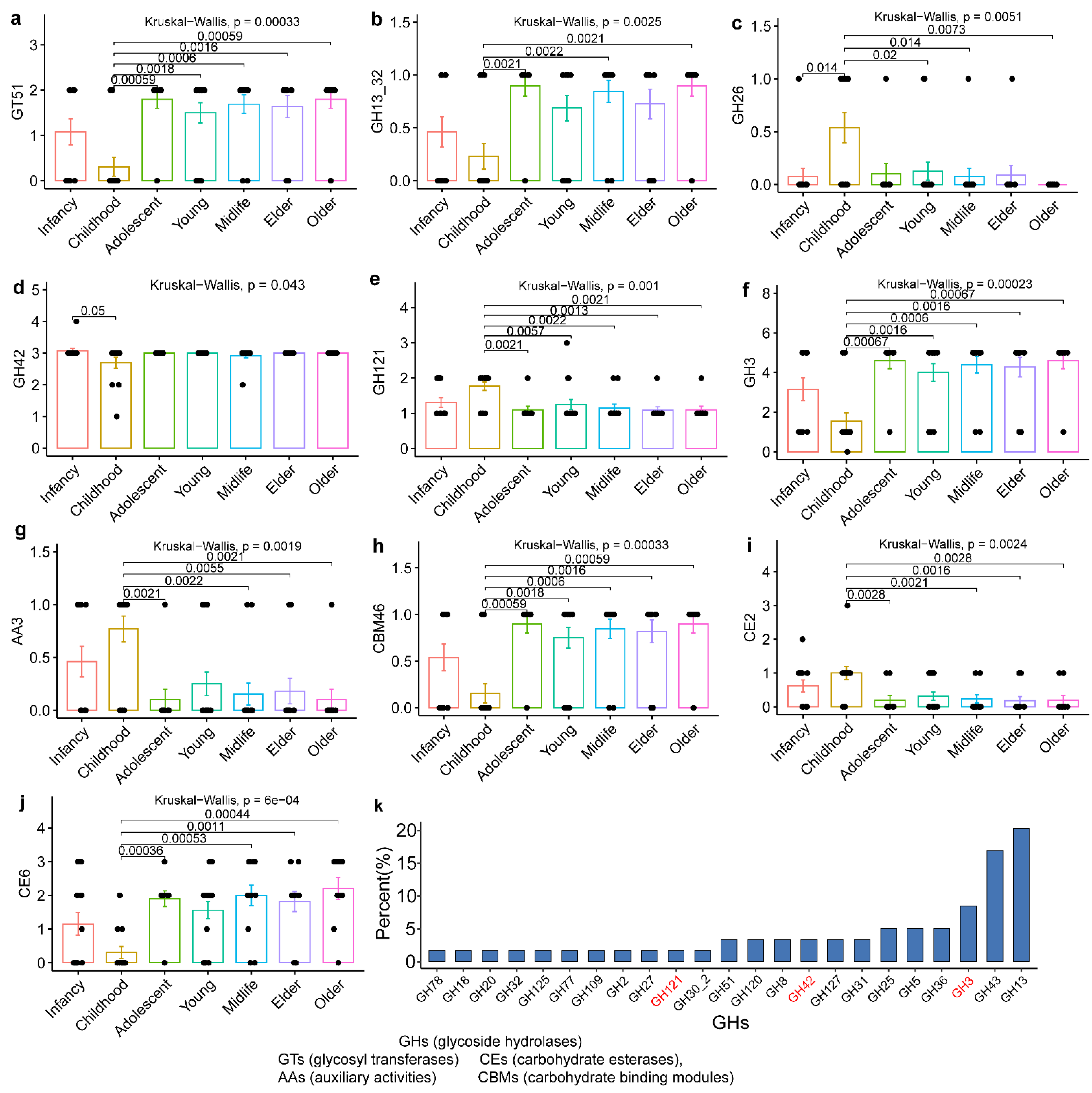
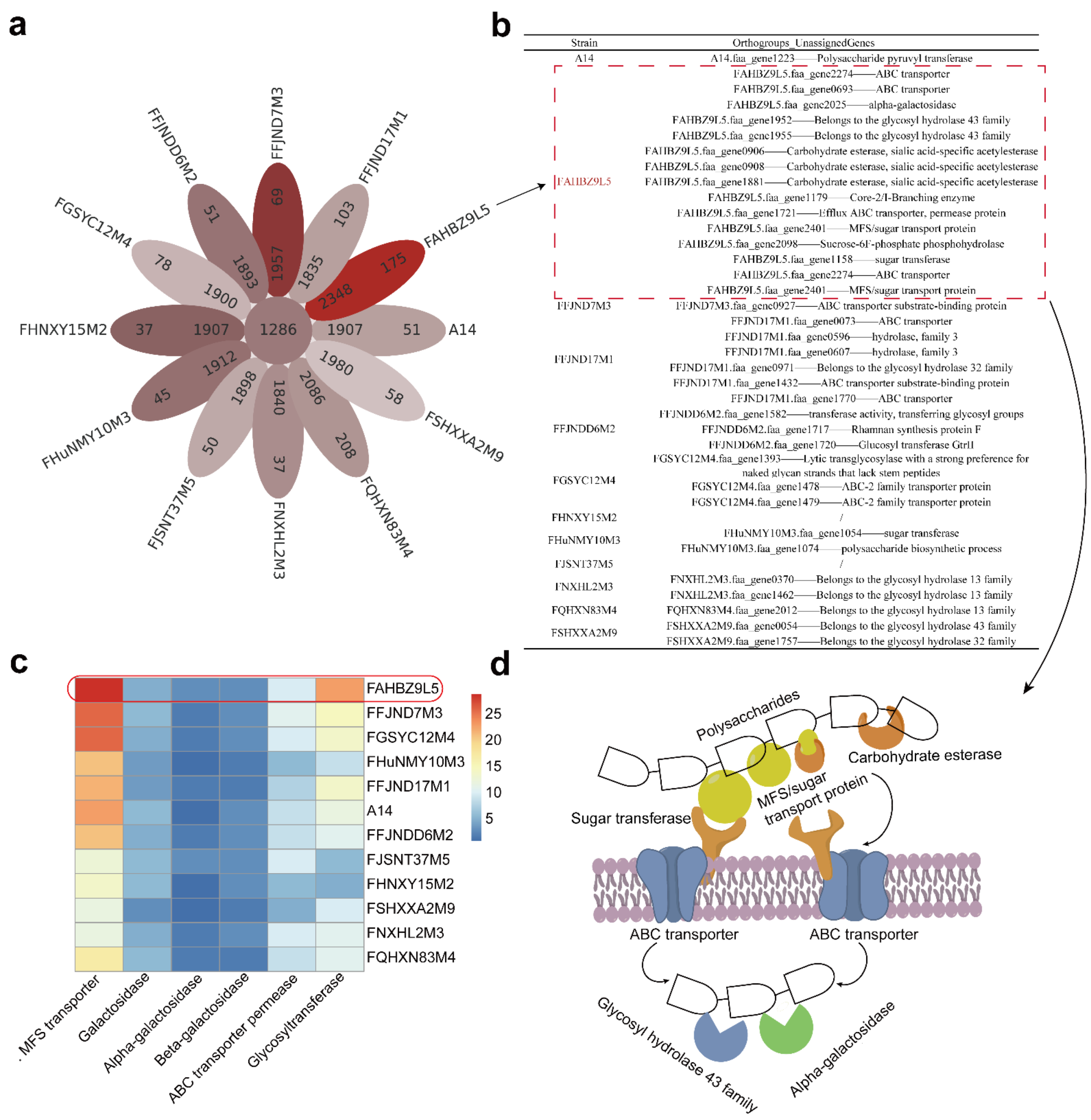
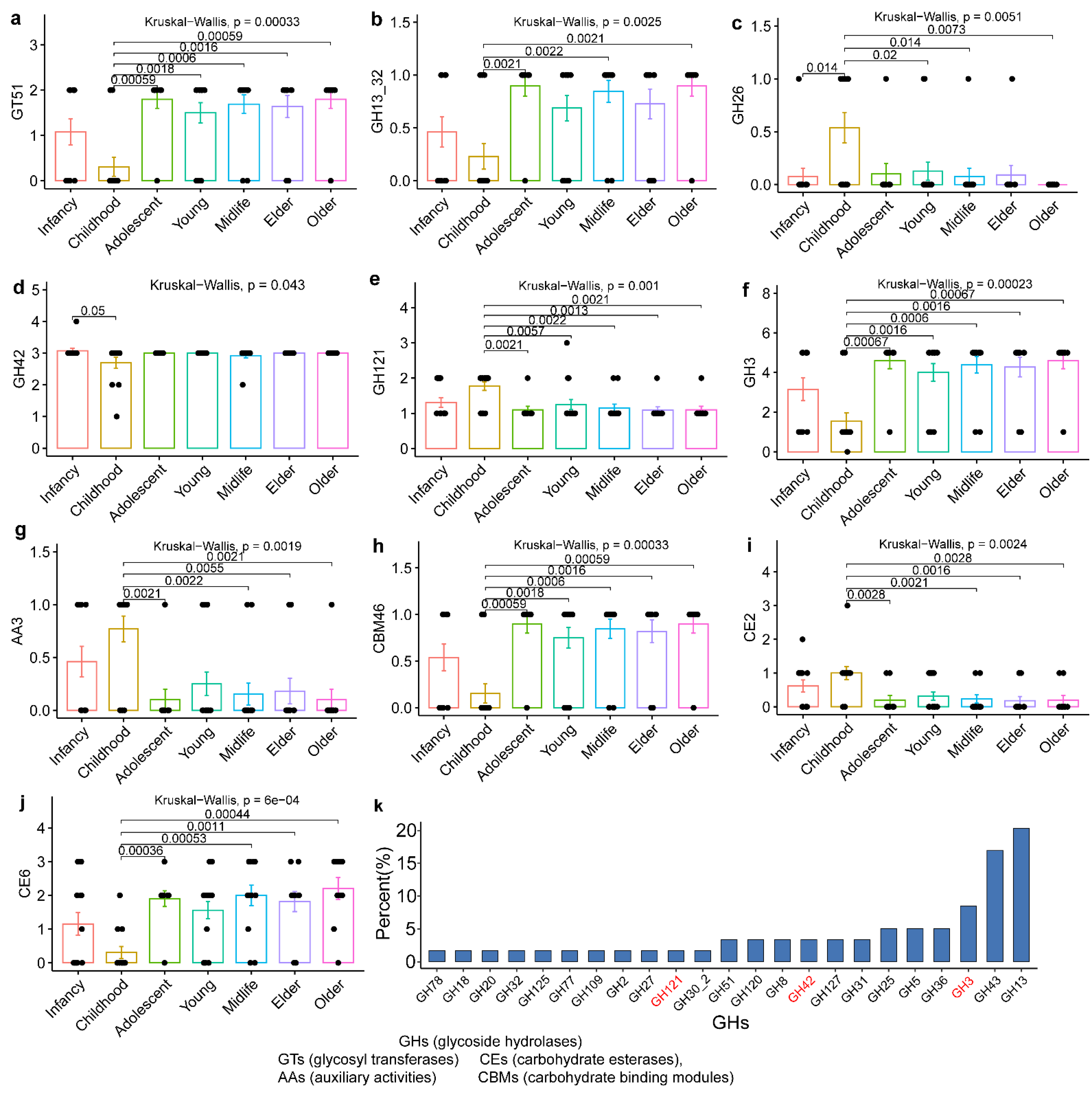
3.3. Evolutionary Genomic Diversity, the GH Enzymes, Orthogroups Unassigned and Antibiotic Resistance Genes of B. pseudocatenulatum
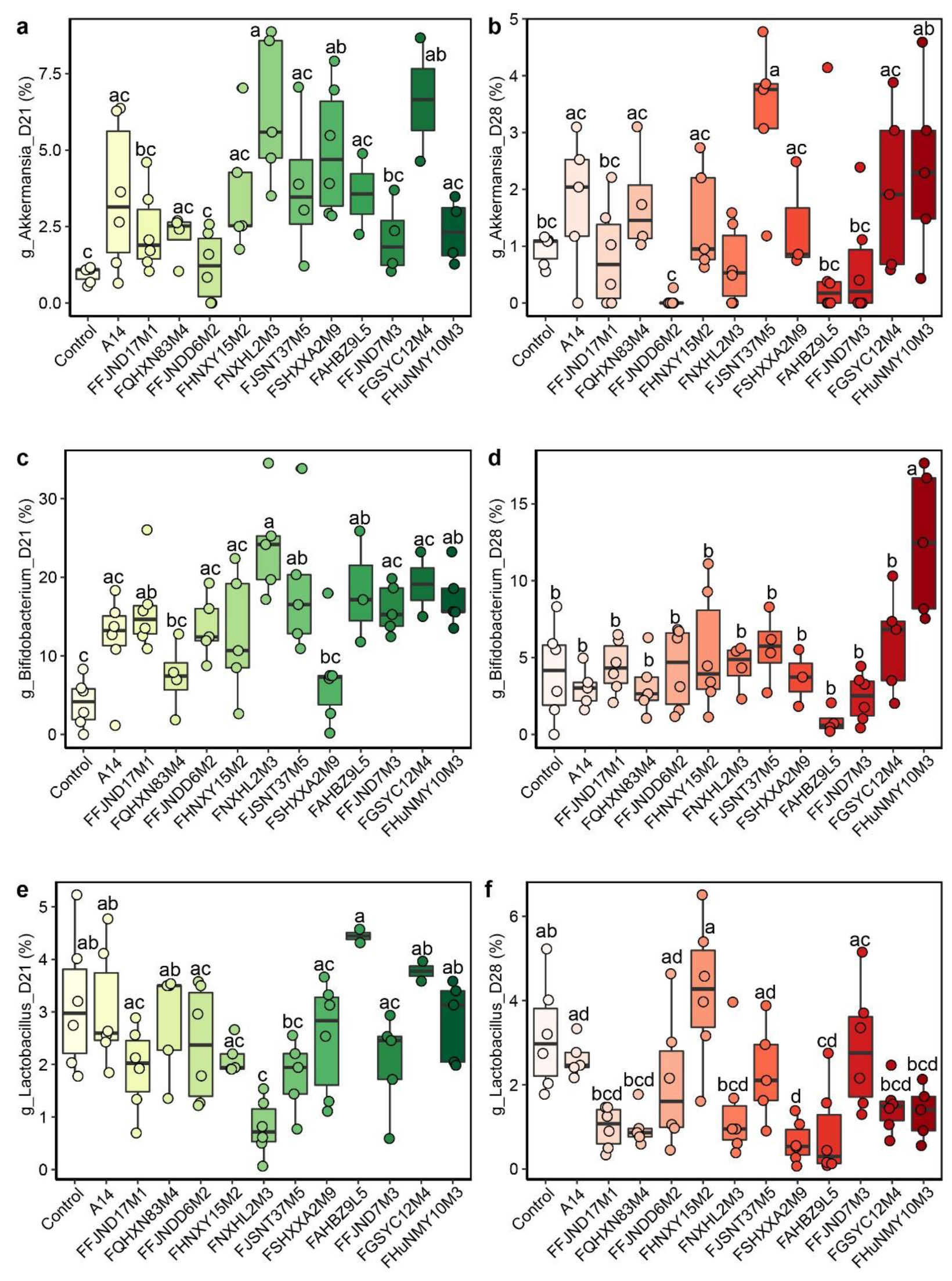
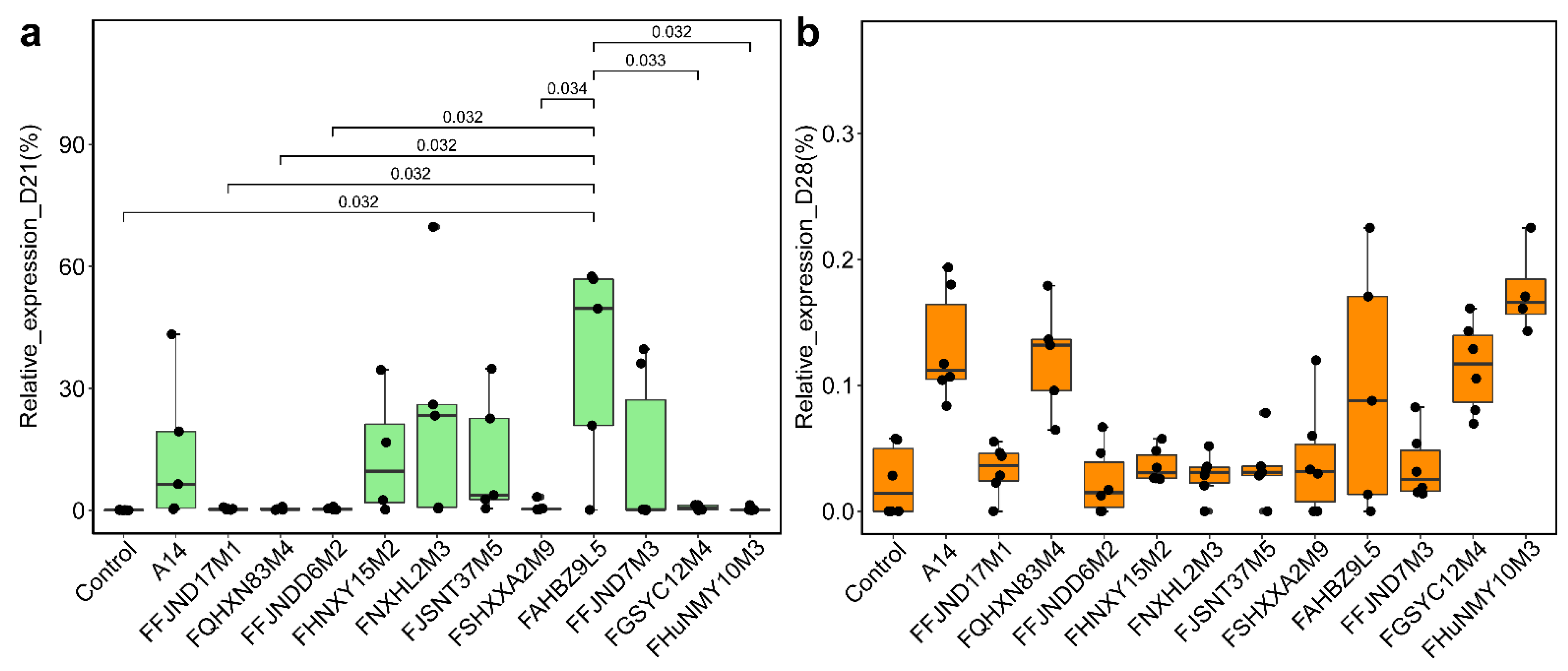
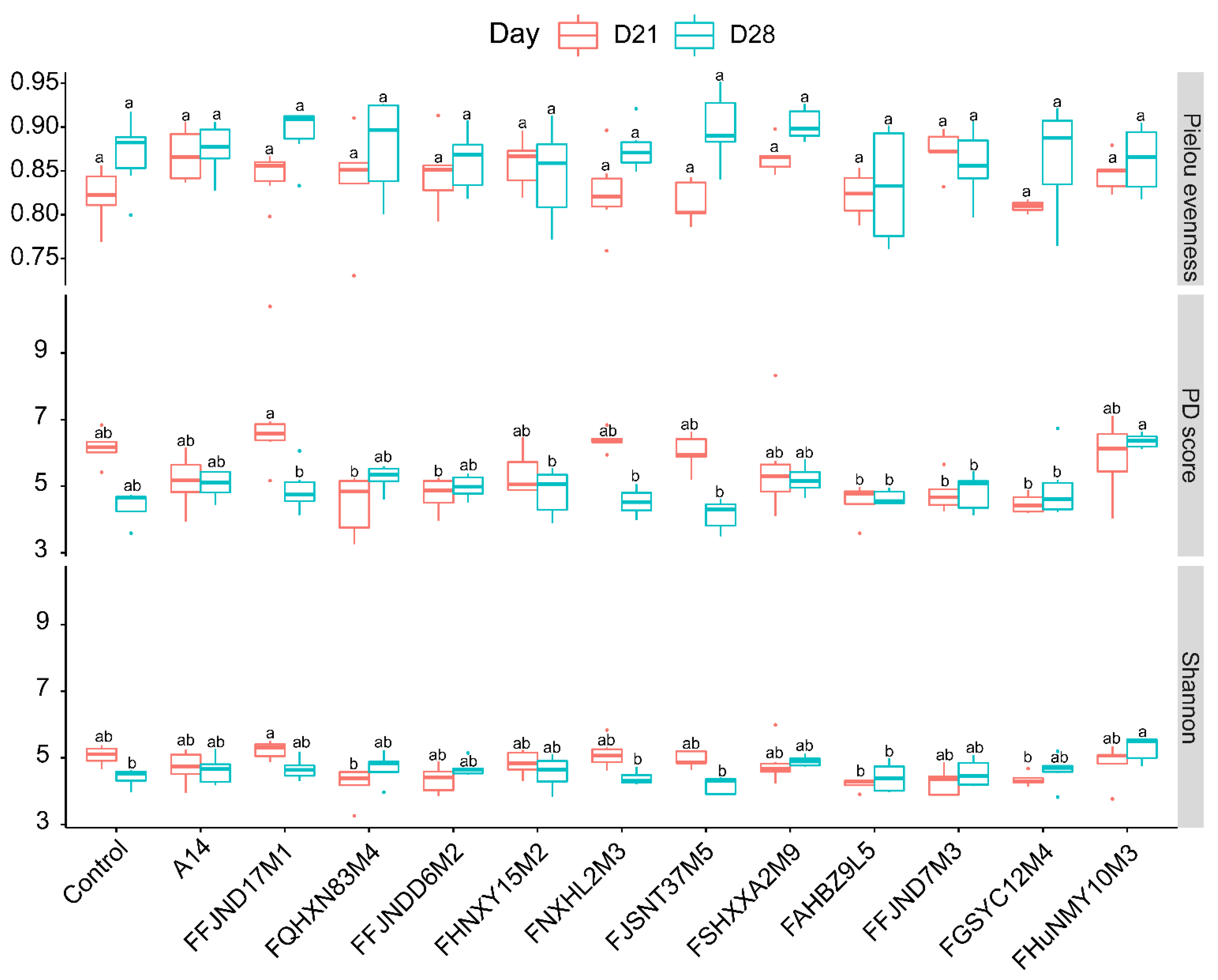
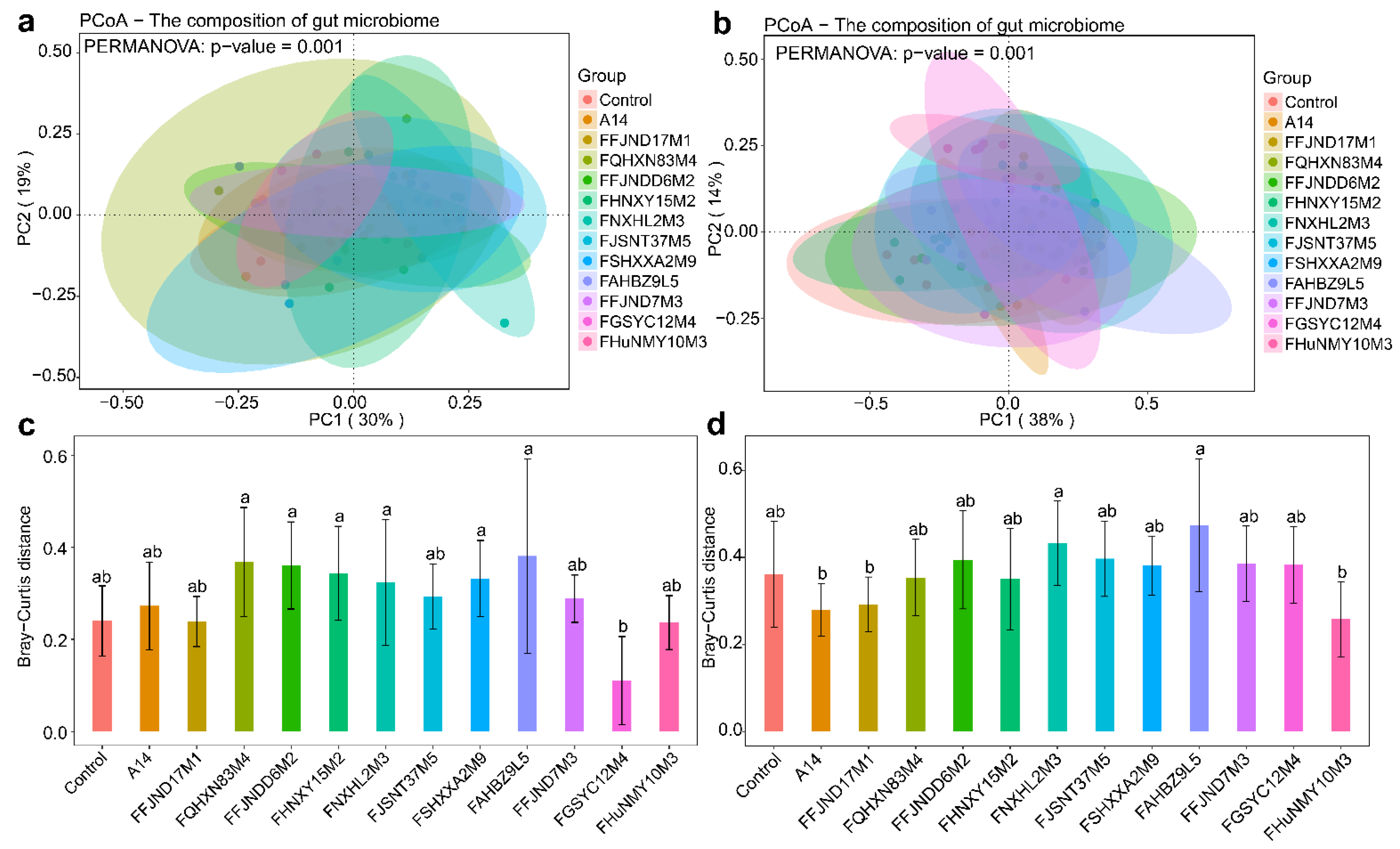
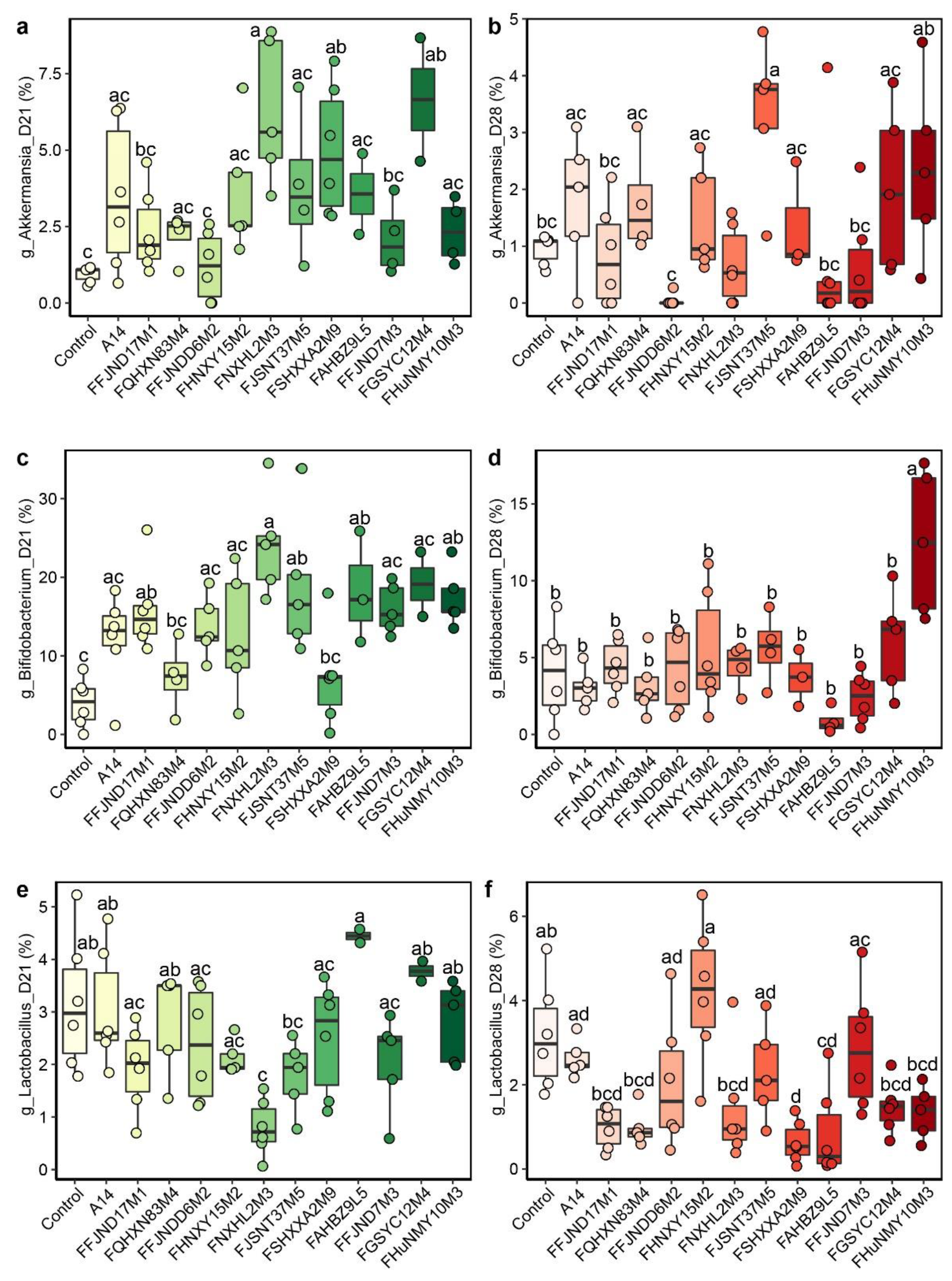
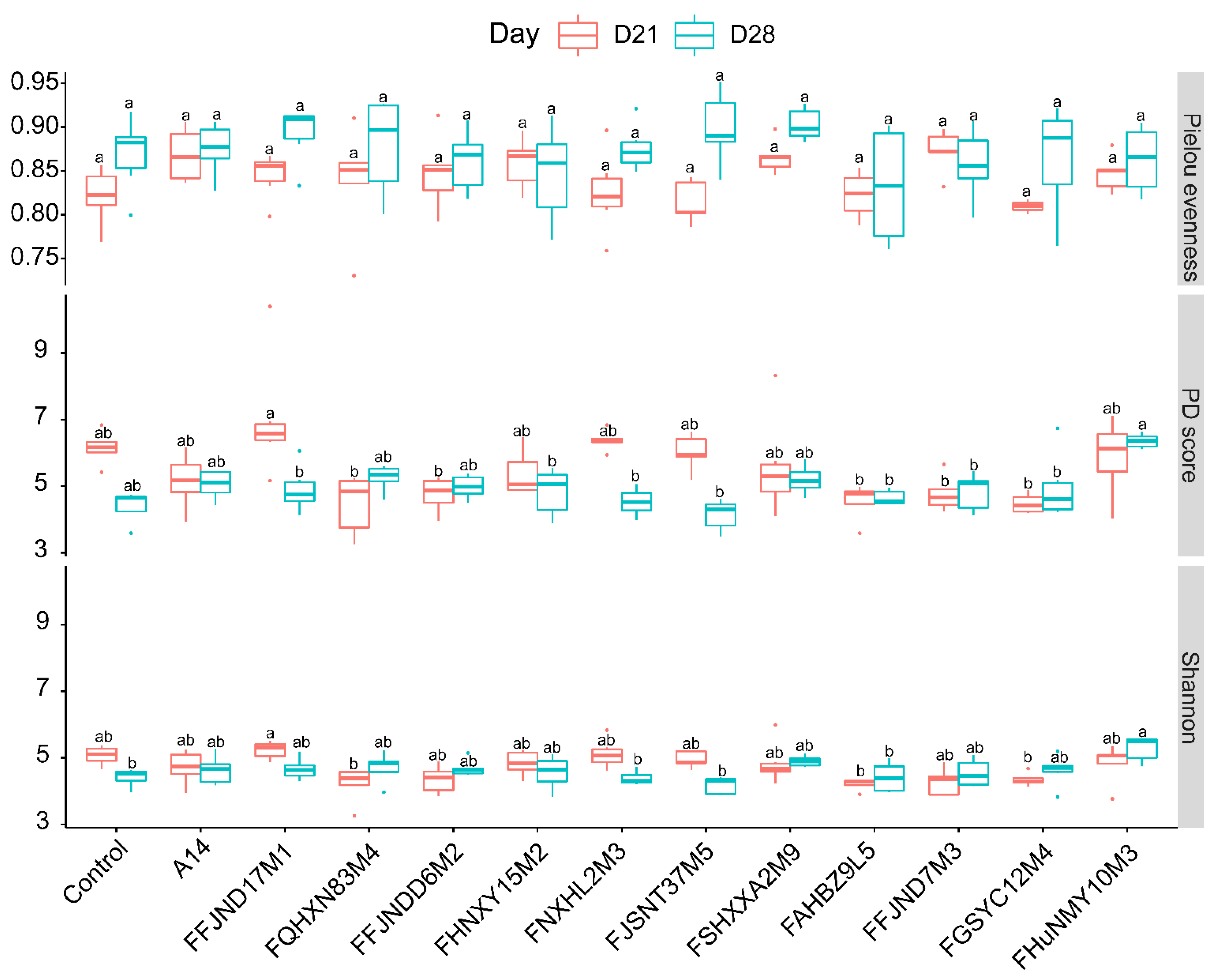
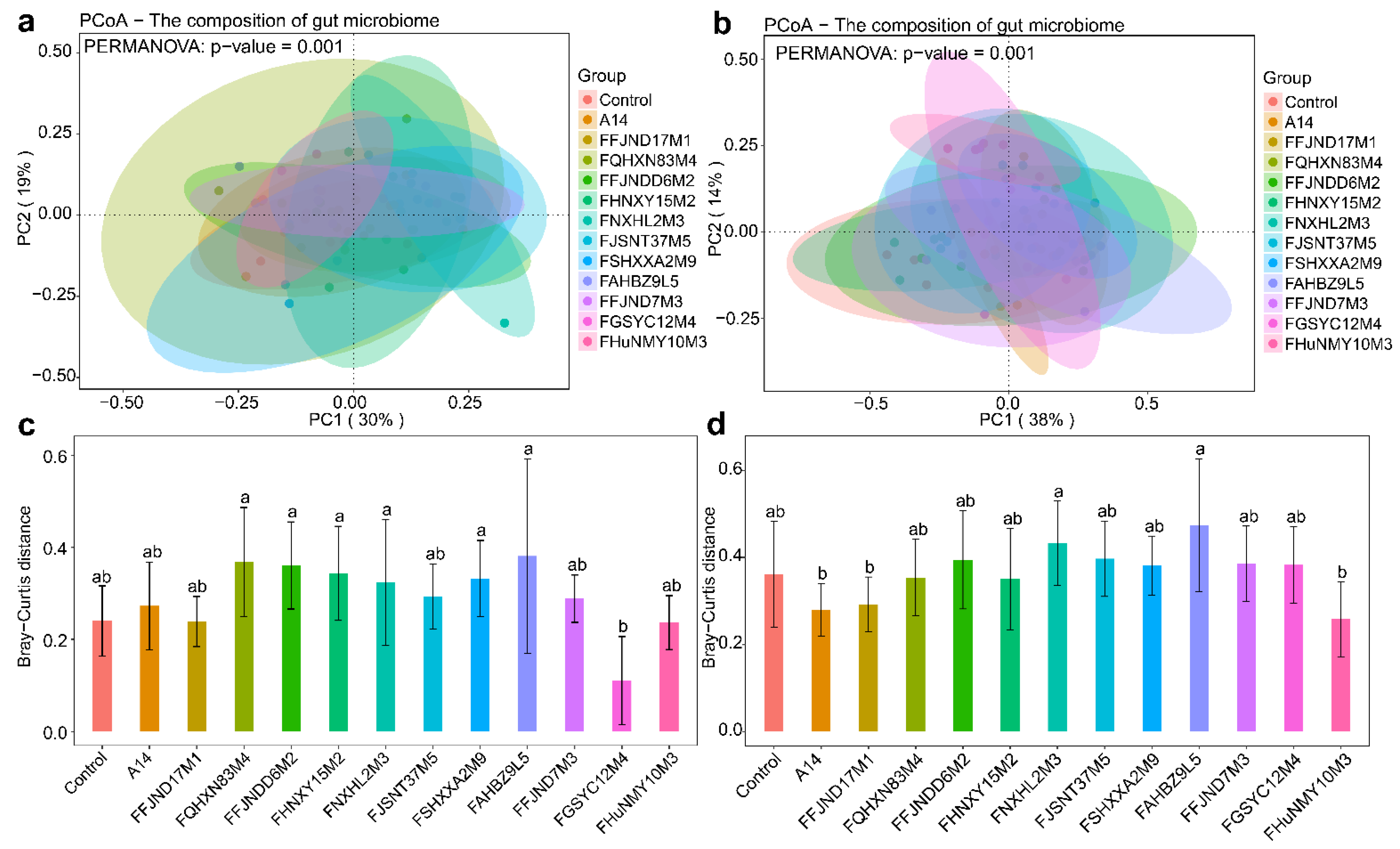
3.4. Effects of B. pseudocatenulatum on the Gut Microbiota in Mice
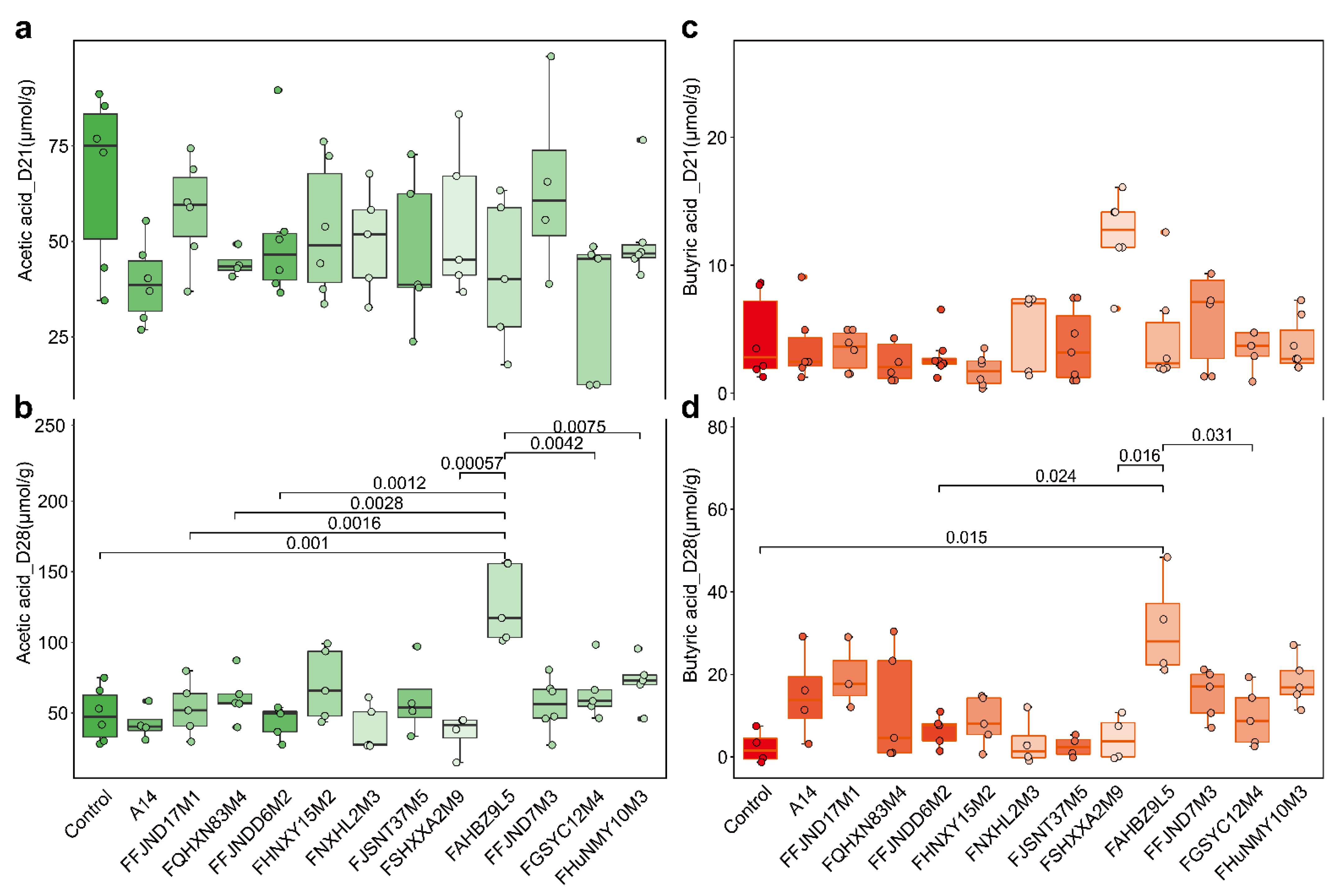
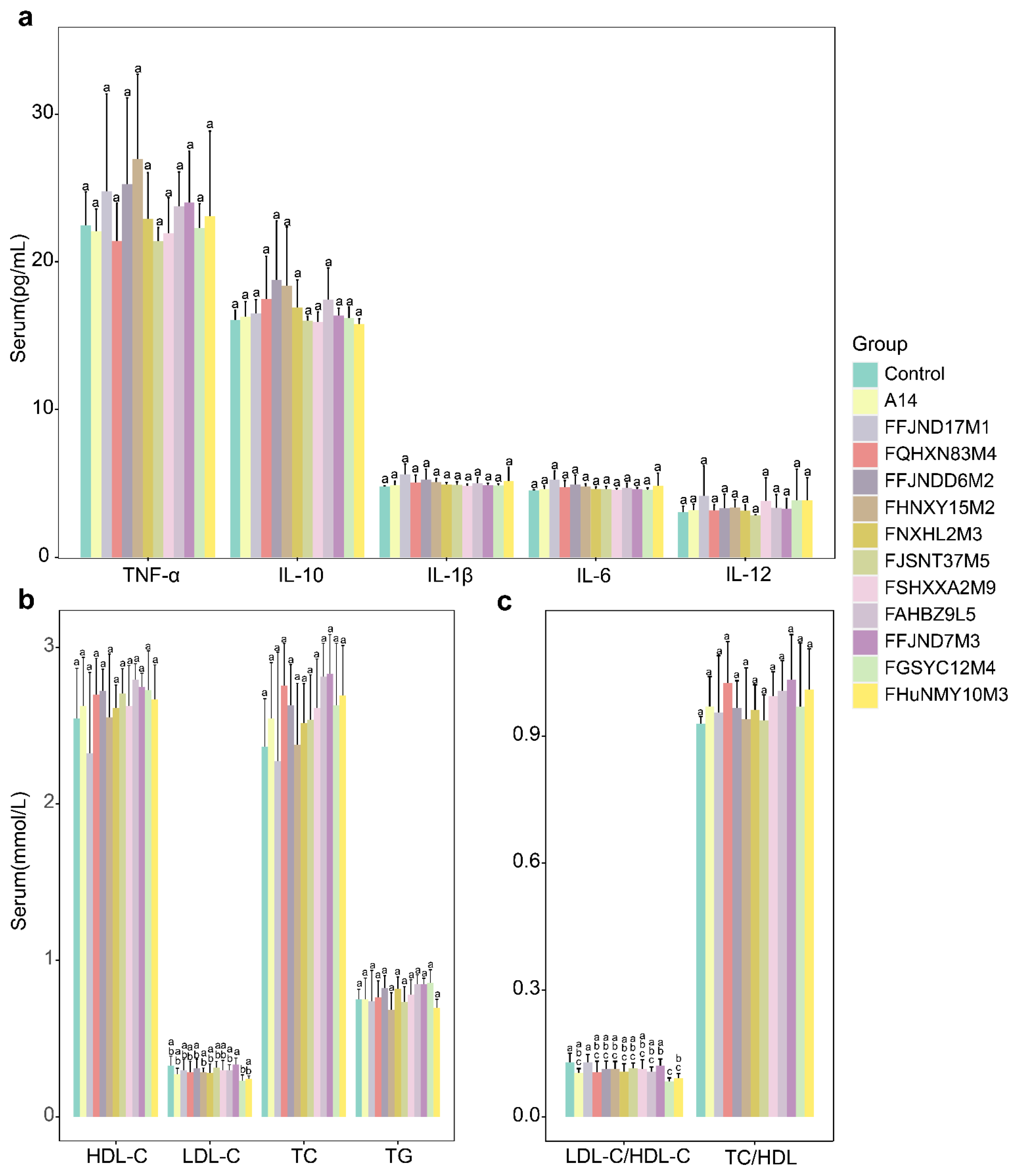
3.5. Effects of B. pseudocatenulatum on Serum Biochemical Indexes and Short Chain Fatty Acids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turroni, F.; Peano, C.; Pass, D.A.; Foroni, E.; Severgnini, M.; Claesson, M.J.; Kerr, C.; Hourihane, J.; Murray, D.; Fuligni, F.; et al. Diversity of Bifidobacteria within the infant gut microbiota. PLoS ONE 2012, 7, e36957. [Google Scholar] [CrossRef] [Green Version]
- Gore, C.; Munro, K.; Lay, C.; Bibiloni, R.; Morris, J.; Woodcock, A.; Custovic, A.; Tannock, G.W. Bifidobacterium pseudocatenulatum is associated with atopic eczema: A nested case-control study investigating the fecal microbiota of infants. J. Allergy Clin. Immunol. 2008, 121, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhang, C.; Wu, H.; Wang, R.; Shen, J.; Wang, L.; Zhao, Y.; Pang, X.; Zhang, X.; Zhao, L.; et al. Genomic microdiversity of Bifidobacterium pseudocatenulatum underlying differential strain-level responses to dietary carbohydrate intervention. mBio 2017, 8, e02348-16. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.; Neef, A.; Sanz, Y. Bifidobacterium pseudocatenulatum CECT 7765 reduces obesity-associated inflammation by restoring the lymphocyte-macrophage balance and gut microbiota structure in high-fat diet-fed mice. PLoS ONE 2015, 10, e0126976. [Google Scholar] [CrossRef]
- Yang, C.; Bo, Y.; Stanton, C.; Ross, R.P.; Jianxin, Z.; Hao, Z.; Wei, C. Bifidobacterium pseudocatenulatum ameliorates DSS-induced colitis by maintaining intestinal mechanical barrier, blocking proinflammatory cytokines, inhibiting TLR4/NF-kappaB signaling, and altering gut microbiota. J. Agric. Food Chem. 2021, 69, 1496–1512. [Google Scholar] [CrossRef]
- Gauffin-Cano, P.; Santacruz, A.; Trejo, F.M.; Sanz, Y. Bifidobacterium CECT 7765 improves metabolic and immunological alterations associated with obesity in high-fat diet-fed mice. Obesity 2013, 21, 2310–2321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Chen, H.; Stanton, C.; Chen, Y.Q.; Zhang, H.; Chen, W. Mining bifidobacteria from the neonatal gastrointestinal tract for conjugated linolenic acid production. Bioengineered 2016, 8, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Gaya, P.; Peirotén, A.; Medina, M.; Álvarez, I.; Landete, J.M. Bifidobacterium pseudocatenulatum INIA P815: The first bacterium able to produce urolithins A and B from ellagic acid. J. Funct. Foods 2018, 45, 95–99. [Google Scholar] [CrossRef]
- Peirotén, A.; Gaya, P.; Álvarez, I.; Bravo, D.; Landete, J.M. Influence of different lignan compounds on enterolignan production by Bifidobacterium and Lactobacillus strains. Int. J. Food Microbiol. 2018, 289, 17–23. [Google Scholar] [CrossRef]
- Duranti, S.; Turroni, F.; Milani, C.; Foroni, E.; Bottacini, F.; Bello, F.D.; Ferrarini, A.; Delledonne, M.; van Sinderen, D.; Ventura, M. Exploration of the genomic diversity and core genome of the Bifidobacterium adolescentis phylogenetic group by means of a polyphasic approach. Appl. Environ. Microbiol. 2013, 79, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokusaeva, K.; Fitzgerald, G.F.; van Sinderen, D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Liu, Q.; Pei, Z.; Wang, L.; Tian, P.; Liu, Z.; Zhao, J.; Zhang, H.; Chen, W. The diversity of the CRISPR-Cas system and prophages present in the genome reveals the co-evolution of Bifidobacterium pseudocatenulatum and phages. Front. Microbiol. 2020, 11, 1088. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Hu, L.; Lu, W.; Wang, L.; Pan, M.; Zhang, H.; Zhao, J.; Chen, W. Assessment of Bifidobacterium species using groEL gene on the basis of Illumina MiSeq high-throughput sequencing. Genes 2017, 8, 336. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Ma, F.; Wang, G.; Wang, Y.; Zhao, J.; Zhang, H.; Chen, W. Bifidobacteria attenuate the development of metabolic disorders, with inter- and intra-species differences. Food Funct. 2018, 9, 3509–3522. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Milani, C.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Sánchez, B.; Margolles, A.; et al. Evaluation of genetic diversity among strains of the human gut commensal Bifidobacterium adolescentis. Sci. Rep. 2016, 6, 23971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Ferrario, C.; Modesto, M.; Mattarelli, P.; Jiří, K.; et al. Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC Genom. 2017, 18, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosi, E.; Monk, J.M.; Aziz, R.K.; Fondi, M.; Nizet, V.; Palsson, B. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 2016, 113, E3801–E3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turroni, F.; Foroni, E.; Pizzetti, P.; Giubellini, V.; Ribbera, A.; Merusi, P.; Cagnasso, P.; Bizzarri, B.; De’Angelis, G.L.; Shanahan, F.; et al. Exploring the diversity of the Bifidobacterial population in the human intestinal tract. Appl. Environ. Microbiol. 2009, 75, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, Y.; Tang, S.; Chen, S.; Gong, S.; Jiang, X.; Wang, L.; Zhang, Y. Dietary diversity and nutrient intake of Han and Dongxiang smallholder farmers in poverty areas of northwest China. Nutrients 2021, 13, 3908. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Milani, C.; Duranti, S.; Mahony, J.; van Sinderen, D.; Ventura, M. Glycan utilization and cross-feeding activities by Bifidobacteria. Trends Microbiol. 2017, 26, 339–350. [Google Scholar] [CrossRef]
- Broek, L.V.D.; Hinz, S.W.A.; Beldman, G.; Vincken, J.-P.; Voragen, A.G.J. Bifidobacterium carbohydrases-their role in breakdown and synthesis of (potential) prebiotics. Mol. Nutr. Food Res. 2008, 52, 146–163. [Google Scholar] [CrossRef]
- Davis, M.Y.; Zhang, H.; Brannan, L.E.; Carman, R.J.; Boone, J.H. Rapid change of fecal microbiome and disappearance of Clostridium difficile in a colonized infant after transition from breast milk to cow milk. Microbiome 2016, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Motherway, M.O.; Kinsella, M.; Fitzgerald, G.F.; van Sinderen, D. Transcriptional and functional characterization of genetic elements involved in galacto-oligosaccharide utilization by Bifidobacterium breve UCC2003. Microb. Biotechnol. 2012, 6, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Kimura, K.; Hatano, H. Diverse galactooligosaccharides consumption by bifidobacteria: Implications of β-galactosidase—LacS operon. Biosci. Biotechnol. Biochem. 2015, 79, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biely, P. Microbial carbohydrate esterases deacetylating plant polysaccharides. Biotechnol. Adv. 2012, 30, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zhang, H.; Wu, H. Glycosyltransferase-mediated sweet modification in oral Streptococci. J. Dent. Res. 2015, 94, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Shigehisa, A.; Watanabe, Y.; Tsukuda, N.; Moriyama-Ohara, K.; Hara, T.; Matsumoto, S.; Tsuji, H.; Matsuki, T. Multiple transporters and glycoside hydrolases are involved in arabinoxylan-derived oligosaccharide utilization in Bifidobacterium pseudocatenulatum. Appl. Environ. Microbiol. 2020, 86, e01782-20. [Google Scholar] [CrossRef]
- Linares-Pastén, J.A.; Falck, P.; Albasri, K.; Kjellström, S.; Adlercreutz, P.; Logan, D.T.; Karlsson, E.N. Three-dimensional structures and functional studies of two GH43 arabinofuranosidases from Weissella sp. strain 142 and Lactobacillus brevis. FEBS J. 2017, 284, 2019–2036. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, G.; Liu, Q.; Wang, L.; Li, H.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. The Comparative Analysis of Genomic Diversity and Genes Involved in Carbohydrate Metabolism of Eighty-Eight Bifidobacterium pseudocatenulatum Isolates from Different Niches of China. Nutrients 2022, 14, 2347. https://doi.org/10.3390/nu14112347
Lin G, Liu Q, Wang L, Li H, Zhao J, Zhang H, Wang G, Chen W. The Comparative Analysis of Genomic Diversity and Genes Involved in Carbohydrate Metabolism of Eighty-Eight Bifidobacterium pseudocatenulatum Isolates from Different Niches of China. Nutrients. 2022; 14(11):2347. https://doi.org/10.3390/nu14112347
Chicago/Turabian StyleLin, Guopeng, Qian Liu, Luyao Wang, Haitao Li, Jianxin Zhao, Hao Zhang, Gang Wang, and Wei Chen. 2022. "The Comparative Analysis of Genomic Diversity and Genes Involved in Carbohydrate Metabolism of Eighty-Eight Bifidobacterium pseudocatenulatum Isolates from Different Niches of China" Nutrients 14, no. 11: 2347. https://doi.org/10.3390/nu14112347
APA StyleLin, G., Liu, Q., Wang, L., Li, H., Zhao, J., Zhang, H., Wang, G., & Chen, W. (2022). The Comparative Analysis of Genomic Diversity and Genes Involved in Carbohydrate Metabolism of Eighty-Eight Bifidobacterium pseudocatenulatum Isolates from Different Niches of China. Nutrients, 14(11), 2347. https://doi.org/10.3390/nu14112347