Abstract
Since the 1980s, chronic kidney disease (CKD) affecting all ages has increased by almost 25%. This increase may be partially attributable to lifestyle changes and increased global consumption of a “western” diet, which is typically energy dense, low in fruits and vegetables, and high in animal protein and ultra-processed foods. These modern food trends have led to an increase in the consumption of advanced glycation end products (AGEs) in conjunction with increased metabolic dysfunction, obesity and diabetes, which facilitates production of endogenous AGEs within the body. When in excess, AGEs can be pathological via both receptor-mediated and non-receptor-mediated pathways. The kidney, as a major site for AGE clearance, is particularly vulnerable to AGE-mediated damage and increases in circulating AGEs align with risk of CKD and all-cause mortality. Furthermore, individuals with significant loss of renal function show increased AGE burden, particularly with uraemia, and there is some evidence that AGE lowering via diet or pharmacological inhibition may be beneficial for CKD. This review discusses the pathways that drive AGE formation and regulation within the body. This includes AGE receptor interactions and pathways of AGE-mediated pathology with a focus on the contribution of diet on endogenous AGE production and dietary AGE consumption to these processes. We then analyse the contribution of AGEs to kidney disease, the evidence for dietary AGEs and endogenously produced AGEs in driving pathogenesis in diabetic and non-diabetic kidney disease and the potential for AGE targeted therapies in kidney disease.
1. The Western Diet as a Risk Factor for Kidney Disease
Chronic kidney disease (CKD) is the 12th global cause of mortality with all age mortality attributable to CKD increasing by 41.5% since the 1980s [1]. Furthermore, patients with CKD have been amongst the most vulnerable to the recent COVID19 pandemic [2] with the risk of COVID19 mortality increasing nearly 4-fold in kidney transplant recipients or those receiving dialysis [3]. With the pathogenesis of CKD often likened to accelerated kidney aging, epidemiological evidence suggests that underlying causes of CKD have shifted in the last 50–100 years from glomerulonephritis and congenital diseases towards diabetes and hypertension [4]. Type 2 diabetes (T2D) alone accounts for more than 40% of new CKD cases [1] and more than 50% of patients entering renal replacement therapy [4]. The modern western diet, high in animal protein, saturated- and trans-fats, sugar and salt, whilst low in fruits, vegetables, fibre and other essential nutrients, has been implicated in this rising incidence of CKD, as a result of direct effects on the kidneys and rising rates of obesity, hypertension and type 2 diabetes [5,6,7].
One major factor that has changed in the diet over this period of rising CKD incidence is the increased consumption of processed and ultra-processed foods [8], which are also linked to increased risk of cancer [9], T2D [10], cardiovascular disease (CVD) [11,12] and all-cause mortality [11,13,14], and ultra-processed food consumption appears to have risen during the COVID19 pandemic and associated lockdowns [15,16]. Modern food production, including the use of high temperatures, high pressure, dehydration, decompression, irradiation, salt, and preservatives to extend shelf life and palatability, significantly alters proteins and lipids, forming post-translational modifications, including advanced glycation end products (AGEs) within foods [17,18]. As the kidney plays a major role in the clearance of AGEs from the body, there has been much debate as to whether consumption of dietary AGEs can precipitate and/or contribute to progression of CKD and, hence, whether lowering dietary AGE intake or targeting this pathway might be of therapeutic benefit.
Beyond dietary AGEs, increased consumption of energy dense, nutrient poor foods, which are high in sugars and salt, and low in essential nutrients, actively contribute to hemodynamic and metabolic abnormalities, culminating in hypertension, obesity, and T2D which, in turn, facilitate endogenous AGE production [19]. Circulating concentrations of AGEs are positively associated with diabetic kidney disease (DKD) [20], loss of renal function in diabetes [21], and all cause and CVD mortality [22,23,24,25], with glycation considered one of the major pathways to end organ complications in diabetes [19].
2. AGE Chemistry
AGEs can be produced via several chemical pathways, with the Maillard reaction being the most well-described, non-enzymatic chemistry. This reaction, named after the French physician and chemist responsible for its discovery, is the covalent non-enzymatic attachment of a reducing sugar to an amine or amino acid (AA) and can proceed via a number of chemical pathways. The carbonyl groups in reducing sugars such as glucose, fructose, ribose and mannose are, by nature, reactive towards amine groups, and this chemical attraction underpins Maillard chemistry [26], forming a Schiff’s base (Figure 1 green). These then undergo further rearrangement to produce the Amadori product or a Heyn’s product, depending on whether the originating sugar contained an aldehyde or ketone group [27]. The resulting Amadori products combine with amine groups on proteins to form AGEs or undergo further degradation into reactive carbonyls (Figure 1). These highly reactive carbonyls also react with amino groups of free amines, peptide or proteins to form AGEs [27] (Figure 1 purple). AGE formation is enhanced by factors including heat, the presence of oxidants, increases in pH, atmospheric pressure and higher concentrations of reducing sugars and AAs. In the early steps of this reaction, the reactions are reversible, but become permanent modifications once AGEs are formed [27].
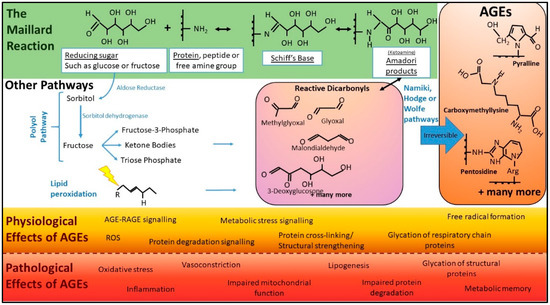
Figure 1.
Basic pathways of AGE chemistry and associated physiology and pathology. In the classical Maillard reaction (green), AGEs are formed when reducing sugars and proteins interact. Initially, reaction intermediates termed Schiff bases are formed. These subsequently rearrange into ketoamines known as Amadori products or early glycation products. These are more stable products than Schiff bases but, at this point in the reaction, the chemistry is still reversible. After further chemistry, the reaction becomes irreversible and forms AGEs (orange). There are several other cellular pathways, including the polyol pathway (shown) and lipid peroxidation (where oxidants, such as free radicals, “attack” (lightning bolt) lipids that contain carbon-carbon double bonds), that can lead to the formation of reactive dicarbonyls, “termed carbonyl stress” (purple) rapidly accelerating the formation of AGEs. Beyond proteins, lipids and DNA can also undergo glycation, leading to AGEs. Pathways such as the polyol pathway only occur inside the body. AGEs have both physiological roles (orange) and pathological effects (red). (Abbreviations: AGEs, advanced glycation end products; ROS, reactive oxygen species).
Maillard chemistry is incredibly diverse. This complexity is in part explained by the many divergent changes to the sugar moiety. For example, sugars may undergo various reactions such as oxidation, dehydration and fragmentation reactions prior to the attachment to amines [28]. Important reactive intermediates of the Maillard reaction are the reactive carbonyls, including, but not limited to, 3-deoxyglucose (3-DG), glyoxal and methylglyoxal (MGO) [29,30,31]. The accumulation of such molecules is known as dicarbonyl stress, and can dramatically accelerate the formation of AGEs, as they are up to 20,000 times more potent glycating agents than glucose [28]. Due to the complexity and variety of starting components, the term AGE refers to a very large heterogeneous population of variable size and structure (Figure 1). These range from low molecular weight, single AGE-modified amino acids to complex high molecular weight AGEs with AGE-crosslinks such as glyoxal lysine dimer (GOLD), 3-deoxyglucosone lysine dimer (DOLD).
3. Factors Regulating AGE Accumulation and Turnover in the Body
In humans and animals, the whole-body AGE load represents the balance between endogenous AGE production, exogenous AGE absorption, and AGE clearance mechanisms.
3.1. Endogenous AGE Production—Beyond the Maillard Reaction
In addition to the Maillard reaction, there are other well-characterised pathways of endogenous AGE production within the body. These include glucose auto-oxidation and lipid peroxidation, generating α-oxoaldehydes, which can subsequently react with monoacids to generate AGEs [32,33] (Figure 1). Additionally, the potent glycating agents and highly reactive dicarbonyls, MGO and 3-DG, are produced in cells from fatty acid catabolism and anaerobic glycolysis [34], and catabolism of ketone bodies [35] (Figure 1). Hence, in states of metabolic or oxidative stress, there is excessive generation of endogenous AGEs.
In the biomedical field, the most commonly measured endogenous AGEs include, carboxymethyl-lysine (CML) and carboxyethyl-lysine (CEL), and cross-linking dimers, such as pentosidine, glyoxal-lysine dimer (GOLD) and methylglyoxal-lysine dimer (MOLD) [36]. AGEs in the skin are also measured both directly by assessment of AGE-modified collagen in skin biopsies [37,38] but increasingly via skin auto-fluorescence, which is a non-invasive point-of-care measurement [39] and considered an indicator of systemic AGE load [40].
Under physiological conditions, AGE formation is most common on long-lived proteins in the circulation and connective tissues [19], such as structural components of cellular basement membranes and proteins including globulin, immunoglobulins and albumin [30,41]. However, conditions such as chronic inflammation, hyperlipidaemia and oxidative stress further accelerate the process of glycation, increasing the likelihood of short-lived proteins also becoming AGE modified [42]. In the context of diabetes and dysregulated glucose metabolism, AGEs such as glycated haemoglobin (HbA1C) and fructosamine-modified albumin are used to monitor long-term glucose control. While chronic conditions, such as diabetes, hypertension, and CKD, are associated with increased levels of circulating AGEs and accumulation of AGE modified proteins [43,44,45], little is understood about AGE homeostasis in healthy individuals and how this may be interrupted to contribute to disease risk.
3.2. Exogenous AGE Sources
AGEs and their highly reactive precursors are produced during food manufacturing. In particular, cooking and food processing conditions favour AGE production. Additionally, AGEs are widely used in the food industry to improve flavour, shelf-life, colour, aroma and texture [46]. High-temperature and dry-cooking methods (frying, baking and broiling) produce higher AGE concentrations than low temperature, aqueous cooking methods such as steaming and boiling [47,48]. Smoking is another exogenous source of AGEs [49]; however, this is outside the scope of this review.
3.2.1. Quantifying AGEs in Commonly Consumed Foods
Several studies have aimed to estimate the AGE load found in commonly consumed foods, with the goal of generating reproducible databases for research purposes; however, quantifying AGEs in food has proven challenging. Studies using immunological methods [47,48,50] suggest that highly industrialised processes produce food with the highest AGE content, such as biscuits and cakes [50], and this is exacerbated for products high in saturated fat [47,48]. In the western diet, however, cooked meats likely constitute the greatest source of dietary AGEs due to both relative AGE content and high consumption rates [48]. More recently, these data have been questioned due to limitations with immunological methods, leading to the establishment of ultra-high performance liquid chromatography-MS/MS (UPLC MSMS)-derived databases of CML [51] and CML, CEL and MG-H1 [52] quantities in commonly consumed foods. Based on these more recent findings, foods such as peanut butter, manufactured biscuits and cakes, and canned and processed meats had the highest quantities of AGEs [52].
3.2.2. Absorption of Dietary AGEs
There is much controversy regarding the quantity and processes used for AGE trafficking across the gastrointestinal (GI) tract. It remains to be fully understood whether dietary AGEs contribute to disease via: (i) direct entry into the circulation, resulting in tissue deposition, inflammation, and increased burden on clearance mechanisms; (ii) impacting intestinal health, permeability and entero-endocrine signalling; (iii) modulation of the GI tract microbiome, or; (iv) promotion of inflammation within the GI tract. It is possible that all these factors act in concert to promote the pathophysiology seen in the context of increased dietary AGE consumption.
The heterogeneity of AGEs that can be produced in food processing is much greater than those produced physiologically [53]. Therefore, well-characterised AGEs such as CML, CEL, pentosidine and pyralline are commonly used as biomarkers to gauge AGE load and uptake from the diet (Table 1). Generally, it is approximated from human and animal studies that between 10–30% of dietary AGEs are absorbed from the GI tract [54,55]. However, extensive analysis of the literature shows little consensus, with some studies suggesting greater uptake in humans [56], particularly in infants [57]. There have been many studies examining the uptake, kinetics, and bio-distribution of orally and intravenously administered AGEs and AGE precursors (Table 1). Despite this, there have been no technologies developed to accurately map this in humans in real-time. Such technologies would strengthen our understanding of the link between dietary AGEs and their associated pathologies.

Table 1.
Uptake, elimination and biodistribution studies of AGEs and their precursor Amadori products.
3.3. AGE Clearance
Another major factor regulating AGE homeostasis is their clearance from the body. At the tissue level, this occurs through cellular proteolytic systems, which endocytose AGEs and break them down via receptor-mediated and non-receptor-mediated pathways into AGE peptides, which are then released back into the circulation [71]. At the systemic level, clearance of AGEs is thought to occur via the liver [72,73] and the kidney [63], where clearance of not only AGEs, but reactive carbonyl precursors, such as MG-H1, 3-DG and glyoxal and AGE-peptides, is important to maintain AGE homeostasis, although trafficking studies in humans are lacking. Animal models suggest that AGEs are filtered by the glomeruli, reabsorbed by proximal tubule cells and further processed and cleared into the urine [63,74]. In humans, AGE concentrations are commonly inversely related to renal function [42,44,75,76]. More recently, Haus et al. demonstrated that in obese but healthy humans, during a 24-h period of hyperglycaemia, plasma concentrations of several AGEs and oxidative products decreased concordant with an increase in the fractional excretion of these products into the urine [77]. Given its role in AGE clearance, the kidney has been highlighted as an important site of AGE mediated pathology [78,79,80,81,82,83,84], with renal function vital for AGE homeostasis, but potentially vulnerable to AGE-mediated damage.
Animal models demonstrate that uptake and clearance of intravenously administered AGEs also occur via the liver by endothelial, Kupffer and parenchymal cells [72]. However, the rate of clearance is dependent on the degree of AGE modification, with minimally AGE-modified bovine serum albumin (AGE-BSA) remaining in the circulation for significantly longer duration [73]. Concordant with the kidney, rodent models suggest that the liver also appears to be vulnerable to AGE-mediated pathology, particularly in the context of high AGE consumption [83,85].
4. AGE Receptors—Facilitators of Clearance and Mediators of Pathology
There are a number of receptors that have been characterised as binding AGEs, including the Receptor for Advanced Glycation End Products (RAGE), AGE Receptor 1 (AGER1; OST-48), AGE Receptor 2 (AGER2; 80K-H), AGE Receptor 3 (AGER3; Galectin 3) and the class A macrophage scavenger receptors types 1 and 2 [86]. The majority of studies focus on RAGE, since AGE binding induces cellular signal transduction. Other AGE receptors may play a role in AGE clearance and detoxification [87], such as AGER1, which has also garnered significant attention.
4.1. RAGE
RAGE, a member of the immunoglobulin superfamily of receptors [88,89], is the most widely studied of the AGE receptors. As a multi-ligand, pattern recognition receptor, RAGE also binds to s100 calgranulins [90], amphoterin/high mobility group box 1 protein (HMGB1) [91], lipopolysaccharide (LPS) [92], β amyloid [93], transthyretin [94], Mac-1 [95], complement 1q [96] and potentially DNA [92]. RAGE is highly expressed on mucous membranes such as those present in the lung and the GI tract, and within the immune system [97]. In the healthy kidney, RAGE localisation and gene expression occurs in the vascular smooth muscle, the epithelia of the proximal and distal tubule and is significantly upregulated in various inflammatory and non-inflammatory disease settings [98,99]. RAGE is significantly upregulated on podocytes in multiple diseases settings including diabetic nephropathy [99,100,101].
The RAGE gene, AGER, can be differentially spliced to form more than 20 different variants [102,103]. The full length form of RAGE (flRAGE) consists of three immunoglobulin-like domains known as the V1(variable), C1 and C2 domains; a transmembrane helix and a short highly charged cytoplasmic domain that is essential for signal transduction (Figure 2) [104]. As well as the full length isoform, there is an N terminal truncated form of the receptor that lacks the AGE binding capacity (Figure 2) [105]. Two secreted forms of RAGE exist, which form the circulating pool: endogenous secretory RAGE (esRAGE), which is a product of AGER transcription and soluble RAGE (sRAGE), which is cleaved from cell membranes by the proteases a-disintegrin and metalloproteinase domain-containing protein 10 (ADAM-10) (Figure 2) [106,107].
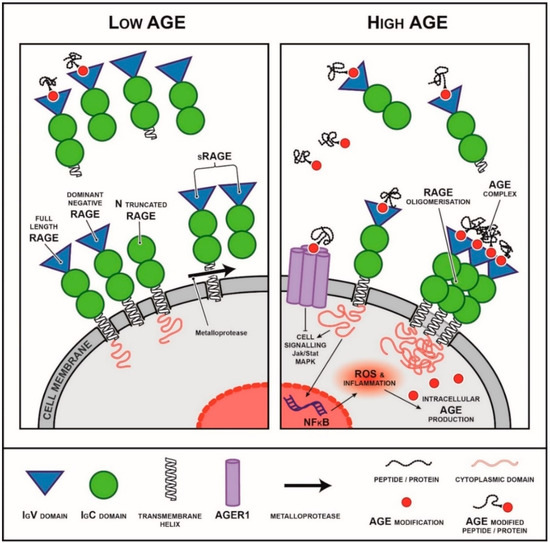
Figure 2.
Isoforms of the receptor for AGEs (RAGE) and their interactions with AGEs and downstream pathways activated. RAGE is a multi-ligand member of the immunoglobulin superfamily of receptors. It can exist as different isoforms including membranous isoforms (full length RAGE, dominant negative RAGE, N truncated RAGE) as well as soluble secreted and cleaved isoforms. The cytoplasmic domain is essential for RAGE signalling. The secreted isoforms are thought to act as decoys, binding to RAGE ligands and preventing them from binding to membranous forms of RAGE activating downstream signalling. In a low AGE environment (left), circulating sRAGE is believed to act as a decoy receptor, binding circulating RAGE ligand such as AGEs and preventing binding to membranous isoforms of RAGE. In a high AGE environment (right), circulating sRAGE levels are commonly decreased or sRAGE capacity is saturated and is no longer sufficient to prevent RAGE downstream signalling.
AGE-flRAGE binding initiates a number of signalling cascades (Figure 2), including mitogen-activated protein kinase (MAPK) [108,109,110], janus kinase/signal transducer and activator of transcription (JAK/STAT) [111,112] and rho GTPases [113,114,115]. Furthermore, RAGE ligand binding results in nuclear translocation of transcriptional factors such as nuclear factor kappa B (NF-κB) [94,116] and early growth response protein 1 (Egr-1) [117]. These pathways, activate downstream pro-inflammatory pathways inducing chemotactic cellular migration, proliferation and apoptosis (Figure 2) [118]. These are in line with the known physiological role for RAGE in host-pathogen defence.
Animal and human data indicate that RAGE expression is likely modulated by environmental factors, including AGEs [119,120] and disease states such as diabetes [98,101]. Whilst increased levels of full length, membrane-bound RAGE are associated with disease progression and poor outcomes in both animal models and humans [100,101,104], circulating soluble forms are inversely correlated with metabolic parameters, including body mass index (BMI), serum triglycerides, HbA1c, insulin resistance [121], and chronic disease [122,123,124,125,126,127], and positively associate with longevity [128,129]. This is supported by animal studies that have conclusively shown that administration of sRAGE or esRAGE is protective or has positive outcomes for a number of disease states [101,130,131,132]. This evidence has led to the hypothesis that soluble forms of RAGE may act as a competitive antagonist for AGEs, modulating their ability to interact with receptors such as membrane-bound RAGE, thereby preventing downstream signalling (Figure 2) [133,134].
4.2. AGER1
AGER1, also known as OST48, is a 48-kDa, type I transmembrane receptor protein [135] that localises to the plasma membrane [136] and endoplasmic reticulum (ER) [137]. The primary role of OST48 is as a subunit of the multiprotein oligosaccharyltransferase complex responsible for the N-linked glycosylation of asparagine residues during protein translation at the ER; therefore, it is expressed in all tissues, but particularly glandular cells (proteinatlas.org) [138,139]. In AGE biology, it is postulated to facilitate AGE clearance by lowering AGE levels in the intra- and extra-cellular environment and facilitating their clearance into the urine [140,141]. Additionally, it appears to be a negative regulator of the inflammatory response in some cell types [142].
In several chronic disease states, such as diabetes, CKD and autoimmune diseases AGER1 levels are down-regulated [82]. Dietary AGEs and other dietary factors appear to influence AGER1 levels. For example, restricting of AGE consumption has been shown to increase AGER1 in peripheral blood mononuclear cells (PBMCs) in CKD patients [140] and in the kidney, spleen and liver of healthy, but aged, mice [143]. Similar increases in PBMC AGER1 have been observed in clinical trials of a Mediterranean diet, low in AGEs [144,145] and a diet high in monounsaturated fatty acids (PUFAs) [146]. However, global over-expression of AGER1 increased urinary AGE clearance and improved insulin effectiveness in experimental diabetes in mice, but resulted in increased tubulointerstitial fibrosis [147]. Similarly, overexpression of AGER1 in the podocytes of mice resulted in glomerulosclerosis and podocyte damage and a decline in GFR, despite increasing renal AGE clearance, this was further exacerbated by diabetes [148].
5. AGE-Mediated Pathology
5.1. AGEs Can Induce Both Receptor Mediated and Non-Receptor Mediated Pathology
AGE-mediated pathology can result from the deposition of AGEs and the subsequent crosslinking of structural proteins in cells and organs and tissues or through AGE-receptor interactions, both of which impair tissue and cellular function. For example, collagen AGE modification and crosslinking of other extracellular matrix proteins leads to structural alterations, including changes in packing density [149] and surface charges [150] and the loss of structural integrity. This results in stiffening of the vasculature and expansion of cellular basement membranes in diabetes and CKD. AGE-RAGE binding activates pro-inflammatory signal transduction cascades increasing cytokine and growth factor expression [19,151,152]. As such, the AGE-RAGE axis is implicated in diabetes complications, CKD and end-stage renal diseases (ESRD), but also many other chronic diseases, including Alzheimer’s disease, atherosclerosis, cataracts, Parkinson’s disease, sarcopenia, vascular dementia and aging [153,154]. In humans, several studies show strong associations between circulating AGEs and inflammatory markers in elderly [155], young (adolescent) [155], and diabetic [156] populations. However, low and high AGE dietary studies in humans have been contradictory in their findings with some authors reporting no effects of dietary AGEs on systemic inflammation [157].
5.2. Dietary AGEs and AGE Pathology
The contribution of dietary AGEs to various pathologies remains to be fully elucidated. In rodent studies, chronic dietary exposure to excess CML results in damage to the glomerulus of the kidney, and albumin in the urine [158], insulin resistance [159,160], and insulin secretory defects [161,162]. In both wild type and T2D models, excess AGE dietary consumption elevates fasting plasma glucose levels, and worsens proteinuria, albuminuria and kidney [79,163,164,165] and liver [85] injury. Conversely, low AGE diets extend lifespan and improve age-related glucose abnormalities and renal outcomes and increase AGER1 expression in C57Bl6K mice [143]. Low AGE diets can also prevent type 1 diabetes (T1D) when administered to pregnant and weaning mothers in non-obese diabetic (NOD) mouse models of T1D [166] and attenuate insulin resistance and improve vascular and renal outcomes [81,167,168] and wound healing [169] in other diabetic models.
In humans, many benefits have been shown with AGE restriction in the diet. In individuals with T2D and renal failure, excessive AGE intake positively correlated with serum biomarkers of oxidative stress, inflammation, endothelial dysfunction, hyperglycaemia and hyperlipidaemia [170,171]. Conversely, dietary AGE restriction in healthy individuals and those with T2D have demonstrated favourable outcomes in circulating 8-isoprostanes and tumor necrosis factor alpha (TNFα) [140,141], improvements in cognitive function [172] and insulin sensitivity [141,173]. The effects of dietary AGEs on insulin sensitivity is vitally important, since abnormalities in glucose homeostasis are potent risk factors for CKD development and progression. In animal models, increasing circulating AGEs [174,175] and consumption of diets high in AGEs [160,162,167,176] irrefutably result in decreased insulin sensitivity, independent of other dietary factors [119,160]. In humans, several studies have demonstrated a relationship between high AGE diets and insulin signalling defects [177] with acute changes in insulin secretion following high AGE meal challenges [178,179]. Additionally, two independent randomised crossover dietary intervention studies have found that low AGE diets improve insulin sensitivity [173,180], and renal function [79] following a 2-week, or 4-week, high and low AGE dietary protocol, respectively. Although a recent study failed to recapitulate these findings with regard to insulin sensitivity [157].
5.3. Dietary AGEs and the Microbiome
One of the most interesting frontiers in AGE biology is their interaction with the microbiome. Many dietary AGEs are not easily absorbed by the small intestine, passing instead to the colon where they are available for metabolism by the colonic microbiome [53]. Only limited in vivo data exists regarding the effects of ingested AGEs on the colonic microbiome. In adolescent males, CML intake was negatively associated with Lactobacilli and positively associated with Enterobacteria following two weeks on a high or low AGE diet (randomised crossover design) [181]. Meanwhile, in dialysis patients, dietary AGE restriction resulted in alterations to the gut microbiome [182]. However, both studies are limited by small study size and understanding the physiological relevance of reported microbial changes is challenging. In mice however, 22 weeks consuming a diet enriched with the AGE MG-H1 induced significant microbial changes in the gut that were associated with metabolic dysregulation and increased systemic inflammation [183]. Similarly, in mice fed a heat-treated high AGE diet for 24 weeks, significant expansion of Helicobacteraceae and contraction of Saccharibacteria populations was observed, which coincided with an increase in gut permeability, increased circulating lipopolysaccharide (LPS), complement activation and onset of changes associated with early CKD. This was attenuated by a diet high in resistant starch [165].
It is reasonable to assume that probiotic or prebiotic supplementation that modulates the microbiome may, in turn, contribute to circulating and tissue AGE burden. However, whether this occurs through modulation of enteroendocrine factors, inflammation or gut-barrier permeability remains to be fully elucidated. Indeed, a recent meta-analysis found that pre-, pro- and symbiotic-supplementation reduces fasting insulin levels, hyperinsulinaemia and circulating AGEs in individuals with diabetes [184], suggesting the link between the microbiome, glucose homeostasis and AGE burden is an important one. However, these biota-based effects were modest, and the authors highlight the challenges of this type of human study where variation is significant and highly dependent on microbiome composition at baseline. A small, randomised control trial in women with T2D found supplementation with the prebiotic resistant dextrin significantly increased serum sRAGE and reduced the serum concentrations of AGEs CML, pentosidine, malondialdehyde (MDA) [185]. However, the study included only a small number of participants and did not assess microbiome composition. Further, the prebiotic supplementation with dextrin also reduced blood glucose concentrations, body weight and energy intake, and so it is almost impossible to determine the causative factors driving changes in circulating AGE concentrations. Another randomised crossover study comparing the effects of prebiotic supplementation on circulating AGEs, in individuals with pre-diabetes, may help further elucidate the contribution of the microbiome to AGE burden within the body [186]. This study has completed recruitment (ACTRN12613000130763) but to our knowledge the data have not been published. This remains an interesting and novel area requiring further investigation.
6. AGEs and Kidney Disease
Regardless of the presence of diabetes, previous studies have established a link between elevated circulating AGEs and a progressive decline in renal function [20,21,75,187,188,189]. Since dialysis does not as effectively remove AGEs [190], individuals receiving haemo- or peritoneal-dialysis show higher circulating AGE concentrations compared to healthy individuals [191,192]. Circulating AGEs also show a positive association with markers of inflammation and oxidative stress in uremic patients [193] and predict cardiovascular disease mortality in stable renal transplant recipients [194]. Moreover, elevated circulating concentrations of sRAGE are positively associated with the incidence of diabetic kidney disease (DKD) [195] and CKD [196,197]. In the ADVANCE study of 3763 individuals with T2D, both circulating AGEs and sRAGE were positively associated with incident CKD, progressive CKD, and mortality which led researchers to propose that the AGE:RAGE axis may represent an important target for the prevention and management of diabetic nephropathy [198].
The relationship between endogenous AGE production and renal function has been demonstrated in studies using skin collagen fluorescence as a biomarker of AGE burden. Skin collagen autofluorescence is significantly increased in CKD patients without diabetes [199,200,201] and predicts those individuals with progressive CKD [199,202]. In individuals with diabetes and established CKD, skin autofluorescence is also inversely associated with estimated glomerular filtration rate eGFR, a measure of renal function [203,204], and positively associated with incidence of CKD [204] and mortality [205]. Furthermore, in young adults with T1D but without established kidney disease, skin autofluorescence and eGFR predicted approximately 25% of variance for DKD risk [206]. In a longitudinal study of individuals with T1D, the Diabetes Control and Complications Trial (DCCT), and its follow-up study, Epidemiology of Diabetes Interventions and Complications (EDIC), skin collagen CML concentrations predicted individuals who developed nephropathy and cardiovascular disease [207,208], and this was independent of glycaemic control. These results indicate that AGE production and its accumulation is implicated in CKD as more than simply a biomarker for glucose homeostasis.
Increases in AGE concentrations in individuals with CKD are likely due to both reduced renal clearance and accelerated endogenous AGE production. In vitro, AGEs (CML and pentosidine) and reactive precursors form more rapidly and to significantly greater levels in serum from uremic patients compared to healthy controls [209]. This suggests that circulating factors that promote or stimulate the rapid formation of AGEs are present in the bloodstream of individuals with reduced renal function. In diabetes, increased circulating and urinary excretion of AGEs appears to predict development and progression of DKD [210], even in young adult populations without established kidney disease [211]. However, in a prospective study of T1D patients with early DKD, there was a decrease in circulating AGE concentrations associated with increased urinary CML clearance. This was associated with future rapid GFR decline [212]. A decrease in circulating AGEs might be the result of glomerular hyperfiltration, which is common in diabetes and may lead to a reduction in low molecular weight AGEs [213]. However, in cross-sectional data from patients with diabetes of long duration, urinary AGEs were positively associated with albuminuria, independent of isotopic GFR [210], suggesting tubular processing and excretion of AGEs is an important determinant of urinary AGE concentrations, as suggested by Kern et al., who also concluded that urinary AGEs are a useful early marker of tubular damage [214]
6.1. Mechanisms by Which AGEs Damage the Kidney
AGEs accumulate in the renal compartment where they mediate kidney damage (Figure 3). Cross-linking of matrix proteins by AGEs leads to stiffness and altered structural function at sites such as the glomerulus, peritubular vasculature and arterioles of the kidney, promoting glomerulosclerosis, atherosclerosis and thickening of the basement membrane [26]. AGE accumulation in the glomerulus is also associated with podocyte epithelial mesenchymal transition [215,216]. Similarly, in vitro exposure to high concentrations of AGEs induces tubular-epithelial-myofibroblast transition via RAGE dependent pathways, contributing to tubulointerstitial fibrosis [217,218,219]. AGE interactions with membrane-bound forms of RAGE, ultimately lead to the induction of a number of pro-inflammatory cytokines and chemo-attractants [220,221,222,223,224,225] via the activation of Nuclear Factor kappa B (NF-κB) and JAK-STAT pathways, promotion of inflammation via interleukin 1 β (IL-1β) and TNFα pathways [226,227], and the stimulation and production of NADPH oxidase and mitochondrial derived ROS [101,228]. These aforementioned pathways are well described in the development of fibrosis, glomerulosclerosis, apoptosis and cell death and are characterised contributors to the progression of DKD and CKD in both humans and pre-clinical models [19]. Accumulation of RAGE ligands, including AGEs, stimulates increased RAGE expression on podocytes in humans and rodent models [98,100,101] and numerous studies have shown RAGE knock-out in diabetic mice improves renal injury [228,229,230].
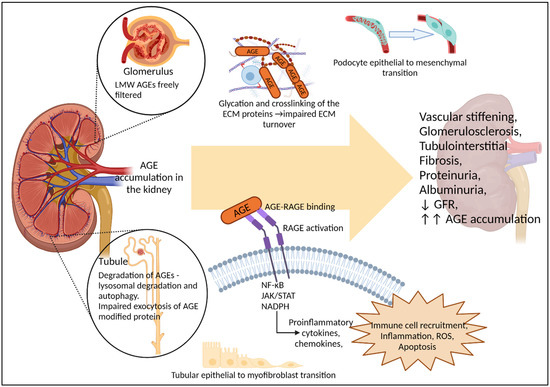
Figure 3.
Renal handling of AGEs and their contribution to renal pathology. In vivo evidence from rodents suggests AGEs accumulate in the kidney, which is also a major site for AGE clearance. Low molecular weight (LMW) AGEs are freely filtered by the glomeruli, while lysosomal degradation and autophagy of AGEs appears to occur within the tubules, with AGE modification impairing exocytosis of proteins by the tubular cells. AGE accumulation in the kidney contributes to a number of pathological pathways, including glycation and crosslinking of structural proteins, dedifferentiation of specialised epithelial cells such as podocytes, and RAGE activation leading to further inflammation, ROS and cellular apoptosis. Together these can contribute to, or exacerbate hemodynamic changes, glomerulosclerosis, tubulointerstitial fibrosis, proteinuria, albuminuria and loss of GFR. As renal function declines, renal capacity to excrete AGEs is reduced, leading to increased AGE burden within the body, as is seen with CKD. However, evidence that AGEs alone, in the absence of diabetes or underlying renal conditions, can induce renal dysfunction has largely come from rodent models. with only limited associative studies in humans.
6.2. Renal Handling of AGEs
In vivo uptake and trafficking studies in healthy rats have shown that intravenously injected low molecular weight AGE-peptides, but not AGE-BSA, are freely filtered at the glomerulus and detected in urine [74]. However, both in vivo and in vitro data suggest that, once filtered, AGEs bind to and/or enter proximal tubule cells [63,74,217,231,232], where their removal is dependent on lysosomal degradation and autophagy [233]. This is supported by in vitro studies in renal proximal tubule cells, which can degrade AGE-modified albumin and release them into the supernatant However, exocytosis of glycated albumin peptide fragments was slow compared to non-glycated fragments, suggesting that renal tubule processing of AGE modified proteins is impeded by glycation [231]. Ultimately, this may lead to increased accumulation of AGEs within proximal tubule cells and disrupt autophagosome lysosomal pathways [234]. Supporting this, Tessier et al. showed that in mice, the kidneys were amongst those organs with the highest levels of dietary AGE accumulation following 30 days on an 13C-CML-BSA enriched diet [69]. Overall, it appears that the transport of free- and/or protein-bound AGEs across the renal filtration barrier and tubular cells may depend on protein size and the degree and site of glycation.
6.3. Exogenous AGEs and Kidney Function
There is a paucity of studies examining the longitudinal effects of high AGE consumption on risk of kidney disease in healthy humans. One prospective study using dietary questionnaires demonstrated a 2-fold increase in CKD risk with high AGE consumption from dietary fat sources [235] even after adjustment for diabetes and hypertension, however, human studies examining the effects of dietary AGEs on direct measures of kidney function are lacking [236]. A small pilot study of 10 healthy participants demonstrated that the degree of glycation of isocaloric protein loads differentially affected renal haemodynamics, with high AGE loads increasing renal perfusion and renal oxygen consumption [237]. In overweight individuals without diabetes, a randomised cross-over study showed that increased AGE consumption for 2 weeks increased systemic inflammation, albuminuria and decreased eGFR, while paradoxically increasing urinary AGE clearance and reducing circulating levels [79]. In agreement, several studies in patients with diabetes [156] and healthy individuals [173] have demonstrated a significant effect of increased dietary AGEs on other determinants of kidney disease such as increased endothelial dysfunction and oxidative stress [156], as well as adverse effects on insulin sensitivity and circulating lipids [173] when compared to low AGE diets. Even a single oral AGE challenge was sufficient to induce endothelial dysfunction and oxidative stress in healthy individuals and in those with diabetes [238]. However, not all studies of high/low AGE dietary interventions in healthy adults have reported changes in endothelial dysfunction and inflammation [239,240]. This is possibly because the AGE content of the high AGE diets used were similar to that of a typical Western diet and may have been comparable to the patients’ diets at baseline.
In animal models, significantly more work has been undertaken to measure the direct effects of exogenous AGEs on the kidney. Animals fed a high CML diet accumulate AGEs preferentially in the kidneys [69,241] and, in models with established kidney disease, such as the remnant kidney model, high AGE diets for a period of 5–13 weeks (study dependent) increased proteinuria [164,242,243], fibrosis and glomerular injury [243]. In healthy rats without kidney injury, a 4-week high fat/high AGE diet increased serum creatinine, suggestive of decreased GFR, which was associated with enhanced kidney CML deposition and markers of oxidative stress and inflammation [244]. In contrast, a more recently published study examining the effects of a high CML diet for 18 months in healthy male mice reported no effects on kidney ageing [245]. This may indicate that where there is no pre-existing kidney injury, a genetic predisposition to kidney disease, or risk factors such as insulin resistance, enrichment of the diet with single AGEs is not a potent driver of renal changes compared to a baked heterogeneous AGE diet.
While the majority of studies have reported on the impact of AGEs on kidney disease risk factors (e.g., diabetes, hypertension), few have assessed effects on kidney function. However, as highlighted above, there is much to be untangled regarding whether kidney changes (functional and structural) are actioned via direct renal uptake and trafficking of ingested AGEs or mediated indirectly by AGE effects on, and renal interactions with, the vascular, GI tract, enteroendocrine systems and the microbiome.
6.4. Therapeutic Targeting of AGEs in Kidney Disease
There are several classes of therapeutic agents aimed at targeting AGEs. A thorough review of these can be found here [246]. Several of these agents have been trialled in experimental models of DKD or CKD and in clinical trials.
6.4.1. Carbonyl Scavengers
Carbonyl scavenging is one approach that has been trialled quite extensively since the early 1990s. Aminoguanidine (Pimagedine), a carbonyl scavenger, was found to lower AGEs and attenuate the DKD-driven rise in albuminuria and prevent mesangial matrix expansion in rats [247]. However, aminoguanidine was found to bind to a number of targets including functional endogenous carbonyls, making it inappropriate as a therapeutic agent [248]. Furthermore, two clinical trials were performed, ACTION I and ACTION II, but they failed to show efficacy and ACTION II was terminated early due to safety concerns and off target effects [248].
6.4.2. Vitamin B and Its Derivatives
The B group vitamins and a number of their derivatives have also been trialled as AGE lowering therapies. These include thiamine, benfotiamine, and pyridoxamine [246]. These agents are thought to reduce AGEs by increasing the activity of the thiamine-dependent enzyme, transketolase. The resulting stimulation of the pentose phosphate pathway should, in theory, reduce glycolytic intermediates. Clinical trials of thiamine and benfotiamine and their efficacy in the context of kidney disease are lacking. However, pyridoxamine, which is thought to act by inhibiting the conversion of Amadori products and by chelation of dicationic metal ions has shown efficacy in animal models of kidney disease [249,250,251] and to effectively reduce CML, CEL and transforming growth factor β (TGFβ) in DKD patients [252]. Unfortunately, a phase III clinical trial examining the efficacy of pyridoxamine in DKD, PIONEERIII, began recruiting in 2014 but was terminated due to financial constraints in 2016. As yet, the study has not been recommenced.
6.4.3. AGE Cross Link Breakers
N-phenacylthiazolium bromide (PTB) was the first reported compound capable of breaking the crosslinks formed by AGEs, leading to the development of a class of compounds known as “cross link breakers” [253]. While PTB demonstrated efficacy for reducing AGEs in diabetic rodents [254], PTB was not effective at preventing or attenuating DKD in rodents [255,256] and due to its in vivo instability further compounds were developed [246]. 4,5-dimethyl-3-phenacylthiazolium chloride (ALT-711®, Alagebrium chloride) is the most well studied of the PTB analogues. Alagebrium has cross link breaking properties but also appears to be a MG scavenger, and exhibit antioxidant and chelating properties. It has demonstrated efficacy in reducing blood pressure, mesangial matrix expansion and tubulointerstitial fibrosis in rodent models of DKD [257,258] and hypertension [259]. Due to loss of the clinical sponsor and patent during the global financial crisis, various Phase II clinical trials both in heart failure (NCT00739687 and NCT00516646) and DKD (NCT00557518) had to be prematurely terminated.
6.4.4. RAGE Blockade
AGE-RAGE blockade is another potential therapeutic target for chronic kidney disease. RAGE knockout in mice prevents or attenuates DKD [101,168,260] and deletion of RAGE from bone marrow derived cells reduced renal functional changes as well as immune infiltration seen with experimental (STZ induced) diabetes [261]. Long term treatment with the RAGE decoy receptor, sRAGE, has been shown to improve measures of DKD in the db/db model of T2D and obesity [101] and RAGE blockade by antibodies was effective at slowing progression of DKD in models of T1D [262] and T2D [263]. More recently, AGE targeted aptamers that prevented AGE-RAGE signalling protected against DKD in a mouse model of T2D and obesity [264]. Similarly, RAGE aptamers given early in diabetes or at diabetes induction showed significant potential attenuating progression of DKD in rats [265] and showed effectiveness in reducing markers of kidney injury in uni-nephrectomised deoxycorticosterone acetate (DOCA)/salt-induced hypertensive mice [266]. Although there is considerable pre-clinical evidence for RAGE blockade as a therapeutic option in kidney disease, to our knowledge no agents have reached the clinical trial phase for kidney disease.
7. Conclusions
AGEs are an important mediator of pathology, particularly in diabetes and kidney disease where AGE homeostasis is unbalanced by impaired clearance and increased endogenous production. Given the evidence presented, dietary AGEs appear to be an important contributor the body’s AGE pool, but may also act to accelerate endogenous AGE production through increased oxidative stress, endothelial dysfunction and by precipitating glucose abnormalities. Critical experiments need to be performed in humans to understand the extent to which dietary AGEs contribute to the onset and progression of CKD in humans, since AGE-lowering strategies show some promise in clinical studies performed to date.
Funding
A.K.F. is supported by a UQ Research Support Fellowship. D.J.B. is supported by a UQ Research Support Fellowship and Mater Foundation. J.M.F. is supported by a Leadership Award from the National health and Medical Research Council of Australia (GNT 2010053). This work was also supported by Mater Foundation.
Acknowledgments
Graphical abstract was created with BioRender.com (accessed on 23 May 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Gur, E.; Levy, D.; Topaz, G.; Naser, R.; Wand, O.; Kitay-Cohen, Y.; Benchetrit, S.; Sarel, E.; Cohen-Hagai, K. Disease severity and renal outcomes of patients with chronic kidney disease infected with COVID-19. Clin. Exp. Nephrol. 2022, 26, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef]
- Odermatt, A. The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am. J. Physiol.-Ren. Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef]
- Hariharan, D.; Vellanki, K.; Kramer, H. The Western Diet and Chronic Kidney Disease. Curr. Hypertens. Rep. 2015, 17, 16. [Google Scholar] [CrossRef]
- Wang, L.; Martínez Steele, E.; Du, M.; Pomeranz, J.L.; O’Connor, L.E.; Herrick, K.A.; Luo, H.; Zhang, X.; Mozaffarian, D.; Zhang, F.F. Trends in Consumption of Ultraprocessed Foods Among US Youths Aged 2–19 Years, 1999–2018. JAMA 2021, 326, 519–530. [Google Scholar] [CrossRef]
- Fiolet, T.; Srour, B.; Sellem, L.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Deschasaux, M.; Fassier, P.; Latino-Martel, P.; Beslay, M.; et al. Consumption of ultra-processed foods and cancer risk: Results from NutriNet-Santé prospective cohort. BMJ 2018, 360, k322. [Google Scholar] [CrossRef]
- Srour, B.; Fezeu, L.K.; Kesse-Guyot, E.; Allès, B.; Debras, C.; Druesne-Pecollo, N.; Chazelas, E.; Deschasaux, M.; Hercberg, S.; Galan, P.; et al. Ultraprocessed Food Consumption and Risk of Type 2 Diabetes Among Participants of the NutriNet-Santé Prospective Cohort. JAMA Intern. Med. 2020, 180, 283–291. [Google Scholar] [CrossRef]
- Suksatan, W.; Moradi, S.; Naeini, F.; Bagheri, R.; Mohammadi, H.; Talebi, S.; Mehrabani, S.; Hojjati Kermani, M.A.; Suzuki, K. Ultra-Processed Food Consumption and Adult Mortality Risk: A Systematic Review and Dose–Response Meta-Analysis of 207,291 Participants. Nutrients 2022, 14, 174. [Google Scholar] [CrossRef]
- Srour, B.; Fezeu, L.K.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Andrianasolo, R.M.; Chazelas, E.; Deschasaux, M.; Hercberg, S.; Galan, P.; et al. Ultra-processed food intake and risk of cardiovascular disease: Prospective cohort study (NutriNet-Santé). BMJ 2019, 365, l1451. [Google Scholar] [CrossRef]
- Schnabel, L.; Kesse-Guyot, E.; Allès, B.; Touvier, M.; Srour, B.; Hercberg, S.; Buscail, C.; Julia, C. Association Between Ultraprocessed Food Consumption and Risk of Mortality Among Middle-aged Adults in France. JAMA Intern. Med. 2019, 179, 490–498. [Google Scholar] [CrossRef]
- Rico-Campà, A.; Martínez-González, M.A.; Alvarez-Alvarez, I.; Mendonça, R.D.D.; de la Fuente-Arrillaga, C.; Gómez-Donoso, C.; Bes-Rastrollo, M. Association between consumption of ultra-processed foods and all cause mortality: SUN prospective cohort study. BMJ 2019, 365, l1949. [Google Scholar] [CrossRef]
- Bonaccio, M.; Costanzo, S.; Ruggiero, E.; Persichillo, M.; Esposito, S.; Olivieri, M.; Di Castelnuovo, A.; Cerletti, C.; Donati, M.B.; de Gaetano, G.; et al. Changes in ultra-processed food consumption during the first Italian lockdown following the COVID-19 pandemic and major correlates: Results from two population-based cohorts. Public Health Nutr. 2021, 24, 3905–3915. [Google Scholar] [CrossRef]
- Ruíz-Roso, M.B.; de Carvalho Padilha, P.; Matilla-Escalante, D.C.; Brun, P.; Ulloa, N.; Acevedo-Correa, D.; Arantes Ferreira Peres, W.; Martorell, M.; Rangel Bousquet Carrilho, T.; de Oliveira Cardoso, L.; et al. Changes of Physical Activity and Ultra-Processed Food Consumption in Adolescents from Different Countries during COVID-19 Pandemic: An Observational Study. Nutrients 2020, 12, 2289. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bugel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2013, 60, 10–37. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, X.; Yang, Z.; Liu, Y.; Qiu, X.; Zeng, Z.; Lu, S.; Liu, Y. Sodium Ions Affect Pyrraline Formation in the Maillard Reaction with Lys-Containing Dipeptides and Tripeptides. Front. Nutr. 2022, 9, 874650. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Koska, J.; Gerstein, H.C.; Beisswenger, P.J.; Reaven, P.D. Advanced Glycation End Products Predict Loss of Renal Function and High-Risk Chronic Kidney Disease in Type 2 Diabetes. Diabetes Care 2022, 45, 684–691. [Google Scholar] [CrossRef]
- Saulnier, P.-J.; Wheelock, K.M.; Howell, S.; Weil, E.J.; Tanamas, S.K.; Knowler, W.C.; Lemley, K.V.; Mauer, M.; Yee, B.; Nelson, R.G.; et al. Advanced Glycation End Products Predict Loss of Renal Function and Correlate with Lesions of Diabetic Kidney Disease in American Indians with Type 2 Diabetes. Diabetes 2016, 65, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Ferrucci, L.; Sun, K.; Beck, J.; Dalal, M.; Varadhan, R.; Walston, J.; Guralnik, J.M.; Fried, L.P. Advanced glycation end products and their circulating receptors predict cardiovascular disease mortality in older community-dwelling women. Aging Clin. Exp. Res. 2009, 21, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Plasma Carboxymethyl-Lysine, an Advanced Glycation End Product, and All-Cause and Cardiovascular Disease Mortality in Older Community-Dwelling Adults. J. Am. Geriatr. Soc. 2009, 57, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.-H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D. Higher Plasma Levels of Advanced Glycation End Products Are Associated with Incident Cardiovascular Disease and All-Cause Mortality in Type 1 Diabetes: A 12-year follow-up study. Diabetes Care 2011, 34, 442–447. [Google Scholar] [CrossRef]
- Machowska, A.; Sun, J.; Qureshi, A.R.; Isoyama, N.; Leurs, P.; Anderstam, B.; Heimburger, O.; Barany, P.; Stenvinkel, P.; Lindholm, B. Plasma Pentosidine and Its Association with Mortality in Patients with Chronic Kidney Disease. PLoS ONE 2016, 11, e0163826. [Google Scholar] [CrossRef]
- Ulrich, P.; Cerami, A. Protein glycation, diabetes, and aging. Recent Prog. Horm. Res. 2001, 56, 1–21. [Google Scholar] [CrossRef]
- Thorpe, S.R.; Baynes, J.W. Maillard reaction products in tissue proteins: New products and new perspectives. Amino Acids 2003, 25, 275–281. [Google Scholar] [CrossRef]
- Thornalley, P.J. Dicarbonyl Intermediates in the Maillard Reaction. Ann. N. Y. Acad. Sci. 2005, 1043, 111–117. [Google Scholar] [CrossRef]
- Skovsted, I.C.; Christensen, M.; Breinholt, J.; Mortensen, S.B. Characterisation of a novel AGE-compound derived from lysine and 3-deoxyglucosone. Cell. Mol. Biol. 1998, 44, 1159–1163. [Google Scholar]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef]
- Wells-Knecht, K.J.; Brinkmann, E.; Wells-Knecht, M.C.; Litchfield, J.E.; Ahmed, M.U.; Reddy, S.; Zyzak, D.V.; Thorpe, S.R.; Baynes, J.W. New biomarkers of Maillard reaction damage to proteins. Nephrol. Dial. Transpl. 1996, 11 (Suppl. 5), 41–47. [Google Scholar] [CrossRef]
- Uribarri, J.; Tuttle, K.R. Advanced glycation end products and nephrotoxicity of high-protein diets. Clin. J. Am. Soc. Nephrol. 2006, 1, 1293–1299. [Google Scholar] [CrossRef]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. J. Diabetes Res. 2007, 2007, 061038. [Google Scholar] [CrossRef]
- Thornalley, P.J. Pharmacology of methylglyoxal: Formation, modification of proteins and nucleic acids, and enzymatic detoxification--a role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Thornalley, P.; Langborg, A.; Minhas, H. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef]
- Monnier, V.M.; Bautista, O.; Kenny, D.; Sell, D.R.; Fogarty, J.; Dahms, W.; Cleary, P.A.; Lachin, J.; Genuth, S. Skin collagen glycation, glycoxidation, and crosslinking are lower in subjects with long-term intensive versus conventional therapy of type 1 diabetes: Relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes 1999, 48, 870–880. [Google Scholar] [CrossRef]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; Monnier, V.M. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef]
- den Dekker, M.A.M.; Zwiers, M.; van den Heuvel, E.R.; de Vos, L.C.; Smit, A.J.; Zeebregts, C.J.; Oudkerk, M.; Vliegenthart, R.; Lefrandt, J.D.; Mulder, D.J. Skin autofluorescence, a non-invasive marker for AGE accumulation, is associated with the degree of atherosclerosis. PLoS ONE 2013, 8, e83084. [Google Scholar] [CrossRef]
- Atzeni, I.M.; van de Zande, S.C.; Westra, J.; Zwerver, J.; Smit, A.J.; Mulder, D.J. The AGE Reader: A non-invasive method to assess long-term tissue damage. Methods 2021, 203, 533–541. [Google Scholar] [CrossRef]
- Gugliucci, A.; Menini, T. Circulating advanced glycation peptides in streptozotocin-induced diabetic rats: Evidence for preferential modification of IgG light chains. Life Sci. 1998, 62, 2141–2150. [Google Scholar] [CrossRef]
- Miyata, T.; Wada, Y.; Cai, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K.; Kurokawa, K.; van Ypersele de Strihou, C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int. 1997, 51, 1170–1181. [Google Scholar] [CrossRef]
- Wagner, Z.; Wittmann, I.; Mazak, I.; Schinzel, R.; Heidland, A.; Kientsch-Engel, R.; Nagy, J. N(epsilon)-(carboxymethyl)lysine levels in patients with type 2 diabetes: Role of renal function. Am. J. Kidney Dis. 2001, 38, 785–791. [Google Scholar] [CrossRef]
- Makita, Z.; Bucala, R.; Rayfield, E.J.; Friedman, E.A.; Kaufman, A.M.; Korbet, S.M.; Barth, R.H.; Winston, J.A.; Fuh, H.; Manogue, K.R.; et al. Reactive glycosylation endproducts in diabetic uraemia and treatment of renal failure. Lancet 1994, 343, 1519–1522. [Google Scholar] [CrossRef]
- Thomas, M.C.; Tsalamandris, C.; MacIsaac, R.; Medley, T.; Kingwell, B.; Cooper, M.E.; Jerums, G. Low-molecular-weight AGEs are associated with GFR and anemia in patients with type 2 diabetes. Kidney Int. 2004, 66, 1167–1172. [Google Scholar] [CrossRef][Green Version]
- O’Brien, J.; Morrissey, P.A. Nutritional and toxicological aspects of the Maillard browning reaction in foods. Crit. Rev. Food Sci. Nutr. 1989, 28, 211–248. [Google Scholar] [CrossRef]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Dhar, I.; Caspar-Bell, G. Role of Advanced Glycation End Products and Its Receptors in the Pathogenesis of Cigarette Smoke-Induced Cardiovascular Disease. Int. J. Angiol. 2015, 24, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.-I.; Furuno, S.; Shirai, H.; Kawakami, M.; Muramatsu, M.; Kobayashi, Y.; Yamagishi, S.-i. Assessment of the Concentrations of Various Advanced Glycation End-Products in Beverages and Foods That Are Commonly Consumed in Japan. PLoS ONE 2015, 10, e0118652. [Google Scholar] [CrossRef] [PubMed]
- Hull, G.L.J.; Woodside, J.V.; Ames, J.M.; Cuskelly, G.J. Nε-(carboxymethyl)lysine content of foods commonly consumed in a Western style diet. Food Chem. 2012, 131, 170–174. [Google Scholar] [CrossRef]
- Scheijen, J.L.J.M.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef]
- He, C.; Sabol, J.; Mitsuhashi, T.; Vlassara, H. Dietary glycotoxins: Inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 1999, 48, 1308–1315. [Google Scholar] [CrossRef]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef]
- Delgado-Andrade, C.; Tessier, F.J.; Niquet-Leridon, C.; Seiquer, I.; Pilar Navarro, M. Study of the urinary and faecal excretion of Nepsilon-carboxymethyllysine in young human volunteers. Amino Acids 2012, 43, 595–602. [Google Scholar] [CrossRef]
- Sebekova, K.; Saavedra, G.; Zumpe, C.; Somoza, V.; Klenovicsova, K.; Birlouez-Aragon, I. Plasma concentration and urinary excretion of N epsilon-(carboxymethyl)lysine in breast milk- and formula-fed infants. Ann. N. Y. Acad. Sci. 2008, 1126, 177–180. [Google Scholar] [CrossRef]
- Faist, V.; Erbersdobler, H.F. Metabolic transit and in vivo effects of melanoidins and precursor compounds deriving from the Maillard reaction. Ann. Nutr. Metab. 2000, 45, 1–12. [Google Scholar] [CrossRef]
- Forster, A.; Kuhne, Y.; Henle, T. Studies on absorption and elimination of dietary maillard reaction products. Ann. N. Y. Acad. Sci. 2005, 1043, 474–481. [Google Scholar] [CrossRef]
- Hultsch, C.; Hellwig, M.; Pawelke, B.; Bergmann, R.; Rode, K.; Pietzsch, J.; Krause, R.; Henle, T. Biodistribution and catabolism of 18F-labeled N-ε-fructoselysine as a model of Amadori products. Nucl. Med. Biol. 2006, 33, 865–873. [Google Scholar] [CrossRef]
- Schwenger, V.; Morath, C.; Schönfelder, K.; Klein, W.; Weigel, K.; Deppisch, R.; Henle, T.; Ritz, E.; Zeier, M. An oral load of the early glycation compound lactuloselysine fails to accumulate in the serum of uraemic patients. Nephrol. Dial. Transpl. 2006, 21, 383–388. [Google Scholar] [CrossRef]
- Liardon, R.; De Weck-Gaudard, D.; Philippossian, G.; Finot, P.A. Identification of N.epsilon.-carboxymethyllysine: A new Maillard reaction product in rat urine. J. Agric. Food Chem. 1987, 35, 427–431. [Google Scholar] [CrossRef]
- Miyata, T.; Ueda, Y.; Horie, K.; Nangaku, M.; Tanaka, S.; de Strihou, C.V.Y.; Kurokawa, K. Renal catabolism of advanced glycation end products: The fate of pentosidine. Kidney Int. 1998, 53, 416–422. [Google Scholar] [CrossRef]
- Bergmann, R.; Helling, R.; Heichert, C.; Scheunemann, M.; Mading, P.; Wittrisch, H.; Johannsen, B.; Henle, T. Radio fluorination and positron emission tomography (PET) as a new approach to study the in vivo distribution and elimination of the advanced glycation endproducts N epsilon-carboxymethyllysine (CML) and N epsilon-carboxyethyllysine (CEL). Nahrung 2001, 45, 182–188. [Google Scholar] [CrossRef]
- Somoza, V.; Wenzel, E.; Weiß, C.; Clawin-Rädecker, I.; Grübel, N.; Erbersdobler, H.F. Dose-dependent utilisation of casein-linked lysinoalanine, N (epsilon)-fructoselysine and N (epsilon)-carboxymethyllysine in rats. Mol. Nutr. Food Res. 2006, 50, 833–841. [Google Scholar] [CrossRef]
- Roncero-Ramos, I.; Delgado-Andrade, C.; Tessier, F.J.; Niquet-Leridon, C.; Strauch, C.; Monnier, V.M.; Navarro, M.P. Metabolic transit of N(epsilon)-carboxymethyl-lysine after consumption of AGEs from bread crust. Food Funct. 2013, 4, 1032–1039. [Google Scholar] [CrossRef]
- Alamir, I.; Niquet-Leridon, C.; Jacolot, P.; Rodriguez, C.; Orosco, M.; Anton, P.M.; Tessier, F.J. Digestibility of extruded proteins and metabolic transit of N epsilon -carboxymethyllysine in rats. Amino Acids 2013, 44, 1441–1449. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Wang, Y.; Hu, S.; Liu, N. Biodistribution and elimination study of fluorine-18 labeled Nepsilon-carboxymethyl-lysine following intragastric and intravenous administration. PLoS ONE 2013, 8, e57897. [Google Scholar] [CrossRef]
- Tessier, F.J.; Niquet-Leridon, C.; Jacolot, P.; Jouquand, C.; Genin, M.; Schmidt, A.M.; Grossin, N.; Boulanger, E. Quantitative assessment of organ distribution of dietary protein-bound (13) C-labeled N(varepsilon) -carboxymethyllysine after a chronic oral exposure in mice. Mol. Nutr. Food Res. 2016, 60, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, A.; Ogura, A.; Tahara, T.; Nozaki, S.; Urano, S.; Hara, M.; Kojima, S.; Kurbangalieva, A.; Onoe, H.; Watanabe, Y.; et al. In vivo imaging of advanced glycation end products (AGEs) of albumin: First observations of significantly reduced clearance and liver deposition properties in mice. Org. Biomol. Chem. 2016, 14, 5755–5760. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes/Metab. Res. Rev. 2001, 17, 436–443. [Google Scholar] [CrossRef]
- Smedsrød, B.; Melkko, J.; Araki, N.; Sano, H.; Horiuchi, S. Advanced glycation end products are eliminated by scavenger-receptor-mediated endocytosis in hepatic sinusoidal Kupffer and endothelial cells. Biochem. J. 1997, 322 Pt 2, 567–573. [Google Scholar] [CrossRef]
- Nagai, R.; Mera, K.; Nakajou, K.; Fujiwara, Y.; Iwao, Y.; Imai, H.; Murata, T.; Otagiri, M. The ligand activity of AGE-proteins to scavenger receptors is dependent on their rate of modification by AGEs. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 1192–1198. [Google Scholar] [CrossRef]
- Gugliucci, A.; Bendayan, M. Renal fate of circulating advanced glycated end products (AGE): Evidence for reabsorption and catabolism of AGE-peptides by renal proximal tubular cells. Diabetologia 1996, 39, 149–160. [Google Scholar] [CrossRef]
- Miyata, T.; Ueda, Y.; Shinzato, T.; Iida, Y.; Tanaka, S.; Kurokawa, K.; van Ypersele de Strihou, C.; Maeda, K. Accumulation of albumin-linked and free-form pentosidine in the circulation of uremic patients with end-stage renal failure: Renal implications in the pathophysiology of pentosidine. J. Am. Soc. Nephrol. 1996, 7, 1198–1206. [Google Scholar] [CrossRef]
- Makita, Z.; Radoff, S.; Rayfield, E.J.; Yang, Z.; Skolnik, E.; Delaney, V.; Friedman, E.A.; Cerami, A.; Vlassara, H. Advanced glycosylation end products in patients with diabetic nephropathy. N. Engl. J. Med. 1991, 325, 836–842. [Google Scholar] [CrossRef]
- Perkins, R.K.; Miranda, E.R.; Karstoft, K.; Beisswenger, P.J.; Solomon, T.P.J.; Haus, J.M. Experimental Hyperglycemia Alters Circulating Concentrations and Renal Clearance of Oxidative and Advanced Glycation End Products in Healthy Obese Humans. Nutrients 2019, 11, 532. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E.; Oldfield, M.D.; Thomas, M.C. Role of Advanced Glycation End Products in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 3), S254–S258. [Google Scholar] [CrossRef]
- Harcourt, B.E.; Sourris, K.C.; Coughlan, M.T.; Walker, K.Z.; Dougherty, S.L.; Andrikopoulos, S.; Morley, A.L.; Thallas-Bonke, V.; Chand, V.; Penfold, S.A.; et al. Targeted reduction of advanced glycation improves renal function in obesity. Kidney Int. 2011, 80, 190–198. [Google Scholar] [CrossRef]
- Tan, A.L.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in diabetic nephropathy. Semin. Nephrol. 2007, 27, 130–143. [Google Scholar] [CrossRef]
- Zheng, F.; He, C.; Cai, W.; Hattori, M.; Steffes, M.; Vlassara, H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes/Metab. Res. Rev. 2002, 18, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.M.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic Toxicity of Advanced Glycation End Products in CKD. J. Am. Soc. Nephrol. 2015, 27, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.; Kozielski, R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H.; et al. Effect of Dietary Advanced Glycation End Products on Mouse Liver. PLoS ONE 2012, 7, e35143. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Steffes, M.; Donnelly, T.; Liu, C.; Fuh, H.; Basgen, J.; Bucala, R.; Vlassara, H. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc. Natl. Acad. Sci. USA 1996, 93, 3902–3907. [Google Scholar] [CrossRef]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Bucciarelli, L.G.; Wendt, T.; Rong, L.; Lalla, E.; Hofmann, M.A.; Goova, M.T.; Taguchi, A.; Yan, S.F.; Yan, S.D.; Stern, D.M.; et al. RAGE is a multiligand receptor of the immunoglobulin superfamily: Implications for homeostasis and chronic disease. Cell. Mol. Life Sci. 2002, 59, 1117–1128. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Vianna, M.; Gerlach, M.; Brett, J.; Ryan, J.; Kao, J.; Esposito, C.; Hegarty, H.; Hurley, W.; Clauss, M.; et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 1992, 267, 14987–14997. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- Park, H.; Adsit, F.G.; Boyington, J.C. The 1.5 A crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J. Biol. Chem. 2010, 285, 40762–40770. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Sousa, M.M.; Yan, S.D.; Stern, D.; Saraiva, M.J. Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 2012, 274, 72–82. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar]
- Soulis, T.; Thallas, V.; Youssef, S.; Gilbert, R.E.; McWilliam, B.G.; Murray-McIntosh, R.P.; Cooper, M.E. Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 1997, 40, 619–628. [Google Scholar] [CrossRef]
- Ritthaler, U.; Deng, Y.; Zhang, Y.; Greten, J.; Abel, M.; Sido, B.; Allenberg, J.; Otto, G.; Roth, H.; Bierhaus, A.; et al. Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am. J. Pathol. 1995, 146, 688–694. [Google Scholar]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’agati, V.D. Expression of Advanced Glycation End Products and Their Cellular Receptor RAGE in Diabetic Nephropathy and Nondiabetic Renal Disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S.; et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 2003, 162, 1123–1137. [Google Scholar] [CrossRef]
- Sugaya, K.; Fukagawa, T.; Matsumoto, K.; Mita, K.; Takahashi, E.; Ando, A.; Inoko, H.; Ikemura, T. Three genes in the human MHC class III region near the junction with the class II: Gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics 1994, 23, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Kalea, A.Z.; Reiniger, N.; Yang, H.; Arriero, M.; Schmidt, A.M.; Hudson, B.I. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009, 23, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.; Nawroth, P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.; Dhawan, V. Receptor for advanced glycation end products (RAGE) in vascular and inflammatory diseases. Int. J. Cardiol. 2013, 168, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Du Yan, S.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2000, 1498, 99–111. [Google Scholar] [CrossRef]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Sturgis, L.; Haidacher, J.; Zhang, X.-N.; Sherwood, S.J.; Bjercke, R.J.; Juhasz, O.; Crow, M.T.; Tilton, R.G.; Denner, L. Requirement for p38 and p44/p42 Mitogen-Activated Protein Kinases in RAGE-Mediated Nuclear Factor-κB Transcriptional Activation and Cytokine Secretion. Diabetes 2001, 50, 1495–1504. [Google Scholar] [CrossRef]
- Grimm, S.; Ott, C.; Horlacher, M.; Weber, D.; Hohn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef]
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for Advanced Glycation End Products (RAGE)-mediated Neurite Outgrowth and Activation of NF-κB Require the Cytoplasmic Domain of the Receptor but Different Downstream Signaling Pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef]
- Sorci, G.; Riuzzi, F.; Arcuri, C.; Giambanco, I.; Donato, R. Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol. Cell. Biol. 2004, 24, 4880–4894. [Google Scholar] [CrossRef]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef]
- Yang, C.W.; Vlassara, H.; Peten, E.P.; He, C.J.; Striker, G.E.; Striker, L.J. Advanced glycation end products up-regulate gene expression found in diabetic glomerular disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9436–9440. [Google Scholar] [CrossRef]
- Zeng, S.; Dun, H.; Ippagunta, N.; Rosario, R.; Zhang, Q.Y.; Lefkowitch, J.; Yan, S.F.; Schmidt, A.M.; Emond, J.C. Receptor for advanced glycation end product (RAGE)-dependent modulation of early growth response-1 in hepatic ischemia/reperfusion injury. J. Hepatol. 2009, 50, 929–936. [Google Scholar] [CrossRef]
- Xie, J.; Mendez, J.D.; Mendez-Valenzuela, V.; Aguilar-Hernandez, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Zheng, F.; Striker, G.E.; Vlassara, H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am. J. Pathol. 2008, 173, 327–336. [Google Scholar] [CrossRef]
- Buchs, A.E.; Kornberg, A.; Zahavi, M.; Aharoni, D.; Zarfati, C.; Rapoport, M.J. Increased expression of tissue factor and receptor for advanced glycation end products in peripheral blood mononuclear cells of patients with type 2 diabetes mellitus with vascular complications. Exp. Diabesity Res. 2004, 5, 163–169. [Google Scholar] [CrossRef]
- Koyama, H.; Shoji, T.; Yokoyama, H.; Motoyama, K.; Mori, K.; Fukumoto, S.; Emoto, M.; Shoji, T.; Tamei, H.; Matsuki, H.; et al. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; van Ree, R.M.; Oterdoom, L.H.; de Vries, A.P.J.; van Son, W.J.; de Jong, P.E.; Navis, G.J.; Zuurman, M.W.; Bierhaus, A.; Gans, R.O.B.; et al. Low Levels of sRAGE Are Associated With Increased Risk for Mortality in Renal Transplant Recipients. Transplantation 2007, 84, 659–663. [Google Scholar] [CrossRef]
- Selvin, E.; Halushka, M.; Rawlings, A.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Astor, B.C. sRAGE and risk of diabetes, cardiovascular disease and death. Diabetes 2013, 62, 2116–2121. [Google Scholar] [CrossRef] [PubMed]
- Loomis, S.J.; Chen, Y.; Sacks, D.B.; Christenson, E.S.; Christenson, R.H.; Rebholz, C.M.; Selvin, E. Cross-sectional Analysis of AGE-CML, sRAGE, and esRAGE with Diabetes and Cardiometabolic Risk Factors in a Community-Based Cohort. Clin. Chem. 2017, 63, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Sironi, A.M.; Lazzerini, G.; Del Turco, S.; Buzzigoli, E.; Casolaro, A.; Natali, A.; Ferrannini, E.; Gastaldelli, A. Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J. Clin. Endocrinol. Metab. 2006, 91, 4628–4634. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Leonardis, D.; Mallamaci, F.; Cutrupi, S.; Pizzini, P.; Gaetano, L.; Tripepi, R.; Tripepi, G.; De Caterina, R.; Zoccali, C. Circulating soluble receptor of advanced glycation end product inversely correlates with atherosclerosis in patients with chronic kidney disease. Kidney Int. 2010, 77, 225–231. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laudenslager, M.; Lazo, M.; Wang, D.; Selvin, E.; Chen, P.-H.; Pankow, J.S.; Clark, J.M. Association between the soluble receptor for advanced glycation end products (sRAGE) and NAFLD in participants in the Atherosclerosis Risk in Communities Study. Dig. Liver Dis. 2021, 53, 873–878. [Google Scholar] [CrossRef]
- Geroldi, D.; Falcone, C.; Minoretti, P.; Emanuele, E.; Arra, M.; D’Angelo, A. High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J. Am. Geriatr. Soc. 2006, 54, 1149–1150. [Google Scholar] [CrossRef]
- Scavello, F.; Tedesco, C.C.; Castiglione, S.; Maciag, A.; Sangalli, E.; Veglia, F.; Spinetti, G.; Puca, A.A.; Raucci, A. Modulation of soluble receptor for advanced glycation end products isoforms and advanced glycation end products in long-living individuals. Biomark Med. 2021, 15, 785–796. [Google Scholar] [CrossRef]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef]
- Bierhaus, A.; Haslbeck, K.M.; Humpert, P.M.; Liliensiek, B.; Dehmer, T.; Morcos, M.; Sayed, A.A.; Andrassy, M.; Schiekofer, S.; Schneider, J.G.; et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J. Clin. Investig. 2004, 114, 1741–1751. [Google Scholar] [CrossRef]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J., Jr.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef]
- Kalea, A.Z.; Schmidt, A.M.; Hudson, B.I. RAGE: A novel biological and genetic marker for vascular disease. Clin. Sci. 2009, 116, 621–637. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Li, Y.M.; Mitsuhashi, T.; Wojciechowicz, D.; Shimizu, N.; Li, J.; Stitt, A.; He, C.; Banerjee, D.; Vlassara, H. Molecular identity and cellular distribution of advanced glycation endproduct receptors: Relationship of p60 to OST-48 and p90 to 80K-H membrane proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 11047–11052. [Google Scholar] [CrossRef]
- Yang, Z.; Makita, Z.; Horii, Y.; Brunelle, S.; Cerami, A.; Sehajpal, P.; Suthanthiran, M.; Vlassara, H. Two novel rat liver membrane proteins that bind advanced glycosylation endproducts: Relationship to macrophage receptor for glucose-modified proteins. J. Exp. Med. 1991, 174, 515–524. [Google Scholar] [CrossRef]
- Kelleher, D.J.; Kreibich, G.; Gilmore, R. Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kd protein. Cell 1992, 69, 55–65. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Blal, H.A.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: Role of the antiinflammatory AGE receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483–4491. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Wallenstein, S.; Striker, G.E.; Vlassara, H. Reduced Oxidant Stress and Extended Lifespan in Mice Exposed to a Low Glycotoxin Diet: Association with Increased AGER1 Expression. Am. J. Pathol. 2007, 170, 1893–1902. [Google Scholar] [CrossRef]
- Lopez-Moreno, J.; Quintana-Navarro, G.M.; Delgado-Lista, J.; Garcia-Rios, A.; Alcala-Diaz, J.F.; Gomez-Delgado, F.; Camargo, A.; Perez-Martinez, P.; Tinahones, F.J.; Striker, G.E.; et al. Mediterranean Diet Supplemented with Coenzyme Q10 Modulates the Postprandial Metabolism of Advanced Glycation End Products in Elderly Men and Women. J. Gerontol. Ser. A 2018, 73, 340–346. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Cardelo, M.P.; de la Cruz, S.; Alcala-Diaz, J.F.; Roncero-Ramos, I.; Guler, I.; Vals-Delgado, C.; López-Moreno, A.; Luque, R.M.; Delgado-Lista, J.; et al. Reduction in Circulating Advanced Glycation End Products by Mediterranean Diet Is Associated with Increased Likelihood of Type 2 Diabetes Remission in Patients with Coronary Heart Disease: From the Cordioprev Study. Mol. Nutr. Food Res. 2021, 65, 1901290. [Google Scholar] [CrossRef]
- Lopez-Moreno, J.; Quintana-Navarro, G.M.; Camargo, A.; Jimenez-Lucena, R.; Delgado-Lista, J.; Marin, C.; Tinahones, F.J.; Striker, G.E.; Roche, H.M.; Perez-Martinez, P.; et al. Dietary fat quantity and quality modifies advanced glycation end products metabolism in patients with metabolic syndrome. Mol. Nutr. Food Res. 2017, 61, 1601029. [Google Scholar] [CrossRef]
- Zhuang, A.; Yap, F.Y.T.; McCarthy, D.; Leung, C.; Sourris, K.C.; Penfold, S.A.; Thallas-Bonke, V.; Coughlan, M.T.; Schulz, B.L.; Forbes, J.M. Globally elevating the AGE clearance receptor, OST48, does not protect against the development of diabetic kidney disease, despite improving insulin secretion. Sci. Rep. 2019, 9, 13664. [Google Scholar] [CrossRef]
- Zhuang, A.; Yap, F.Y.T.; Borg, D.J.; McCarthy, D.; Fotheringham, A.; Leung, S.; Penfold, S.A.; Sourris, K.C.; Coughlan, M.T.; Schulz, B.L.; et al. The AGE receptor, OST48 drives podocyte foot process effacement and basement membrane expansion (alters structural composition). Endocrinol. Diabetes Metab. 2021, 4, e00278. [Google Scholar] [CrossRef]
- Bai, P.; Phua, K.; Hardt, T.; Cernadas, M.; Brodsky, B. Glycation alters collagen fibril organization. Connect. Tissue Res. 1992, 28, 1–12. [Google Scholar] [CrossRef]
- Hadley, J.C.; Meek, K.M.; Malik, N.S. Glycation changes the charge distribution of type I collagen fibrils. Glycoconj. J. 1998, 15, 835–840. [Google Scholar] [CrossRef]
- Barlovic, D.P.; Soro-Paavonen, A.; Jandeleit-Dahm, K.A. RAGE biology, atherosclerosis and diabetes. Clin. Sci. 2011, 121, 43–55. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Luevano-Contreras, C.; Chapman-Novakofski, K. Dietary advanced glycation end products and aging. Nutrients 2010, 2, 1247–1265. [Google Scholar] [CrossRef] [PubMed]
- Bengmark, S. Impact of nutrition on ageing and disease. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating Glycotoxins and Dietary Advanced Glycation Endproducts: Two Links to Inflammatory Response, Oxidative Stress, and Aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Cai, W.; Crandall, J.; Goldberg, T.; Oberstein, R.; Dardaine, V.; Peppa, M.; Rayfield, E.J. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 15596–15601. [Google Scholar] [CrossRef] [PubMed]
- Linkens, A.M.A.; Houben, A.J.; Niessen, P.M.; Wijckmans, N.; de Goei, E.; Van den Eynde, M.D.; Scheijen, J.; Waarenburg, M.; Mari, A.; Berendschot, T.T.; et al. A 4-week high-AGE diet does not impair glucose metabolism and vascular function in obese individuals. JCI Insight 2022, 7, e156950. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cowan, S.P.; Andrikopoulos, S.; Morley, A.L.; Ward, L.C.; Walker, K.Z.; Cooper, M.E.; Coughlan, M.T. Glucose homeostasis can be differentially modulated by varying individual components of a western diet. J. Nutr. Biochem. 2013, 24, 1251–1257. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, C.; Zhang, X.H.; Zheng, F.; Cai, W.; Vlassara, H.; Ma, Z.A. Advanced Glycation End Products Inhibit Glucose-Stimulated Insulin Secretion through Nitric Oxide-Dependent Inhibition of Cytochrome c Oxidase and Adenosine Triphosphate Synthesis. Endocrinology 2009, 150, 2569–2576. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Yap, F.Y.; Tong, D.C.; Andrikopoulos, S.; Gasser, A.; Thallas-Bonke, V.; Webster, D.E.; Miyazaki, J.; Kay, T.W.; Slattery, R.M.; et al. Advanced glycation end products are direct modulators of beta-cell function. Diabetes 2011, 60, 2523–2532. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Forbes, J.M. Temporal increases in urinary carboxymethyllysine correlate with albuminuria development in diabetes. Am. J. Nephrol. 2011, 34, 9–17. [Google Scholar] [CrossRef]
- Sebekova, K.; Hofmann, T.; Boor, P.; SebekovaJr, K.; Ulicna, O.; Erbersdobler, H.F.; Baynes, J.W.; Thorpe, S.R.; Heidland, A.; Somoza, V. Renal effects of oral maillard reaction product load in the form of bread crusts in healthy and subtotally nephrectomized rats. Ann. N. Y. Acad. Sci. 2005, 1043, 482–491. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; Pasquale, C.D.; Thallas-Bonke, V.; Nguyen, T.-V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef]
- Peppa, M.; He, C.; Hattori, M.; McEvoy, R.; Zheng, F.; Vlassara, H. Fetal or neonatal low-glycotoxin environment prevents autoimmune diabetes in NOD mice. Diabetes 2003, 52, 1441–1448. [Google Scholar] [CrossRef]
- Hofmann, S.M.; Dong, H.J.; Li, Z.; Cai, W.; Altomonte, J.; Thung, S.N.; Zeng, F.; Fisher, E.A.; Vlassara, H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes 2002, 51, 2082–2089. [Google Scholar] [CrossRef]
- Tan, A.L.; Sourris, K.C.; Harcourt, B.E.; Thallas-Bonke, V.; Penfold, S.; Andrikopoulos, S.; Thomas, M.C.; O’Brien, R.C.; Bierhaus, A.; Cooper, M.E.; et al. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010, 298, F763–F770. [Google Scholar] [CrossRef]
- Peppa, M.; Brem, H.; Ehrlich, P.; Zhang, J.G.; Cai, W.; Li, Z.; Croitoru, A.; Thung, S.; Vlassara, H. Adverse effects of dietary glycotoxins on wound healing in genetically diabetic mice. Diabetes 2003, 52, 2805–2813. [Google Scholar] [CrossRef]
- Uribarri, J.; Peppa, M.; Cai, W.; Goldberg, T.; Lu, M.; Baliga, S.; Vassalotti, J.A.; Vlassara, H. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am. J. Kidney Dis. 2003, 42, 532–538. [Google Scholar] [CrossRef]
- Chao, P.C.; Huang, C.N.; Hsu, C.C.; Yin, M.C.; Guo, Y.R. Association of dietary AGEs with circulating AGEs, glycated LDL, IL-1alpha and MCP-1 levels in type 2 diabetic patients. Eur. J. Nutr. 2010, 49, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Lotan, R.; Ganmore, I.; Livny, A.; Itzhaki, N.; Waserman, M.; Shelly, S.; Zacharia, M.; Moshier, E.; Uribarri, J.; Beisswenger, P.; et al. Effect of Advanced Glycation End Products on Cognition in Older Adults with Type 2 Diabetes: Results from a Pilot Clinical Trial. J. Alzheimer’s Dis. 2021, 82, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Birlouez-Aragon, I.; Saavedra, G.; Tessier, F.J.; Galinier, A.; Ait-Ameur, L.; Lacoste, F.; Niamba, C.N.; Alt, N.; Somoza, V.; Lecerf, J.M. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am. J. Clin. Nutr. 2010, 91, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef]
- Hagiwara, S.; Gohda, T.; Tanimoto, M.; Ito, T.; Murakoshi, M.; Ohara, I.; Yamazaki, T.; Matsumoto, M.; Horikoshi, S.; Funabiki, K. Effects of pyridoxamine (K-163) on glucose intolerance and obesity in high-fat diet C57BL/6J mice. Metabolism 2009, 58, 934–945. [Google Scholar] [CrossRef]
- Sandu, O.; Song, K.; Cai, W.; Zheng, F.; Uribarri, J.; Vlassara, H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes 2005, 54, 2314–2319. [Google Scholar] [CrossRef]
- Mark, A.B.; Poulsen, M.W.; Andersen, S.; Andersen, J.M.; Bak, M.J.; Ritz, C.; Holst, J.J.; Nielsen, J.; de Courten, B.; Dragsted, L.O.; et al. Consumption of a Diet Low in Advanced Glycation End Products for 4 Weeks Improves Insulin Sensitivity in Overweight Women. Diabetes Care 2014, 37, 88–95. [Google Scholar] [CrossRef]
- Vaaler, S.; Hanssen, K.F.; Aagenaes, O. The effect of cooking upon the blood glucose response to ingested carrots and potatoes. Diabetes Care 1984, 7, 221–223. [Google Scholar] [CrossRef]
- Rizkalla, S.W.; Laromiguiere, M.; Champ, M.; Bruzzo, F.; Boillot, J.; Slama, G. Effect of baking process on postprandial metabolic consequences: Randomized trials in normal and type 2 diabetic subjects. Eur. J. Clin. Nutr. 2007, 61, 175–183. [Google Scholar] [CrossRef]
- de Courten, B.; de Courten, M.P.; Soldatos, G.; Dougherty, S.L.; Straznicky, N.; Schlaich, M.; Sourris, K.C.; Chand, V.; Scheijen, J.L.; Kingwell, B.A.; et al. Diet low in advanced glycation end products increases insulin sensitivity in healthy overweight individuals: A double-blind, randomized, crossover trial. Am. J. Clin. Nutr. 2016, 103, 1426–1433. [Google Scholar] [CrossRef]
- Seiquer, I.; Rubio, L.A.; Peinado, M.J.; Delgado-Andrade, C.; Navarro, M.P. Maillard reaction products modulate gut microbiota composition in adolescents. Mol. Nutr. Food Res. 2014, 58, 1552–1560. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.S.; Ferreira Alves, G.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F.; et al. Effects of Exogenous Dietary Advanced Glycation End Products on the Cross-Talk Mechanisms Linking Microbiota to Metabolic Inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef]
- Bock, P.M.; Telo, G.H.; Ramalho, R.; Sbaraini, M.; Leivas, G.; Martins, A.F.; Schaan, B.D. The effect of probiotics, prebiotics or synbiotics on metabolic outcomes in individuals with diabetes: A systematic review and meta-analysis. Diabetologia 2021, 64, 26–41. [Google Scholar] [CrossRef]
- Farhangi, M.A.; Dehghan, P.; Namazi, N. Prebiotic supplementation modulates advanced glycation end-products (AGEs), soluble receptor for AGEs (sRAGE), and cardiometabolic risk factors through improving metabolic endotoxemia: A randomized-controlled clinical trial. Eur. J. Nutr. 2020, 59, 3009–3021. [Google Scholar] [CrossRef]
- Kellow, N.J.; Coughlan, M.T.; Savige, G.S.; Reid, C.M. Effect of dietary prebiotic supplementation on advanced glycation, insulin resistance and inflammatory biomarkers in adults with pre-diabetes: A study protocol for a double-blind placebo-controlled randomised crossover clinical trial. BMC Endocr. Disord. 2014, 14, 55. [Google Scholar] [CrossRef]
- Semba, R.D.; Fink, J.C.; Sun, K.; Bandinelli, S.; Guralnik, J.M.; Ferrucci, L. Carboxymethyl-lysine, an advanced glycation end product, and decline of renal function in older community-dwelling adults. Eur. J. Nutr. 2009, 48, 38–44. [Google Scholar] [CrossRef][Green Version]
- Hou, F.F.; Ren, H.; OwenJr, W.F.; Guo, Z.J.; Chen, P.Y.; Schmidt, A.M.; Miyata, T.; Zhang, X. Enhanced expression of receptor for advanced glycation end products in chronic kidney disease. J. Am. Soc. Nephrol. 2004, 15, 1889–1896. [Google Scholar] [CrossRef]
- Semba, R.D.; Fink, J.C.; Sun, K.; Windham, B.G.; Ferrucci, L. Serum Carboxymethyl-Lysine, a Dominant Advanced Glycation End Product, Is Associated with Chronic Kidney Disease: The Baltimore Longitudinal Study of Aging. J. Ren. Nutr. 2010, 20, 74–81. [Google Scholar] [CrossRef]
- Gerdemann, A.; Lemke, H.D.; Nothdurft, A.; Heidland, A.; Münch, G.; Bahner, U.; Schinzel, R. Low-molecular but not high-molecular advanced glycation end products (AGEs) are removed by high-flux dialysis. Clin. Nephrol. 2000, 54, 276–283. [Google Scholar]
- Peppa, M.; Uribarri, J.; Cai, W.; Lu, M.; Vlassara, H. Glycoxidation and inflammation in renal failure patients. Am. J. Kidney Dis. 2004, 43, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Agalou, S.; Ahmed, N.; Dawnay, A.; Thornalley, P.J. Removal of advanced glycation end products in clinical renal failure by peritoneal dialysis and haemodialysis. Biochem. Soc. Trans. 2003, 31, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Friedlander, M.; Capeillere-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor, C.G.; Gomes-Neto, A.W.; van Londen, M.; Gans, R.O.B.; Nolte, I.M.; Berger, S.P.; Navis, G.J.; Rodrigo, R.; Leuvenink, H.G.D.; Schalkwijk, C.G.; et al. Circulating Advanced Glycation Endproducts and Long-Term Risk of Cardiovascular Mortality in Kidney Transplant Recipients. Clin. J. Am. Soc. Nephrol. 2019, 14, 1512–1520. [Google Scholar] [CrossRef]
- Klein, R.; Horak, K.; Lee, K.E.; Danforth, L.; Cruickshanks, K.J.; Tsai, M.Y.; Gangnon, R.E.; Klein, B.E.K. The Relationship of Serum Soluble Receptor for Advanced Glycation End Products (sRAGE) and Carboxymethyl Lysine (CML) to the Incidence of Diabetic Nephropathy in Persons with Type 1 Diabetes. Diabetes Care 2017, 40, e117–e119. [Google Scholar] [CrossRef]
- Dalal, M.; Semba, R.D.; Sun, K.; Crasto, C.; Varadhan, R.; Bandinelli, S.; Fink, J.C.; Guralnik, J.M.; Ferrucci, L. Endogenous secretory receptor for advanced glycation end products and chronic kidney disease in the elderly population. Am. J. Nephrol. 2011, 33, 313–318. [Google Scholar] [CrossRef]
- Rebholz, C.M.; Astor, B.C.; Grams, M.E.; Halushka, M.K.; Lazo, M.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Selvin, E. Association of plasma levels of soluble receptor for advanced glycation end products and risk of kidney disease: The Atherosclerosis Risk in Communities study. Nephrol. Dial. Transpl. 2015, 30, 77–83. [Google Scholar] [CrossRef]
- Thomas, M.C.; Woodward, M.; Neal, B.; Li, Q.; Pickering, R.; Marre, M.; Williams, B.; Perkovic, V.; Cooper, M.E.; Zoungas, S.; et al. The Relationship Between Levels of Advanced Glycation End-Products and Their Soluble Receptor and Adverse Outcomes in Adults with Type 2 Diabetes. Diabetes Care 2015, 38, 1891–1897. [Google Scholar] [CrossRef]
- McIntyre, N.J.; Fluck, R.J.; McIntyre, C.W.; Taal, M.W. Skin Autofluorescence and the Association with Renal and Cardiovascular Risk Factors in Chronic Kidney Disease Stage 3. Clin. J. Am. Soc. Nephrol. 2011, 6, 2356–2363. [Google Scholar] [CrossRef]
- Tanaka, K.; Tani, Y.; Asai, J.; Nemoto, F.; Kusano, Y.; Suzuki, H.; Hayashi, Y.; Asahi, K.; Katoh, T.; Miyata, T.; et al. Skin autofluorescence is associated with renal function and cardiovascular diseases in pre-dialysis chronic kidney disease patients. Nephrol. Dial. Transpl. 2011, 26, 214–220. [Google Scholar] [CrossRef]
- Smit, A.J.; Gerrits, E.G. Skin autofluorescence as a measure of advanced glycation endproduct deposition: A novel risk marker in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2010, 19, 527–533. [Google Scholar] [CrossRef]
- Tanaka, K.; Nakayama, M.; Kanno, M.; Kimura, H.; Watanabe, K.; Tani, Y.; Kusano, Y.; Suzuki, H.; Hayashi, Y.; Asahi, K.; et al. Skin autofluorescence is associated with the progression of chronic kidney disease: A prospective observational study. PLoS ONE 2013, 8, e83799. [Google Scholar] [CrossRef]
- Genevieve, M.; Vivot, A.; Gonzalez, C.; Raffaitin, C.; Barberger-Gateau, P.; Gin, H.; Rigalleau, V. Skin autofluorescence is associated with past glycaemic control and complications in type 1 diabetes mellitus. Diabetes Metab. 2013, 39, 349–354. [Google Scholar] [CrossRef]
- Sugisawa, E.; Miura, J.; Iwamoto, Y.; Uchigata, Y. Skin autofluorescence reflects integration of past long-term glycemic control in patients with type 1 diabetes. Diabetes Care 2013, 36, 2339–2345. [Google Scholar] [CrossRef][Green Version]
- Meerwaldt, R.; Hartog, J.W.L.; Graaff, R.; Huisman, R.J.; Links, T.P.; den Hollander, N.C.; Thorpe, S.R.; Baynes, J.W.; Navis, G.; Gans, R.O.B.; et al. Skin Autofluorescence, a Measure of Cumulative Metabolic Stress and Advanced Glycation End Products, Predicts Mortality in Hemodialysis Patients. J. Am. Soc. Nephrol. 2005, 16, 3687–3693. [Google Scholar] [CrossRef]
- Forbes, J.M.; Le Bagge, S.; Righi, S.; Fotheringham, A.K.; Gallo, L.A.; McCarthy, D.A.; Leung, S.; Baskerville, T.; Nisbett, J.; Morton, A.; et al. Advanced glycation end products as predictors of renal function in youth with type 1 diabetes. Sci. Rep. 2021, 11, 9422. [Google Scholar] [CrossRef]
- Yu, Y.; Thorpe, S.R.; Jenkins, A.J.; Shaw, J.N.; Sochaski, M.A.; McGee, D.; Aston, C.E.; Orchard, T.J.; Silvers, N.; Peng, Y.G.; et al. Advanced glycation end-products and methionine sulphoxide in skin collagen of patients with type 1 diabetes. Diabetologia 2006, 49, 2488–2498. [Google Scholar] [CrossRef]
- Orchard, T.J.; Lyons, T.J.; Cleary, P.A.; Braffett, B.H.; Maynard, J.; Cowie, C.; Gubitosi-Klug, R.A.; Way, J.; Anderson, K.; Barnie, A.; et al. The Association of Skin Intrinsic Fluorescence with Type 1 Diabetes Complications in the DCCT/EDIC Study. Diabetes Care 2013, 36, 3146–3153. [Google Scholar] [CrossRef]
- Weiss, M.F.; Erhard, P.; Kader-Attia, F.A.; Wu, Y.C.; Deoreo, P.B.; Araki, A.; Glomb, M.A.; Monnier, V.M. Mechanisms for the formation of glycoxidation products in end-stage renal disease. Kidney Int. 2000, 57, 2571–2585. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Patel, S.K.; Jerums, G.; Penfold, S.A.; Nguyen, T.-V.; Sourris, K.C.; Panagiotopoulos, S.; Srivastava, P.M.; Cooper, M.E.; Burrell, L.M.; et al. Advanced glycation urinary protein-bound biomarkers and severity of diabetic nephropathy in man. Am. J. Nephrol. 2011, 34, 347–355. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Howell, S.K.; Russell, G.B.; Miller, M.E.; Rich, S.S.; Mauer, M. Early Progression of Diabetic Nephropathy Correlates with Methylglyoxal-Derived Advanced Glycation End Products. Diabetes Care 2013, 36, 3234–3239. [Google Scholar] [CrossRef]
- Perkins, B.A. Abstract #0097 Advanced glycation end products and other markers of protein damage in early diabetic nephropathy in type 1 diabetes. In Proceedings of the International Diabetes Federation, World Diabetes Congress, Melbourne, Australia, 3 December 2013. [Google Scholar]
- Thomas, M.C.; Forbes, J.M.; MacIsaac, R.; Jerums, G.; Cooper, M.E. Low-Molecular Weight Advanced Glycation End Products: Markers of Tissue AGE Accumulation and More? Ann. N. Y. Acad. Sci. 2005, 1043, 644–654. [Google Scholar] [CrossRef]
- Kern, E.F.O.; Erhard, P.; Sun, W.; Genuth, S.; Weiss, M.F. Early Urinary Markers of Diabetic Kidney Disease: A Nested Case-Control Study From the Diabetes Control and Complications Trial (DCCT). Am. J. Kidney Dis. 2010, 55, 824–834. [Google Scholar] [CrossRef]
- Nishad, R.; Tahaseen, V.; Kavvuri, R.; Motrapu, M.; Singh, A.K.; Peddi, K.; Pasupulati, A.K. Advanced-Glycation End-Products Induce Podocyte Injury and Contribute to Proteinuria. Front. Med. 2021, 8, 685447. [Google Scholar] [CrossRef]
- Kumar, P.A.; Welsh, G.I.; Raghu, G.; Menon, R.K.; Saleem, M.A.; Reddy, G.B. Carboxymethyl lysine induces EMT in podocytes through transcription factor ZEB2: Implications for podocyte depletion and proteinuria in diabetes mellitus. Arch. Biochem. Biophys. 2016, 590, 10–19. [Google Scholar] [CrossRef][Green Version]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Li, J.H.; Wang, W.; Huang, X.R.; Oldfield, M.; Schmidt, A.M.; Cooper, M.E.; Lan, H.Y. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am. J. Pathol. 2004, 164, 1389–1397. [Google Scholar] [CrossRef]
- Burns, W.C.; Twigg, S.M.; Forbes, J.M.; Pete, J.; Tikellis, C.; Thallas-Bonke, V.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Connective Tissue Growth Factor Plays an Important Role in Advanced Glycation End Product–Induced Tubular Epithelial-to-Mesenchymal Transition: Implications for Diabetic Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 2484. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Haruki, H.; Kanda, T. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood–nerve barrier by stimulating the release of TGF-β and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia 2011, 54, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.E. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet 1998, 352, 213–219. [Google Scholar] [CrossRef]
- Sourris, K.C.; Harcourt, B.E.; Forbes, J.M. A new perspective on therapeutic inhibition of advanced glycation in diabetic microvascular complications: Common downstream endpoints achieved through disparate therapeutic approaches? Am. J. Nephrol. 2009, 30, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.M.; Chen, M.M.; Joly, A.H.; Chakrapani, S.D.; Tsubaki, J.; Kim, H.S.; Oh, Y.; Rosenfeld, R.G. Advanced glycosylation end products up-regulate connective tissue growth factor (insulin-like growth factor-binding protein-related protein 2) in human fibroblasts: A potential mechanism for expansion of extracellular matrix in diabetes mellitus. Endocrinology 2001, 142, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Isoda, K.; Folco, E.; Marwali, M.R.; Ohsuzu, F.; Libby, P. Glycated LDL increases monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated chemotaxis. Atherosclerosis 2008, 198, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hori, O.; Chen, J.X.; Li, J.F.; Crandall, J.; Zhang, J.; Cao, R.; Yan, S.D.; Brett, J.; Stern, D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Investig. 1995, 96, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.-M.; Yamamoto, Y.; Doi, T.; Kato, I.; Harashima, A.; Yonekura, H.; Watanabe, T.; Shinohara, H.; Takeuchi, M.; Tsuneyama, K. RAGE control of diabetic nephropathy in a mouse model: Effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 2006, 55, 2510–2522. [Google Scholar] [CrossRef]
- Sourris, K.C.; Morley, A.L.; Koitka, A.; Samuel, P.; Coughlan, M.T.; Penfold, S.A.; Thomas, M.C.; Bierhaus, A.; Nawroth, P.P.; Yamamoto, H.; et al. Receptor for AGEs (RAGE) blockade may exert its renoprotective effects in patients with diabetic nephropathy via induction of the angiotensin II type 2 (AT2) receptor. Diabetologia 2010, 53, 2442–2451. [Google Scholar] [CrossRef]
- Ozdemir, A.M.; Hopfer, U.; Rosca, M.V.; Fan, X.J.; Monnier, V.M.; Weiss, M.F. Effects of Advanced Glycation End Product Modification on Proximal Tubule Epithelial Cell Processing of Albumin. Am. J. Nephrol. 2008, 28, 14–24. [Google Scholar] [CrossRef]
- Gallicchio, M.A.; Bach, L.A. Uptake of advanced glycation end products by proximal tubule epithelial cells via macropinocytosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 2922–2932. [Google Scholar] [CrossRef][Green Version]
- Takahashi, A.; Takabatake, Y.; Kimura, T.; Maejima, I.; Namba, T.; Yamamoto, T.; Matsuda, J.; Minami, S.; Kaimori, J.-Y.; Matsui, I.; et al. Autophagy Inhibits the Accumulation of Advanced Glycation End Products by Promoting Lysosomal Biogenesis and Function in the Kidney Proximal Tubules. Diabetes 2017, 66, 1359–1372. [Google Scholar] [CrossRef]
- Liu, W.J.; Shen, T.T.; Chen, R.H.; Wu, H.-L.; Wang, Y.J.; Deng, J.K.; Chen, Q.H.; Pan, Q.; Huang Fu, C.-M.; Tao, J.-L.; et al. Autophagy-Lysosome Pathway in Renal Tubular Epithelial Cells Is Disrupted by Advanced Glycation End Products in Diabetic Nephropathy. J. Biol. Chem. 2015, 290, 20499–20510. [Google Scholar] [CrossRef]
- Ejtahed, H.-S.; Angoorani, P.; Asghari, G.; Mirmiran, P.; Azizi, F. Dietary Advanced Glycation End Products and Risk of Chronic Kidney Disease. J. Ren. Nutr. 2016, 26, 308–314. [Google Scholar] [CrossRef]
- Clarke, R.; Dordevic, A.; Tan, S.; Ryan, L.; Coughlan, M. Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials. Nutrients 2016, 8, 125. [Google Scholar] [CrossRef]
- Normand, G.; Lemoine, S.; Villien, M.; Le Bars, D.; Merida, I.; Irace, Z.; Troalen, T.; Costes, N.; Juillard, L. AGE Content of a Protein Load Is Responsible for Renal Performances: A Pilot Study. Diabetes Care 2018, 41, 1292–1294. [Google Scholar] [CrossRef]
- Uribarri, J.; Stirban, A.; Sander, D.; Cai, W.; Negrean, M.; Buenting, C.E.; Koschinsky, T.; Vlassara, H. Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and nondiabetic subjects. Diabetes Care 2007, 30, 2579–2582. [Google Scholar] [CrossRef]
- Semba, R.D.; Gebauer, S.K.; Baer, D.J.; Sun, K.; Turner, R.; Silber, H.A.; Talegawkar, S.; Ferrucci, L.; Novotny, J.A. Dietary intake of advanced glycation end products did not affect endothelial function and inflammation in healthy adults in a randomized controlled trial. J. Nutr. 2014, 144, 1037–1042. [Google Scholar] [CrossRef]
- Baye, E.; de Courten, M.P.; Walker, K.; Ranasinha, S.; Earnest, A.; Forbes, J.M.; de Courten, B. Effect of dietary advanced glycation end products on inflammation and cardiovascular risks in healthy overweight adults: A randomised crossover trial. Sci. Rep. 2017, 7, 4123. [Google Scholar] [CrossRef]
- Li, M.; Zeng, M.; He, Z.; Zheng, Z.; Qin, F.; Tao, G.; Zhang, S.; Chen, J. Increased Accumulation of Protein-bound N-(carboxymethyl)lysine in Tissues of Healthy Rats after Chronic Oral N-(carboxymethyl)lysine. J. Agric. Food. Chem. 2015, 63, 1658–1663. [Google Scholar] [CrossRef]
- Sebekova, K.; Faist, V.; Hofmann, T.; Schinzel, R.; Heidland, A. Effects of a diet rich in advanced glycation end products in the rat remnant kidney model. Am. J. Kidney Dis. 2003, 41, S48–S51. [Google Scholar] [CrossRef]
- Feng, J.X.; Hou, F.F.; Liang, M.; Wang, G.B.; Zhang, X.; Li, H.Y.; Xie, D.; Tian, J.W.; Liu, Z.Q. Restricted intake of dietary advanced glycation end products retards renal progression in the remnant kidney model. Kidney Int. 2007, 71, 901–911. [Google Scholar] [CrossRef]
- de Assis, A.M.; Rech, A.; Longoni, A.; da Silva Morrone, M.; de Bittencourt Pasquali, M.A.; Perry, M.L.; Souza, D.O.; Moreira, J.C. Dietary n-3 polyunsaturated fatty acids revert renal responses induced by a combination of 2 protocols that increase the amounts of advanced glycation end product in rats. Nutr. Res. 2015, 35, 512–522. [Google Scholar] [CrossRef]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.-M.; Cauffiez, C.; et al. Knockout of receptor for advanced glycation end-products attenuates age-related renal lesions. Aging Cell 2019, 18, e12850. [Google Scholar] [CrossRef]
- Borg, D.J.; Forbes, J.M. Targeting advanced glycation with pharmaceutical agents: Where are we now? Glycoconj. J. 2016, 33, 653–670. [Google Scholar] [CrossRef]
- Soulis-Liparota, T.; Cooper, M.; Papazoglou, D.; Clarke, B.; Jerums, G. Retardation by Aminoguanidine of Development of Albuminuria, Mesangial Expansion, and Tissue Fluorescence in Streptozocin-Induced Diabetic Rat. Diabetes 1991, 40, 1328–1334. [Google Scholar] [CrossRef]
- Thornalley, P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Alderson, N.L.; Chachich, M.E.; Youssef, N.N.; Beattie, R.J.; Nachtigal, M.; Thorpe, S.R.; Baynes, J.W. The AGE inhibitor pyridoxamine inhibits lipemia and development of renal and vascular disease in Zucker obese rats. Kidney Int. 2003, 63, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zeng, Y.j.; Plati, A.R.; Elliot, S.J.; Berho, M.; Potier, M.; Striker, L.J.; Striker, G.E. Combined AGE inhibition and ACEi decreases the progression of established diabetic nephropathy in B6 db/db mice. Kidney Int. 2006, 70, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Vasan, S.; Zhang, X.; Zhang, X.; Kapurniotu, A.; Bernhagen, J.; Teichberg, S.; Basgen, J.; Wagle, D.; Shih, D.; Terlecky, I.; et al. An agent cleaving glucose-derived protein crosslinks in vitro and in vivo. Nature 1996, 382, 275–278. [Google Scholar] [CrossRef]
- Cooper, M.E.; Thallas, V.; Forbes, J.; Scalbert, E.; Sastra, S.; Darby, I.; Soulis, T. The cross-link breaker, N-phenacylthiazolium bromide prevents vascular advanced glycation end-product accumulation. Diabetologia 2000, 43, 660–664. [Google Scholar] [CrossRef]
- Schwedler, S.B.; Verbeke, P.; Bakala, H.; Weiss, M.F.; Vilar, J.; Depreux, P.; Fourmaintraux, E.; Striker, L.J.; Striker, G.E. N-phenacylthiazolium bromide decreases renal and increases urinary advanced glycation end products excretion without ameliorating diabetic nephropathy in C57BL/6 mice. Diabetes Obes. Metab. 2001, 3, 230–239. [Google Scholar] [CrossRef]
- Oturai, P.S.; Christensen, M.; Rolin, B.; Pedersen, K.E.; Mortensen, S.B.; Boel, E. Effects of advanced glycation end-product inhibition and cross-link breakage in diabetic rats. Metab.-Clin. Exp. 2000, 49, 996–1000. [Google Scholar] [CrossRef]
- Thallas-Bonke, V.; Lindschau, C.; Rizkalla, B.; Bach, L.A.; Boner, G.; Meier, M.; Haller, H.; Cooper, M.E.; Forbes, J.M. Attenuation of Extracellular Matrix Accumulation in Diabetic Nephropathy by the Advanced Glycation End Product Cross-Link Breaker ALT-711 via a Protein Kinase C-α−Dependent Pathway. Diabetes 2004, 53, 2921–2930. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thallas, V.; Thomas, M.C.; Founds, H.W.; Burns, W.C.; Jerums, G.; Cooper, M.E. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J. 2003, 17, 1762–1764. [Google Scholar] [CrossRef]
- Shapiro, B.P.; Owan, T.E.; Mohammed, S.F.; Meyer, D.M.; Mills, L.D.; Schalkwijk, C.G.; Redfield, M.M. Advanced glycation end products accumulate in vascular smooth muscle and modify vascular but not ventricular properties in elderly hypertensive canines. Circulation 2008, 118, 1002–1010. [Google Scholar] [CrossRef]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the Receptor for Advanced Glycation End Products Reduces Glomerulosclerosis and Preserves Renal Function in the Diabetic OVE26 Mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef]
- Tesch, G.; Sourris, K.C.; Summers, S.A.; McCarthy, D.; Ward, M.S.; Borg, D.J.; Gallo, L.A.; Fotheringham, A.K.; Pettit, A.R.; Yap, F.Y.T.; et al. Deletion of bone-marrow-derived receptor for AGEs (RAGE) improves renal function in an experimental mouse model of diabetes. Diabetologia 2014, 57, 1977–1985. [Google Scholar] [CrossRef]
- Jensen, L.J.N.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Rasch, R.; Flyvbjerg, A. Renal effects of a neutralising RAGE-antibody in long-term streptozotocin-diabetic mice. J. Endocrinol. 2006, 188, 493–501. [Google Scholar] [CrossRef]
- Flyvbjerg, A.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Mogensen, T.H.; Paludan, S.R.; Rasch, R. Long-Term Renal Effects of a Neutralizing RAGE Antibody in Obese Type 2 Diabetic Mice. Diabetes 2004, 53, 166–172. [Google Scholar] [CrossRef]
- Kaida, Y.; Fukami, K.; Matsui, T.; Higashimoto, Y.; Nishino, Y.; Obara, N.; Nakayama, Y.; Ando, R.; Toyonaga, M.; Ueda, S.; et al. DNA Aptamer Raised Against AGEs Blocks the Progression of Experimental Diabetic Nephropathy. Diabetes 2013, 62, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.-I. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamagishi, S.-I.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).