
Ameliorative Effects of Gut Microbial Metabolite Urolithin A on Pancreatic Diseases

 ,
, 

 ,
, 
 and
and
Abstract
:1. Introduction
2. Urolithin A
2.1. Intestinal Microbial Metabolite Urolithin A
2.2. Pharmacokinetics of Urolithin A
2.3. Biological Activity of Urolithin A
2.3.1. Anti-Inflammatory and Improved Oxidative Stress
2.3.2. Anti-Aging
2.3.3. Regulation of Metabolic Homeostasis
2.3.4. Improve Alzheimer’s Disease and Cognitive Impairment
3. Pathogenic Mechanisms of Pancreatic Diseases
3.1. Pancreatitis
3.2. Pancreatic Cancer
3.3. Diabetes Mellitus
4. Effects of Urolithin A on Pancreatic Diseases
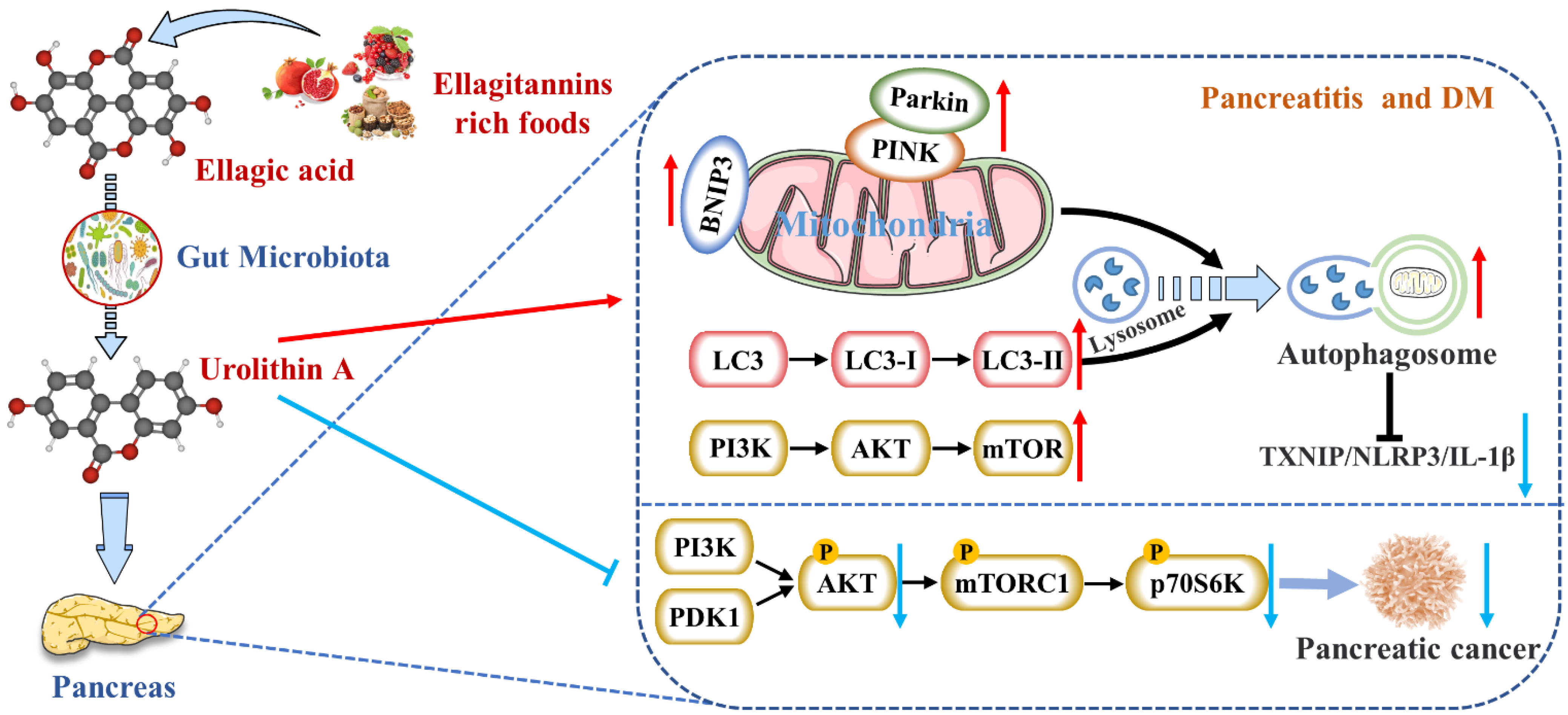
4.1. Reduces the Expression of Pancreatic Inflammatory Factors
4.2. Activates Autophagy and Maintains Mitochondrial Function in the Pancreas
4.3. Inhibits Endoplasmic Reticulum Stress in the Pancreas
4.4. Inhibits the Occurrence and Development of Pancreatic Tumors
4.5. Protects Pancreatic β Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delbarco-Trillo, J.; Harelimana, I.H.; Goodwin, T.E.; Drea, C.M. Chemical Differences Between Voided and Bladder Urine in the Aye-Aye (Daubentonia madagascariensis): Implications for Olfactory Communication Studies. Am. J. Primatol. 2013, 75, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Schooneman, M.G.; Vaz, F.M.; Houten, S.M.; Soeters, M.R. Acylcarnitines Reflecting or Inflicting Insulin Resistance? Diabetes 2013, 62, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; D’Amico, D.; Andreux, P.A.; Fouassier, A.M.; Blanco-Bose, W.; Evans, M.; Aebischer, P.; Auwerx, J.; Rinsch, C. Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell Rep. Med. 2022, 3, 100633. [Google Scholar] [CrossRef]
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem. Toxicol. 2017, 108, 289–297. [Google Scholar] [CrossRef]
- Kimmel, R.A.; Meyer, D. Molecular Regulation of Pancreas Development in Zebrafish. Methods Cell Biol. 2010, 100, 261–280. [Google Scholar]
- Zhou, Q.; Melton, D.A. Pancreas regeneration. Nature 2018, 557, 351–358. [Google Scholar] [CrossRef]
- Li, J.; Zheng, Y.; Yan, P.; Song, M.; Wang, S.; Sun, L.; Liu, Z.; Ma, S.; Izpisua Belmonte, J.C.; Chan, P.; et al. A single-cell transcriptomic atlas of primate pancreatic islet aging. Natl. Sci. Rev. 2021, 8, 127. [Google Scholar] [CrossRef]
- Evtyugin, D.D.; Magina, S.; Evtuguin, D.V. Recent Advances in the Production and Applications of Ellagic Acid and Its Derivatives: A Review. Molecules 2020, 25, 2745. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, S.; Mao, B.; Zhang, Q.; Zhao, J.; Zhang, H.; Tang, X.; Chen, W. Ellagic acid and intestinal microflora metabolite urolithin A: A review on its sources, metabolic distribution, health benefits, and biotransformation. Crit. Rev. Food Sci. Nutr. 2022, 1–23. [Google Scholar] [CrossRef]
- Carlos Espin, J.; Larrosa, M.; Teresa Garcia-Conesa, M.; Tomas-Barberan, F. Biological Significance of Urolithins, the Gut Microbial Ellagic Acid-Derived Metabolites, The Evidence So Far. Evid.-Based Complement. Altern. Med. 2013, 2013, 270418. [Google Scholar]
- Cerdá, B.; Periago, P.; Espín, A.J.C.; Tomás-Barberán, F.A. Identification of urolithin A as a metabolite produced by human colon microflora from ellagic acid and related compounds. J. Agric. Food Chem. 2005, 53, 5571–5576. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Giménez-Bastida, J.A.; Cortés-Martín, A.; Ávila-Gálvez, M.; Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C.; González-Sarrías, A. Urolithins: A Comprehensive Update on their Metabolism, Bioactivity, and Associated Gut Microbiota. Mol. Nutr. Food Res. 2022, e2101019. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Martín, A.; Selma, M.V.; Tomás-Barberán, F.A.; González-Sarrías, A.; Espín, J.C. Where to Look into the Puzzle of Polyphenols and Health? The Postbiotics and Gut Microbiota Associated with Human Metabotypes. Mol. Nutr. Food Res. 2020, 64, 1900952. [Google Scholar] [CrossRef]
- Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Interactions of gut microbiota with dietary polyphenols and consequences to human health. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 471–476. [Google Scholar] [CrossRef]
- Selma, M.V.; Romo-Vaquero, M.; García-Villalba, R.; González-Sarrías, A.; Tomás-Barberán, F.A.; Espín, J.C. The human gut microbial ecology associated with overweight and obesity determines ellagic acid metabolism. Food Funct. 2016, 7, 1769–1774. [Google Scholar] [CrossRef]
- Selma, M.V.; Beltrán, D.; García-Villalba, R.; Espín, J.C.; Tomás-Barberán, F.A. Description of urolithin production capacity from ellagic acid of two human intestinal Gordonibacter species. Food Funct. 2014, 5, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Selma, M.V.; González-Sarrías, A.; Salas-Salvadó, J.; Andres-Lacueva, C.; Alasalvar, C.; Örem, A.; Tomás-Barberán, F.A.; Espín, J.C. The gut microbiota metabolism of pomegranate or walnut ellagitannins yields two urolithin-metabotypes that correlate with cardiometabolic risk biomarkers: Comparison between normoweight, overweight-obesity and metabolic syndrome. Clin. Nutr. 2018, 37, 897–905. [Google Scholar] [CrossRef]
- Gaya, P.; Peiroten, A.; Medina, M.; Alvarez, I.; Landete, J.M. Bifidobacterium pseudocatenulatum INIA P815, The first bacterium able to produce urolithins A and B from ellagic acid. J. Funct. Foods 2018, 45, 95–99. [Google Scholar] [CrossRef]
- García-Villalba, R.; Beltrán, D.; Espín, J.C.; Selma, M.V.; Tomás-Barberán, F.A. Time Course Production of Urolithins from Ellagic Acid by Human Gut Microbiota. J. Agric. Food Chem. 2013, 61, 8797–8806. [Google Scholar] [CrossRef]
- Yan, L.; Yin, P.; Ma, C.; Liu, Y. Method Development and Validation for Pharmacokinetic and Tissue Distributions of Ellagic Acid Using Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS). Molecules 2014, 19, 18923–18935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ávila-Gálvez, M.A.; Giménez-Bastida, J.A.; González-Sarrías, A.; Espín, J.C. Tissue deconjugation of urolithin A glucuronide to free urolithin A in systemic inflammation. Food Funct. 2019, 10, 3135–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DaSilva, N.A.; Nahar, P.P.; Ma, H.; Eid, A.; Wei, Z.; Meschwitz, S.; Zawia, N.H.; Slitt, A.L.; Seeram, N.P. Pomegranate ellagitannin-gut microbial-derived metabolites, urolithins, inhibit neuroinflammation in vitro. Nutr. Neurosci. 2019, 22, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Cásedas, G.; Les, F.; Choya-Foces, C.; Hugo, M.; López, V. The Metabolite Urolithin-A Ameliorates Oxidative Stress in Neuro-2a Cells, Becoming a Potential Neuroprotective Agent. Antioxidants 2020, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, T.; Liao, J.; Shen, K.; Chen, X.; Xu, Z.; Tian, W.; Wang, Y.; Jin, B.; Pan, H. Protective effect of urolithin a on cisplatin-induced nephrotoxicity in mice via modulation of inflammation and oxidative stress. Food Chem. Toxicol. 2019, 129, 108–114. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C-elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Chen, P.; Chen, F.; Lei, J.; Li, Q.; Zhou, B. Activation of the miR-34a-Mediated SIRT1/mTOR Signaling Pathway by Urolithin A Attenuates d-Galactose-Induced Brain Aging in Mice. Neurotherapeutics 2019, 16, 1269–1282. [Google Scholar] [CrossRef]
- Liu, C.F.; Li, X.L.; Zhang, Z.L.; Qiu, L.; Ding, S.X.; Xue, J.X.; Zhao, G.P.; Li, J. Antiaging Effects of Urolithin A on Replicative Senescent Human Skin Fibroblasts. Rejuvenation Res. 2019, 22, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Luan, P.; D’Amico, D.; Andreux, P.A.; Laurila, P.-P.; Wohlwend, M.; Li, H.; de Lima, T.I.; Place, N.; Rinsch, C.; Zanou, N.; et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci. Transl. Med. 2021, 13, eabb0319. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Y.; Henning, S.M.; Chan, B.; Long, J.; Zhong, J.; Acin-Perez, R.; Petcherski, A.; Shirihai, O.; Heber, D.; et al. Ellagic Acid and Its Microbial Metabolite Urolithin A Alleviate Diet-Induced Insulin Resistance in Mice. Mol. Nutr. Food Res. 2020, 64, 2000091. [Google Scholar] [CrossRef]
- Han, Q.A.; Su, D.; Shi, C.; Liu, P.; Wang, Y.; Zhu, B.; Xia, X. Urolithin A attenuated ox-LDL-induced cholesterol accumulation in macrophages partly through regulating miR-33a and ERK/AMPK/SREBP1 signaling pathways. Food Funct. 2020, 11, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Shi, X.C.; Xie, B.C.; Zhu, M.Q.; Chen, Y.; Chu, X.Y.; Cai, G.H.; Liu, M.; Yang, S.Z.; Mitchell, G.A.; et al. Urolithin A exerts antiobesity effects through enhancing adipose tissue thermogenesis in mice. PLoS Biol. 2020, 18, e3000688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflamm. 2019, 16, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.-J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202. [Google Scholar] [CrossRef]
- Singh, R.; Chandrashekharappa, S.; Bodduluri, H.; Baby, B.V.; Hegde, B.; Kotla, N.G.; Hiwale, A.A.; Saiyed, T.; Patel, P.; Vijay-Kumar, M.; et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun. 2019, 10, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.Q.; Wang, Z.Q.; Yan, S.M.; Shi, B.L. Protective effect of chitosan against growth inhibition and pancreatic oxidative stress in weaned piglets. J. Appl. Anim. Res. 2022, 50, 93–96. [Google Scholar] [CrossRef]
- Kloppel, G. Pathomorphology of acute pancreatitis. Ann. Ital. Chir. 1995, 66, 149–154. [Google Scholar]
- Valverde-Lopez, F.; Martinez-Cara, J.G.; Redondo-Cerezo, E. Acute pancreatitis. Med. Clin. 2022; in press. [Google Scholar] [CrossRef]
- Petrov, M.S.; Yadav, D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 175–184. [Google Scholar] [CrossRef]
- Boxhoorn, L.; Voermans, R.P.; Bouwense, S.A.; Bruno, M.J.; Verdonk, R.C.; Boermeester, M.A.; van Santvoort, H.C.; Besselink, M.G. Acute pancreatitis. Lancet 2020, 396, 726–734. [Google Scholar] [CrossRef]
- Xiao, A.Y.; Tan, M.L.Y.; Wu, L.M.; Asrani, V.M.; Windsor, J.A.; Yadav, D.; Petrov, M.S. Global incidence and mortality of pancreatic diseases: A systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol. Hepatol. 2016, 1, 45–55. [Google Scholar] [CrossRef]
- Lee, P.J.; Papachristou, G.I. New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Pendharkar, S.A.; Mathew, J.; Petrov, M.S. Age- and sex-specific prevalence of diabetes associated with diseases of the exocrine pancreas: A population-based study. Dig. Liver Dis. 2017, 49, 540–544. [Google Scholar] [CrossRef]
- Vrolyk, V.; Singh, B. Animal models to study the role of pulmonary intravascular macrophages in spontaneous and induced acute pancreatitis. Cell Tissue Res. 2020, 380, 207–222. [Google Scholar] [CrossRef]
- Jiang, X.; Zheng, Y.W.; Bao, S.H.; Zhang, H.L.; Chen, R.J.; Yao, Q.; Kou, L.F. Drug discovery and formulation development for acute pancreatitis. Drug Deliv. 2020, 27, 1562–1580. [Google Scholar] [CrossRef]
- Tang, Y.; Sun, M.; Liu, Z. Phytochemicals with protective effects against acute pancreatitis: A review of recent literature. Pharm. Biol. 2022, 60, 479–490. [Google Scholar] [CrossRef]
- Grisham, M.B. NF-kappaB activation in acute pancreatitis: Protective, detrimental, or inconsequential? Gastroenterology 1999, 116, 489–492. [Google Scholar] [CrossRef]
- Saluja, A.; Dudeja, V.; Dawra, R.; Sah, R.P. Early Intra-Acinar Events in Pathogenesis of Pancreatitis. Gastroenterology 2019, 156, 1979–1993. [Google Scholar] [CrossRef]
- Zhang F-h Sun Y-h Fan K-l Dong X-b Han, N.; Zhao, H.; Kong, L. Protective effects of heme oxygenase-1 against severe acute pancreatitis via inhibition of tumor necrosis factor-alpha and augmentation of interleukin-10. BMC Gastroenterol. 2017, 17, 100. [Google Scholar]
- Cho, H.-Y.; Marzec, J.; Kleeberger, S.R. Functional polymorphisms in Nrf2: Implications for human disease. Free Radic. Biol. Med. 2015, 88, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, D.; Timmons, L.; Benson, J.T.; Dierkhising, R.A.; Chari, S.T. Incidence, Prevalence, and Survival of Chronic Pancreatitis: A Population-Based Study. Am. J. Gastroenterol. 2011, 106, 2192–2199. [Google Scholar] [CrossRef]
- Whitcomb, D.C.; Frulloni, L.; Garg, P.; Greer, J.B.; Schneider, A.; Yadav, D.; Shimosegawa, T. Chronic pancreatitis: An international draft consensus proposal for a new mechanistic definition. Pancreatology 2016, 16, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Olesen, S.S.; Mortensen, L.H.; Zinck, E.; Becker, U.; Drewes, A.M.; Nøjgaard, C.; Novovic, S.; Yadav, D.; Tolstrup, J.S. Time trends in incidence and prevalence of chronic pancreatitis: A 25-year population-based nationwide study. United Eur. Gastroenterol. J. 2021, 9, 82–90. [Google Scholar] [CrossRef]
- Braganza, J.M.; Lee, S.H.; McCloy, R.F.; McMahon, M.J. Chronic pancreatitis. Lancet 2011, 377, 1184–1197. [Google Scholar] [CrossRef]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Primers 2017, 3, 17060. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Poettler, M.; Lan, B.; Gruetzmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Deshpande, N.; Nagathihalli, N. Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress. Cancers 2021, 13, 4434. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.N.; Maher, M.E.; Tran, N.H.; Van De Hey, D.R.; Foley, T.M.; Yueh, A.E.; Leystra, A.A.; Pasch, C.A.; Jeffrey, J.J.; Clipson, L.; et al. PIK3CA mutations can initiate pancreatic tumorigenesis and are targetable with PI3K inhibitors. Oncogenesis 2015, 4, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlieman, M.G.; Fahy, B.N.; Ramsamooj, R.; Beckett, L.; Bold, R.J. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br. J. Cancer 2003, 89, 2110–2115. [Google Scholar] [CrossRef] [PubMed]
- Jazirehi, A.R.; Wenn, P.B.; Damavand, M. Therapeutic implications of targeting the PI3Kinase/AKT/mTOR signaling module in melanoma therapy. Am. J. Cancer Res. 2012, 2, 178–191. [Google Scholar]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Biancur, D.E.; Parker, S.J.; Yamamoto, K.; Banh, R.S.; Paulo, J.A.; Mancias, J.D.; Kimmelman, A.C. Autophagy is required for proper cysteine homeostasis in pancreatic cancer through regulation of SLC7A11. Proc. Natl. Acad. Sci. USA 2021, 118, e2021475118. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; O’Neill, E.; Gordon-Weeks, A.; Mukherjee, S.; McKenna, W.G.; Muschel, R.J. Pancreatic ductal adenocarcinoma: From genetics to biology to radiobiology to oncoimmunology and all the way back to the clinic. Biochim. Biophys. Acta-Rev. Cancer 2015, 1855, 61–82. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- IDF Diabetes Atlas, 10th ed. Available online: https://www.diabetesatlas.org (accessed on 10 March 2022).
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for Insulin Resistance, Common Threads and Missing Links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, X.Y.; Xie, C.; Yang, M.S. Association between Type 2 Diabetes and CDKN2A/B: A meta-analysis study. Mol. Biol. Rep. 2012, 39, 1609–1616. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. Beta-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef] [Green Version]
- Quan, W.; Lim, Y.-M.; Lee, M.-S. Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic beta-cells. Exp. Mol. Med. 2012, 44, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Kamata, K.; Mizukami, H.; Inaba, W.; Tsuboi, K.; Tateishi, Y.; Yoshida, T.; Yagihashi, S. Islet amyloid with macrophage migration correlates with augmented beta-cell deficits in type 2 diabetic patients. Amyloid 2014, 21, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.J.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [Green Version]
- Marselli, L.; Bugliani, M.; Suleiman, M.; Olimpico, F.; Masini, M.; Petrini, M.; Boggi, U.; Filipponi, F.; Syed, F.; Marchetti, P. Beta-Cell inflammation in human type 2 diabetes and the role of autophagy. Diabetes Obes. Metab. 2013, 15, 130–136. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Sandovici, I.; Hammerle, C.M.; Cooper, W.N.; Smith, N.H.; Tarry-Adkins, J.L.; Dunmore, B.J.; Bauer, J.; Andrews, S.R.; Yeo, G.S.; Ozanne, S.E.; et al. Ageing is associated with molecular signatures of inflammation and type 2 diabetes in rat pancreatic islets. Diabetologia 2016, 59, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Janjuha, S.; Singh, S.P.; Tsakmaki, A.; Gharavy, S.N.M.; Murawala, P.; Konantz, J.; Birke, S.; Hodson, D.J.; Rutter, G.A.; Bewick, G.A.; et al. Age-related islet inflammation marks the proliferative decline of pancreatic beta-cells in zebrafish. Elife 2018, 7, e32965. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Shimosegawa, T. Ellagic Acid Inhibits Pancreatic Fibrosis in Male Wistar Bonn/Kobori Rats. Dig. Dis. Sci. 2009, 54, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Satoh, M.; Kikuta, K.; Suzuki, N.; Satoh, K.; Shimosegawa, T. Ellagic acid blocks activation of pancreatic stellate cells. Biochem. Pharmacol. 2005, 70, 869–878. [Google Scholar] [CrossRef]
- Yilmaz, E.E.; Bozdag, Z.; Ibiloglu, I.; Arikanoglu, Z.; Yazgan, U.C.; Kaplan, I.; Gumus, M.; Atamanalp, S.S. Therapeutic effects of ellagic acid on L-arginin induced acute pancreatitis. Acta Cir. Bras. 2016, 31, 396–401. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Aisker, G.; Dong, H.; Halemahebai, G.; Zhang, Y.; Tian, L. Urolithin A suppresses glucolipotoxicity-induced ER stress and TXNIP/NLRP3/IL-1beta inflammation signal in pancreatic beta cells by regulating AMPK and autophagy. Phytomedicine 2021, 93, 153741. [Google Scholar] [CrossRef]
- Mehra, S.; Srinivasan, S.; Dai, X.; Dosch, A.; Singh, S.; Bianchi, A.; Silva, I.D.C.; Datta, J.; VanSaun, M.N.; Merchant, N.; et al. Protective Effects of Urolithin A on Alcoholic Chronic Pancreatitis via Inhibiting PI3K/AKT/mTOR Signaling Pathway. Pancreas 2021, 50, 1082. [Google Scholar]
- Tuohetaerbaike, B.; Zhang, Y.; Tian, Y.; Zhang, N.N.; Kang, J.; Mao, X.; Zhang, Y.; Li, X. Pancreas protective effects of Urolithin A on type 2 diabetic mice induced by high fat and streptozotocin via regulating autophagy and AKT/mTOR signaling pathway. J. Ethnopharmacol. 2020, 250, 112479. [Google Scholar] [CrossRef]
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A., III; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a Novel Natural Compound to Target PI3K/AKT/mTOR Pathway in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Jala, V.; Honnenahally, K.; Castellanos, J.; Vermula, P.K.; VanSaun, M.; Merchant, N.; Nagathihalli, N. Urolithin A prevents pancreatic tumor growth and increases survival by inhibiting PI3K/PDK1 and STAT3 signaling. Cancer Res. 2017, 77, 5259. [Google Scholar]
- Fatima, N.; Hafizur, R.M.; Hameed, A.; Ahmed, S.; Nisar, M.; Kabir, N. Ellagic acid in Emblica officinalis exerts anti-diabetic activity through the action on beta-cells of pancreas. Eur. J. Nutr. 2017, 56, 591–601. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; González-Sarrías, A.; Yáñez-Gascón, M.J.; Selma, M.V.; Azorín-Ortuño, M.; Toti, S.; Tomas-Barberan, F.; Dolara, P.; Espín, J.C. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J. Nutr. Biochem. 2010, 21, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yan, Z.B.; Cheng, Y.G.; Zhong, M.W.; Liu, S.Z.; Zhang, G.Y.; Hu, S.Y. Deactivation of the NLRP3 inflammasome in infiltrating macrophages by duodenal-jejunal bypass surgery mediates improvement of beta cell function in type 2 diabetes. Metabolism 2018, 81, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vezza, T.; Díaz-Pozo, P.; Canet, F.; de Marañón, A.M.; Abad-Jiménez, Z.; García-Gargallo, C.; Roldan, I.; Solá, E.; Bañuls, C.; López-Domènech, S.; et al. The Role of Mitochondrial Dynamic Dysfunction in Age-Associated Type 2 Diabetes. World J. Men’s Health 2022, 40. [Google Scholar] [CrossRef]
- De Tata, V. Age-related impairment of pancreatic Beta-cell function: Pathophysiological and cellular mechanisms. Front. Endocrinol. 2014, 5, 138. [Google Scholar] [CrossRef] [Green Version]
- Abuarab, N.; Munsey, T.S.; Jiang, L.-H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.S.; Joffraud, M.; Boutant, M.; Ratajczak, J.; Gao, A.W.; Maclachlan, C.; Hernandez-Alvarez, M.I.; Raymond, F.; Metairon, S.; Descombes, P.; et al. Mfn1 Deficiency in the Liver Protects Against Diet-Induced Insulin Resistance and Enhances the Hypoglycemic Effect of Metformin. Diabetes 2016, 65, 3552–3560. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Halemahebai, G.; Tian, L.; Dong, H.; Aisker, G. Urolithin A: A pomegranate metabolite, protects pancreatic beta cells from apoptosis by activating autophagy. J. Ethnopharmacol. 2021, 272, 113628. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, V.; Tran, T.; Zhu, S.; Choi, S.H.; Gamo, A.M.; Li, S.; Fu, Q.; Cunado, M.D.; Roland, J.; Hull, M.; et al. A small molecule UPR modulator for diabetes identified by high throughput screening. Acta Pharm. Sin. B 2021, 11, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Mihailidou, C.; Chatzistamou, I.; Papavassiliou, A.G.; Kiaris, H. Modulation of Pancreatic Islets’ Function and Survival During Aging Involves the Differential Regulation of Endoplasmic Reticulum Stress by p21 and CHOP. Antioxid. Redox Signal. 2017, 27, 185–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeraraghavan, J.; Natarajan, M.; Lagisetty, P.; Awasthi, V.; Herman, T.S.; Aravindan, N. Impact of Curcumin, Raspberry Extract, and Neem Leaf Extract on Rel Protein-Regulated Cell Death/Radiosensitization in Pancreatic Cancer Cells. Pancreas 2011, 40, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lu, C.L.; Tang, R.B.; Pan, Y.M.; Bao, S.H.; Qiu, Y.D.; Xie, M. Ellagic acid inhibits the proliferation of human pancreatic carcinoma PANC-1 cells in vitro and in vivo. Oncotarget 2017, 8, 12301–12310. [Google Scholar] [CrossRef] [Green Version]
- Tomas-Barberan, F.A.; Gonzalez-Sarrias, A.; Garcia-Villalba, R.; Nunez-Sanchez, M.A.; Selma, M.V.; Garcia-Conesa, M.T.; Espin, J.C. Urolithins, the rescue of “old” metabolites to understand a “new” concept, Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol. Nutr. Food Res. 2017, 61, 1500901. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the Point of Inhibition: A Comparative Review of PI3K/AKT/mTOR Pathway Inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.H.; Xue, A.; Baxter, R.C.; Pavlakis, N.; Smith, R.C. Upstream and Downstream Co-inhibition of Mitogen-Activated Protein Kinase and PI3K/Akt/mTOR Pathways in Pancreatic Ductal Adenocarcinoma. Neoplasia 2016, 18, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640. [Google Scholar] [CrossRef] [Green Version]
- Savi, M.; Bocchi, L.; Mena, P.; Dall’Asta, M.; Crozier, A.; Brighenti, F.; Stilli, D.; Del Rio, D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2017, 16, 80. [Google Scholar] [CrossRef]
- Albasher, G.; Alkahtani, S.; Al-Harbi, L.N. Urolithin A prevents streptozotocin-induced diabetic cardiomyopathy in rats by activating SIRT1. Saudi J. Biol. Sci. 2022, 29, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Li, K.; Bian, J.; Liu, H.; Zhai, X.; El-Omar, E.; Han, L.; Gong, L.; Wang, M. Urolithin A Attenuates Diabetes-Associated Cognitive Impairment by Ameliorating Intestinal Barrier Dysfunction via N-glycan Biosynthesis Pathway. Mol. Nutr. Food Res. 2022, 66, 2100863. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, S.; Li, K.; Wang, X.; Li, X.; An, M.; Yu, X.; Long, X.; Zhong, R.; Liu, Q.; et al. Urolithin A ameliorates diabetic retinopathy via activation of the Nrf2/HO-1 pathway. Endocr. J. 2022, EJ21-0490. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, C.; Zheng, G.-H.; Qiu, Z. Emblic Leafflower (Phyllanthus emblica L.) Fruits Ameliorate Vascular Smooth Muscle Cell Dysfunction in Hyperglycemia: An Underlying Mechanism Involved in Ellagitannin Metabolite Urolithin A. Evid.-Based Complement. Altern. Med. 2018, 2018, 8478943. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hamid, A.A.M.; El-Firgany, A.E.-D.L. Hydroxychloroquine hindering of diabetic isletopathy carries its signature on the inflammatory cytokines. J. Mol. Histol. 2016, 47, 183–193. [Google Scholar] [CrossRef]
- Tuduri, E.; Soriano, S.; Almagro, L.; Garcia-Heredia, A.; Rafacho, A.; Alonso-Magdalena, P.; Nadal, A.; Quesada, I. The Effects of Aging on Male Mouse Pancreatic beta-Cell Function Involve Multiple Events in the Regulation of Secretion, Influence of Insulin Sensitivity. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 405–415. [Google Scholar] [CrossRef]
- Pan, F.; He, X.; Feng, J.; Cui, W.; Gao, L.; Li, M.; Yang, H.; Wang, C.; Hu, Y. Peptidome analysis reveals the involvement of endogenous peptides in mouse pancreatic dysfunction with aging. J. Cell. Physiol. 2019, 234, 14090–14099. [Google Scholar] [CrossRef]
- Djedjibegovic, J.; Marjanovic, A.; Panieri, E.; Saso, L. Ellagic Acid-Derived Urolithins as Modulators of Oxidative Stress. Oxid. Med. Cell Longev. 2020, 2020, 5194508. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Zuccari, G. Oxidative Stress, Antioxidant Capabilities, and Bioavailability, Ellagic Acid or Urolithins? Antioxidants 2020, 9, 707. [Google Scholar] [CrossRef]
- Mena, P.; Dall’Asta, M.; Calani, L.; Brighenti, F.; Del Rio, D. Gastrointestinal stability of urolithins: An in vitro approach. Eur. J. Nutr. 2017, 56, 99–106. [Google Scholar] [CrossRef]
- Baquero, F.; Nombela, C. The microbiome as a human organ. Clin. Microbiol. Infect. 2012, 18, 2–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
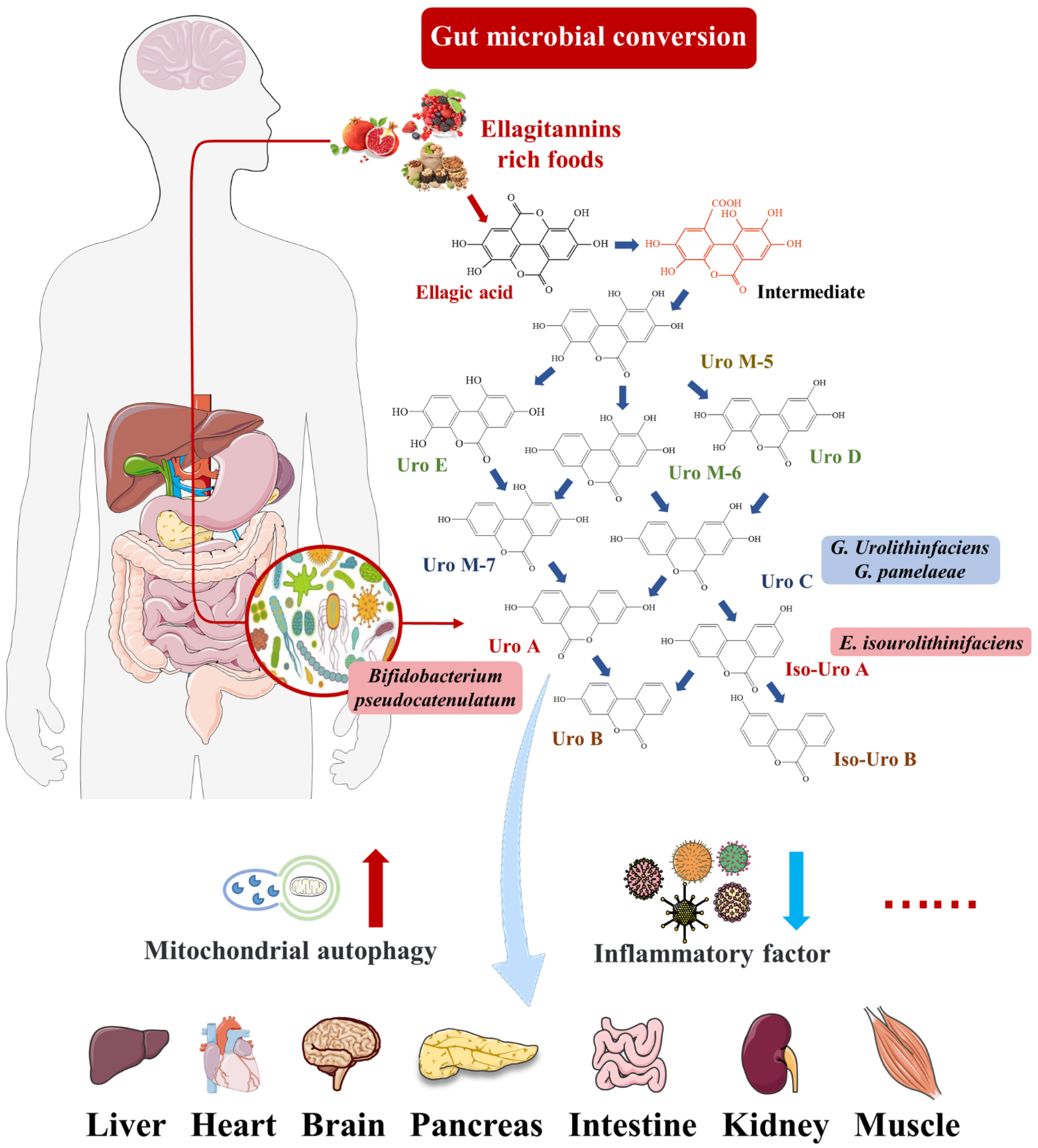
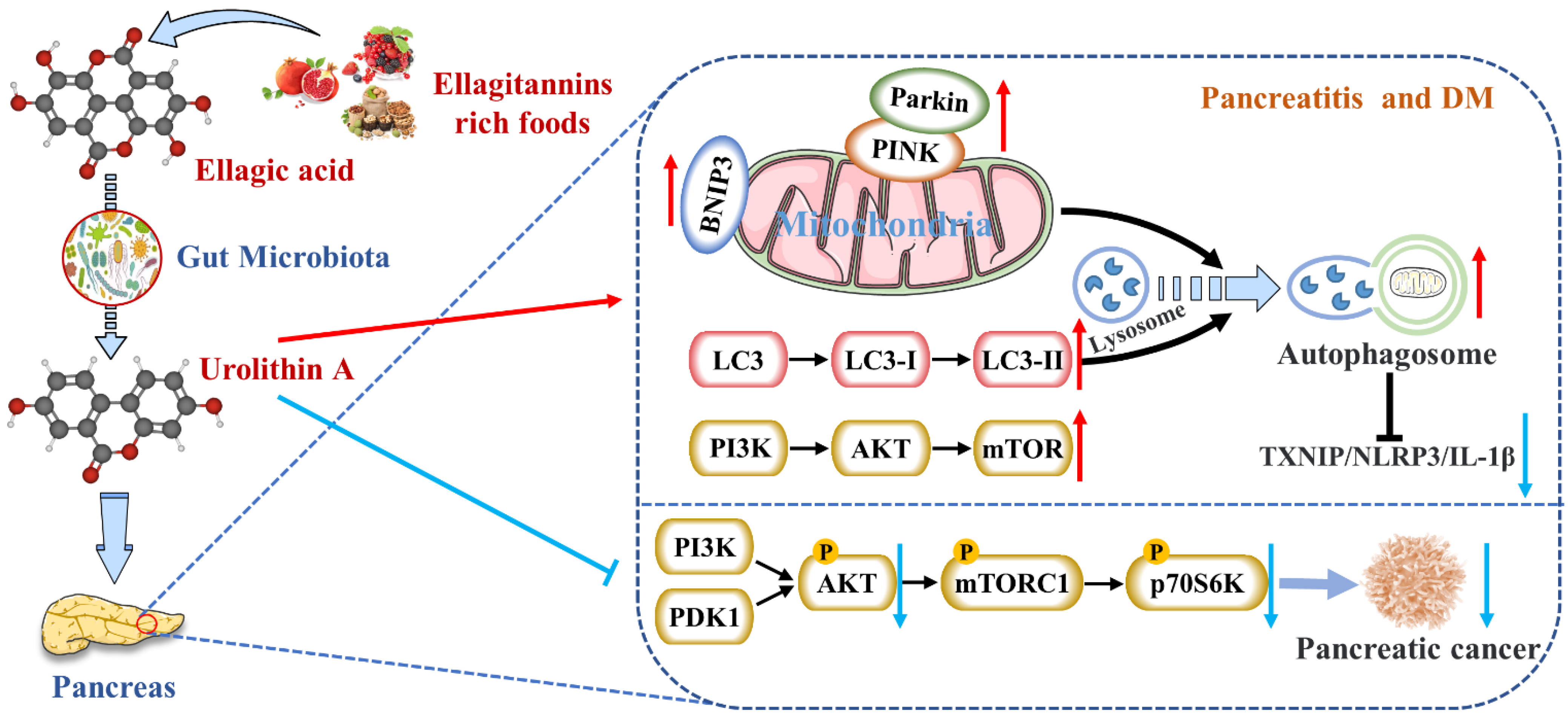

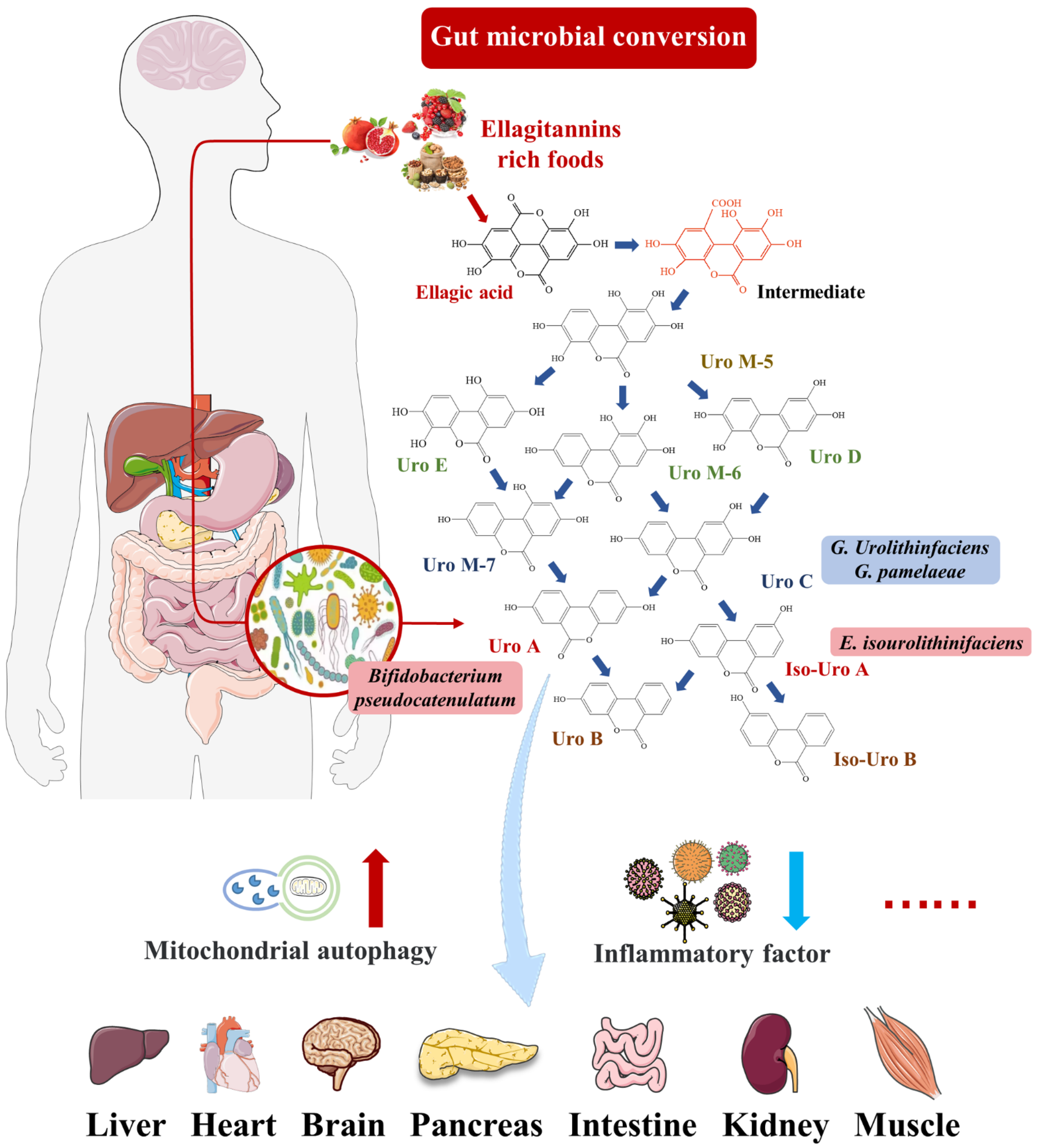{kind=link}
{kind=link}
| Disease Model | Treatment | Metabolic Response | Ref. |
|---|---|---|---|
| Spontaneous CP in male Wistar Bonn/Kobori rats | 100 mg/kg BW/day orally administered with EA for 10 weeks |
| [85] |
| PSCs were isolated from rat pancreas tissue to culture activated, myofibroblast-like phenotype | 1–25 μg/mL EA |
| [86] |
| L-arginine induced AP in rats | 85 mg/kg orally administered with EA |
| [87] |
| MIN6 β-cell inflammations were induced using 25 mM glucose and 0.5 mM palmitic acid | Uro A |
| [88] |
| Alcohol-associated CP in C56BL6/J mice | Administered during the last 3 weeks of alcohol-associated CP induction |
| [89] |
| DM in male C57BL/6 mice was achieved by a HFD and intraperitoneal STZ injections | 50 mg/kg BW/day orally administered with Uro A for 8 weeks |
| [90] |
| Human PDAC cell lines; PDAC mice were achieved by injecting PANC1 cells into the flank of 6-week-old Fox1-nu/nu mice | 0–100 μM; 20 mg/kg BW/day (5 days/week) orally administered with Uro A |
| [91] |
| PKT (Ptf1acre/+; LSL-KrasG12D; Tgfbr2fl/fl) mice, an aggressive genetically engineered PDAC mouse model | Orally administered with Uro A for 5 weeks |
| [92] |
| Neonatal STZ-induced non-obese T2DM rats | 25–100 mg/kg BW orally administered with EA |
| [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Xiao, Y.; Bian, J.; Han, L.; He, C.; El-Omar, E.; Gong, L.; Wang, M. Ameliorative Effects of Gut Microbial Metabolite Urolithin A on Pancreatic Diseases. Nutrients 2022, 14, 2549. https://doi.org/10.3390/nu14122549
Li K, Xiao Y, Bian J, Han L, He C, El-Omar E, Gong L, Wang M. Ameliorative Effects of Gut Microbial Metabolite Urolithin A on Pancreatic Diseases. Nutrients. 2022; 14(12):2549. https://doi.org/10.3390/nu14122549
Chicago/Turabian StyleLi, Kailin, Yao Xiao, Ji Bian, Lin Han, Caian He, Emad El-Omar, Lan Gong, and Min Wang. 2022. "Ameliorative Effects of Gut Microbial Metabolite Urolithin A on Pancreatic Diseases" Nutrients 14, no. 12: 2549. https://doi.org/10.3390/nu14122549
APA StyleLi, K., Xiao, Y., Bian, J., Han, L., He, C., El-Omar, E., Gong, L., & Wang, M. (2022). Ameliorative Effects of Gut Microbial Metabolite Urolithin A on Pancreatic Diseases. Nutrients, 14(12), 2549. https://doi.org/10.3390/nu14122549