
Metabolic Consequences of Glucocorticoid Exposure before Birth



Abstract
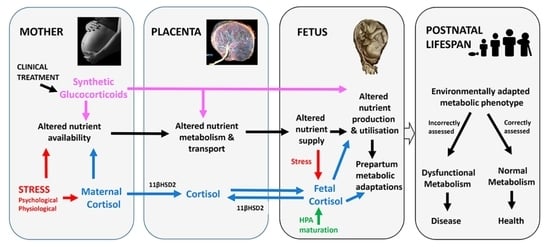
1. Introduction
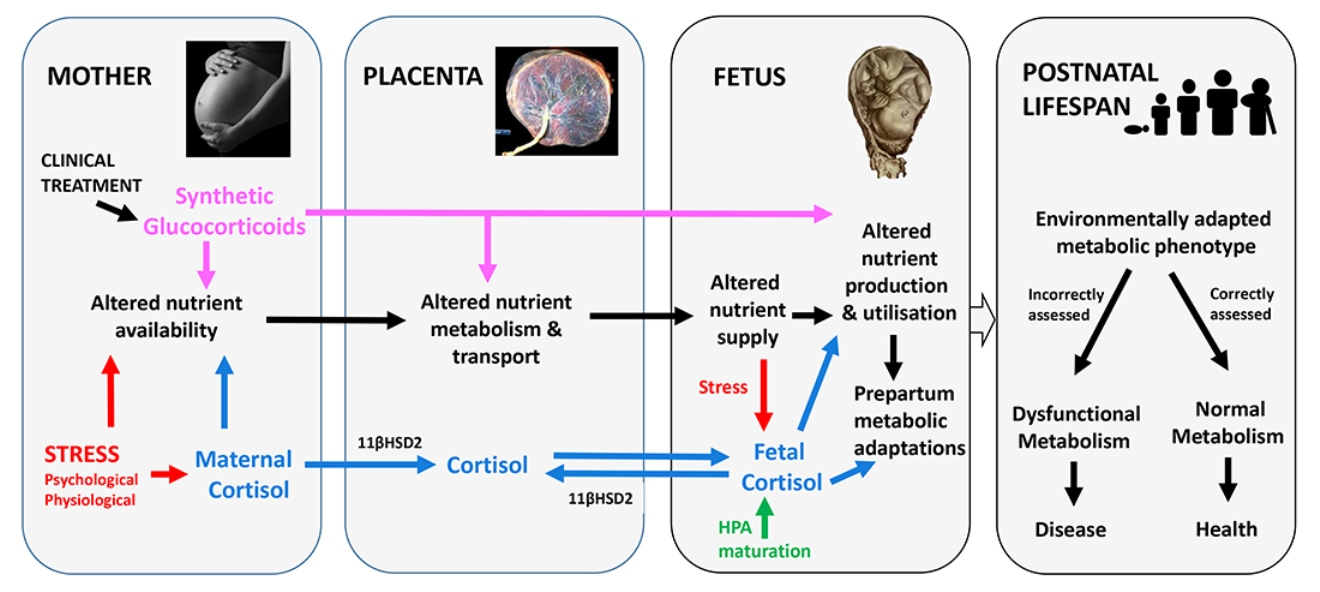
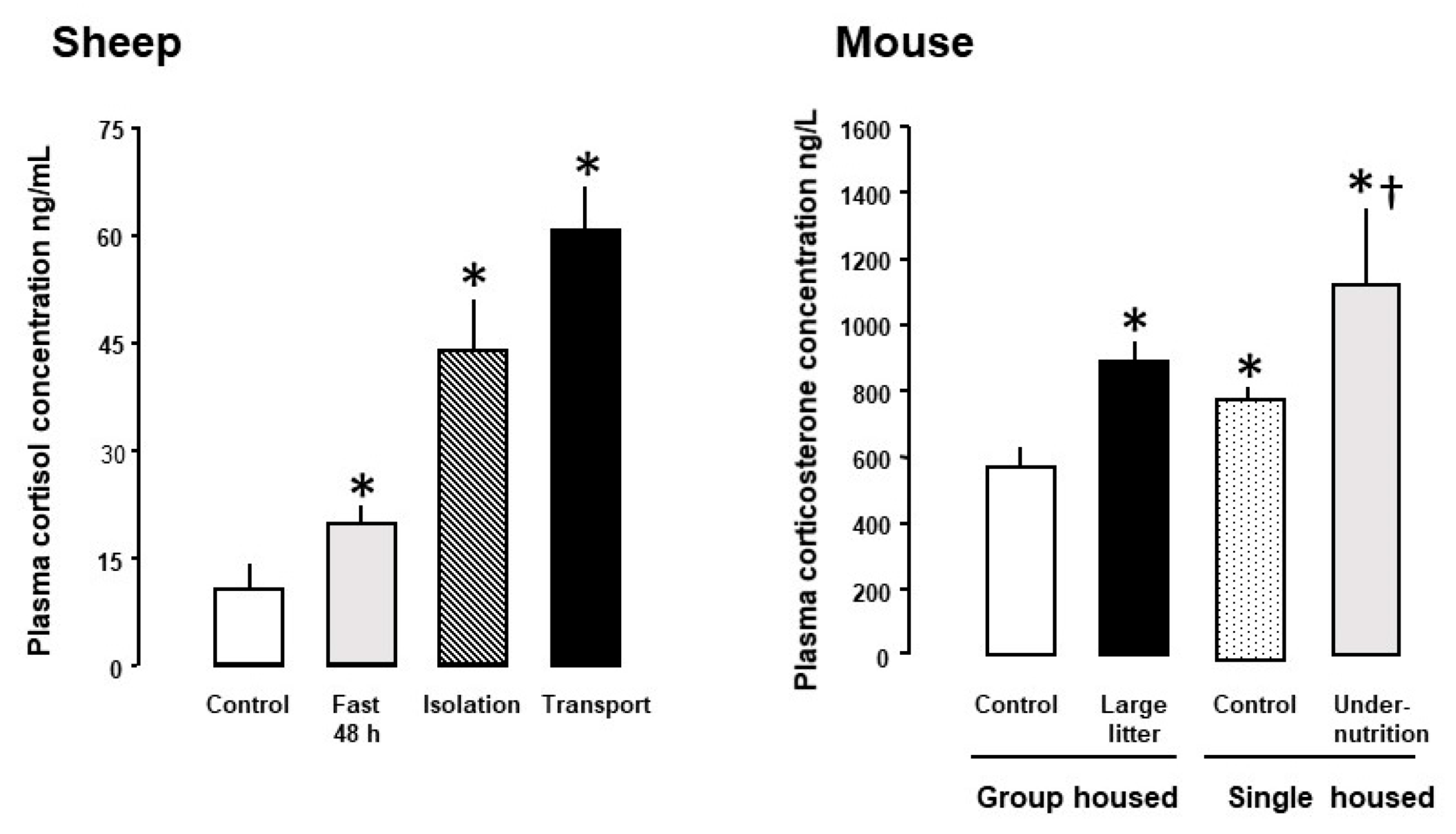
2. Feto-Placental Glucocorticoid Exposure
3. Effects of Glucocorticoids on Feto-Placental Metabolism
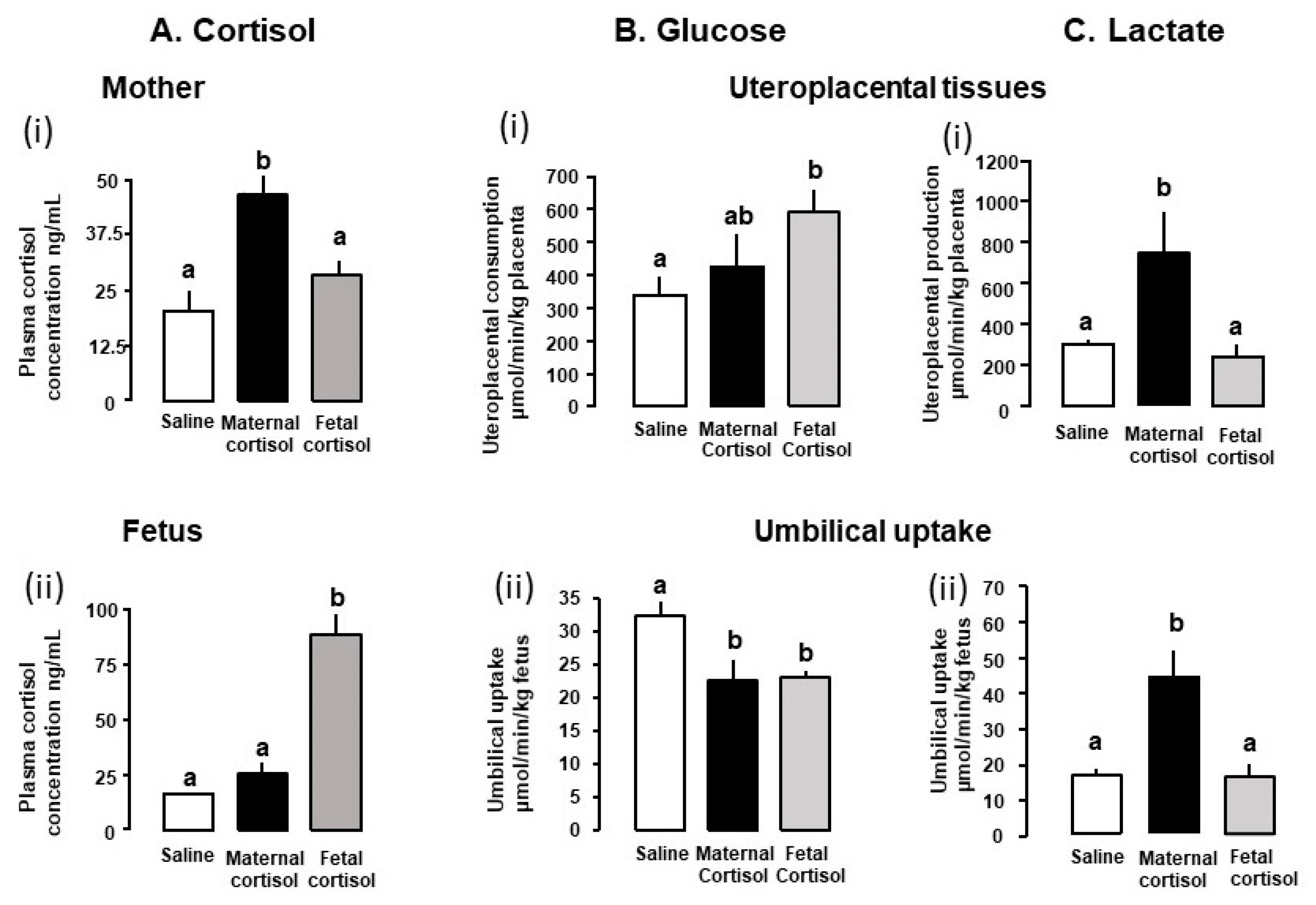
3.1. Glucose Metabolism
3.2. Metabolism of Lactate and Other Carbohydrates
3.3. Amino Acid Metabolism
3.4. Oxygen Transfer and Utilisation
3.5. Bioenergetics and Mitochondrial Function
4. Mechanisms of Glucocorticoid Action
4.1. Molecular and Cellular Effects
4.2. Other Indirect Systemic Effects
4.2.1. Maternal Nutritional State
4.2.2. Placental Development
4.2.3. Other Metabolic Hormones
5. Effects of the Glucocortocid-Induced Metabolic Changes on Fetal Development
6. Postnatal Metabolic Consequences of Prenatal Glucorticoid Overexposure
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munck, A.; Guyre, P.M. Glucocorticoid physiology, pharmacology and stress. Adv. Exp. Med. Biol. 1986, 196, 81–96. [Google Scholar] [PubMed]
- Fowden, A.L.; Forhead, A.J. Glucocorticoids as regulatory signals in intrauterine development. Exp. Physiol. 2015, 100, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Li, J.; Forhead, A.J. Glucocorticoids and the preparation for life after birth: Are there long term consequences of the life insurance? Proc. Nut. Soc. 1998, 57, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bates, K.; Herzog, E.D. Maternal-fetal circadian communication during pregnancy. Front. Endocrinol. 2020, 11, 198. [Google Scholar] [CrossRef]
- Edwards, P.D.; Boonstra, R. Glucocorticoids and CBG during pregnancy in mammals: Diversity, pattern and function. Gen. Comp. Endocrinol. 2018, 259, 122–130. [Google Scholar] [CrossRef]
- Simonetta, G.; Walker, D.W.; McMillen, I.C. Effect of feeding on the diurnal rhythm of plasma cortisol and adrenocorticotrophic hormones concentrations in the pregnant ewe and sheep fetus. Exp. Physiol. 1991, 76, 219–229. [Google Scholar] [CrossRef]
- Pofi, R.; Tomlinson, J.W. Glucocorticoids in pregnancy. Obstet. Med. 2020, 13, 62–69. [Google Scholar] [CrossRef]
- Hennessy, D.P.; Coghlan, J.P.; Hardy, K.J.; Scoggins, B.A.; Wintour, E.M. The origin of cortisol in the blood of fetal sheep. J. Endocrinol. 1982, 95, 71–79. [Google Scholar] [CrossRef]
- Fowden, A.L.; Silver, M. The effects of food withdrawal on uterine contractile activity and on plasma cortisol concentrations in ewes and their fetuses during late gestation. In The Physiological Development of the Fetus and Newborn; Jones, C.T., Nathanielsz, P.W., Eds.; Academic Press: London, UK, 1985; pp. 157–161. [Google Scholar]
- Dreiling, M.; Schiffner, R.; Bischoff, S.; Rupprecht, S.; Kroegel, N.; Schubert, H.; Witte, O.W.; Schwab, M.; Rakers, F. Impact of chronic stress during early gestation on maternal-fetal stress transfer and fetal stress sensitivity in sheep. Stress 2018, 21, 1–10. [Google Scholar] [CrossRef]
- Smith, R.F.; Dodson, H. Hormone interaction within the hypothalamus and pituitary with respect to stress and reproduction in sheep. Dom. Anim. Endocrinol. 2002, 23, 75–85. [Google Scholar] [CrossRef]
- Musial, B.; Vaughan, O.R.; Fernandez-Twinn, D.; Voshol, P.; Ozanne, S.E.; Fowden, A.L.; Sferruzzi-Perri, A.N. A Western-style diet alters maternal metabolic physiology with consequences for fetal nutrient acquisition in mice. J. Physiol. 2017, 595, 4875–4892. [Google Scholar] [CrossRef]
- Sferruzzi-Perri, A.N.; Vaughan, O.R.; Coan, P.M.; Suciu, M.C.; Darbyshire, R.; Constancia, M.; Burton, G.J.; Fowden, A.L. Placental specific Igf2 deficiency alters developmental adaptations to undernutrition in mice. Endocrinology 2011, 152, 3202–3212. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Sferruzzi-Perri, A.N.; Fowden, A.L. Maternal corticosterone regulates nutrient allocation to fetal growth in mice. J. Physiol. 2012, 590, 5529–5540. [Google Scholar] [CrossRef]
- Cottrell, E.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar]
- Cottrell, E.C.; Seckl, J.R.; Holmes, M.C.; Wrywoll, C. Foetal and placental 11βHSD2: A hub for developmental programming. Acta Physiol. 2014, 210, 288–295. [Google Scholar] [CrossRef]
- Gardner, D.S.; Giussani, D.A.; Fowden, A.L. Hind limb glucose and lactate metabolism during umbilical cord compression and acute hypoxemia in the ovine fetus during late gestation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R954–R964. [Google Scholar] [CrossRef]
- Thorn, S.R.; Brown, L.D.; Rozance, P.J.; Hay, W.W.; Friedman, J.E. Increased hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetes 2013, 62, 65–73. [Google Scholar] [CrossRef]
- Sug-Tang, A.; Bocking, A.D.; Brooks, A.N.; Hooper, S.; White, S.E.; Jacobs, R.A.; Fraher, L.J.; Challis, J.R. Effects of restricting uteroplacental blood flow on concentrations of corticotrophin-releasing hormone, adrenocorticotropin, cortisol and prostaglandin E2 in the sheep fetus during late gestation. Can. J. Physiol. Pharmacol. 1992, 70, 1396–1402. [Google Scholar] [CrossRef]
- Fletcher, A.J.W.; Goodfellow, M.R.; Forhead, A.J.; Gardner, D.S.; McGarrigle, H.H.G.; Fowden, A.L.; Giussani, D.A. Low doses of dexamethasone suppress pituitary-adrenal function but augment the glycaemic response to acute hypoxia in fetal sheep during late gestation. Pediatr. Res. 2000, 47, 684–691. [Google Scholar] [CrossRef]
- Fletcher, A.J.W.; Ma, X.H.; Wu, W.X.; Nathanielsz, P.W.; McGarrigle, H.H.G.; Fowden, A.L.; Giussani, D.A. Antenatal glucocorticoids reset the level of baseline and hypoxia-induced pituitary-adrenal activity in the sheep fetus during late gestation. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E311–E319. [Google Scholar] [CrossRef]
- Barnes, J.J.; Comline, R.S.; Silver, M. Effects of cortisol on liver glycogen concentrations in hypophysectomized, adrenalectomized and normal foetal lambs during late or prolonged gestation. J. Physiol. 1978, 275, 567–579. [Google Scholar] [CrossRef]
- Fowden, A.L.; Silver, M. Comparative development of the pituitary-adrenal axis in the fetal foal and lamb. Repro. Dom. Anim. 1995, 30, 170–177. [Google Scholar] [CrossRef]
- Edwards, L.J.; McMillen, I.C. Impact of maternal undernutrition during the periconceptional period, fetal number, and fetal sex on the development of the hypothalmo-pituitary-adrenal axis in sheep during late gestation. Biol. Reprod. 2002, 66, 1562–1569. [Google Scholar] [CrossRef]
- Bloomfield, F.H.; Oliver, M.H.; Hawkins, P.; Holloway, A.C.; Campbell, M.; Gluckman, P.D.; Harding, J.E.; Challis, J.R.G. Periconceptional undernutrition in sheep accelerates maturation of the fetal hypothalamic-pituitary-adrenal axis in late gestation. Endocrinology 2004, 145, 4278–4285. [Google Scholar] [CrossRef]
- Gardner, D.S.; Jamall, E.; Fletcher, A.J.W.; Fowden, A.L.; Giussani, D.A. Adrenocortical responsiveness is blunted in twin relative to singleton ovine fetuses. J. Physiol. 2004, 557, 1021–1032. [Google Scholar] [CrossRef]
- Wood, C.E. Development and programming of the hypothalamic-pituitary-adrenal axis. Clin. Obstet. Gynecol. 2013, 56, 610–621. [Google Scholar] [CrossRef]
- Wood, C.E. Insensitivity of near term fetal sheep to cortisol: Possible relation to the control of parturition. Endocrinology 1988, 122, 1565–1572. [Google Scholar] [CrossRef]
- Jeffray, T.M.; Berdusco, E.T.M.; Wallace, M.; Fowden, A.L.; Challis, J.R.G. Effects of incremental cortisol and adrenalectomy (ADX) on plasma corticosteroid binding capacity (CBC) in fetal sheep. Can. J. Physiol. Pharm. 1995, 73, 1568–1573. [Google Scholar] [CrossRef]
- Kemp, M.W.; Newnham, J.P.; Challis, J.R.G.; Jobe, A.H.; Stock, S.J. The clinical use of corticosteroids in pregnancy. Hum. Reprod. Update 2012, 22, 240–259. [Google Scholar] [CrossRef]
- Kim, Y.J. Glucocoticoid therapy in assisted reproduction. Clin. Exp. Reprod. Med. 2021, 48, 295–302. [Google Scholar] [CrossRef]
- Hou, S. Pregnancy in renal transplant patients. Adv. Chronic Kidney Dis. 2013, 20, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, F.C.; Meschia, G. Principal substrates of fetal metabolism. Physiol. Rev. 1987, 58, 499–527. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L. Comparative aspects of fetal carbohydrate metabolism. Equine Vet. J. 1997, 29, 19–25. [Google Scholar] [CrossRef]
- Dilsworth, M.R.; Sibley, C.P. Review: Transport across the placenta in mice and women. Placenta 2012, 34, S35–S39. [Google Scholar]
- Wesolowski, S.R.; Hay, W.W. Role of placental insufficiency and intrauterine growth restriction on activation of fetal hepatic glucose production. Mol. Cell. Endocrinol. 2016, 435, 61–68. [Google Scholar] [CrossRef]
- Girard, J. Gluconeogenesis in late fetal and early neonatal life. Biol. Neonate 1986, 50, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Audette, M.C.; Challis, J.R.G.; Jones, R.J.; Sibley, C.P.; Matthews, S.G. Antenatal dexamethasone treatment in midgestation reduces system A-mediated transport in the late-gestation murine placenta. Endocrinology 2011, 152, 3561–3570. [Google Scholar] [CrossRef][Green Version]
- Vaughan, O.R.; Sferruzzi-Perri, A.N.; Coan, P.M.; Fowden, A.L. Adaptations in placental phenotype depend on the route and timing of maternal dexamethasone administration in mice. Biol. Reprod. 2013, 89, 80. [Google Scholar] [CrossRef]
- Tye, L.M.; Burton, A.F. Glycogen deposition in fetal mouse tissues and the effect of dexamethasone. Biol. Neonate 1980, 38, 265–269. [Google Scholar] [CrossRef]
- Ivy, J.R.; Carter, R.N.; Zhao, J.-F.; Buckley, C.; Urquijo, H.; Rog-Zielinska, E.A.; Panting, E.; Hrabalkova, L.; Nicholson, C.; Agnew, E.J.; et al. Glucocorticoids regulate mitochondrial fatty acid oxidation in fetal cardiomyocytes. J. Physiol. 2021, 599, 4901–4924. [Google Scholar] [CrossRef]
- O’Connell, B.A.; Moritz, K.M.; Roberts, C.T.; Walker, D.W.; Dickinson, H. The placental response to excess maternal glucocorticoid exposure differs between male and female conceptus in spiny mice. Biol. Reprod. 2011, 85, 1040–1047. [Google Scholar] [CrossRef]
- O’Connell, B.A.; Moritz, K.M.; Walker, D.W.; Dickinson, H. Treatment of pregnant spiny mice at mid gestation with a synthetic glucocorticoid has a sex-dependent effects on placental glycogen stores. Placenta 2013, 34, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Gokulakrishnan, G.; Estrada, I.J.; Sosa, H.A.; Fiorotto, M.L. In utero glucocorticoid exposure reduces fetal skeletal muscle mass in rats independent of the effects on maternal nutrition. Am. J. Physiol Regul. Integr. Comp. Physiol. 2012, 302, R1143–R1152. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Shibazaki, Y.; Taniuchi, Y.; Oya, A.; Asakura, H.; Koshino, T.; Araki, T. Effect of dexamethasone on mitochondrial maturation in fetal rat brain. Am. J. Obstet. Gynecol. 2002, 186, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Klepac, R. Effect of dexamethasone on glycogen deposition in pregnant rats and their fetuses. Exp. Clin. Endocrinol. 1985, 86, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, Y.; Takeba, Y.; Kumai, T.; Matsumoto, N.; Mizuno, M.; Murano, K.; Asoh, K.; Takagi, M.; Yamamoto, H.; Kobayashi, S. Antenatal glucocorticoid therapy increases cardiac alpha-enolase levels in fetus and neonate rats. Life Sci. 2009, 85, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.W.; Wang, C.; Tao, J.; Walsh, S.A.; Sawatzke, A.B.; Hu, S.; Sunderland, J.L.; Segar, J.L.; Pronto, L.L.B. Effect of insulin and dexamethasone on fetal assimilation of maternal glucose. Endocrinology 2011, 152, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Fang, M.; Zhuang, S.; Qiao, Y.; Huang, W.; Gong, Q.; Xu, D.; Zhang, Y.; Wang, H. Prenatal dexamethasone exposure exerts sex-specific effect on placental oxygen and nutrient transport ascribed to differential expression of IGF2. Ann. Transl. Med. 2020, 8, 233. [Google Scholar] [CrossRef]
- Dai, Y.; Kou, H.; Gui, S.; Guo, X.; Liu, H.; Gong, Z.; Sun, X.; Wang, H.; Guo, Y. Prenatal dexamethasone exposure induced pancreatic β-cell dysfunction and glucose intolerance of male offspring rates: Role of epigenetic repression of ACE2. Sci. Total Environ. 2022, 826, 154095. [Google Scholar] [CrossRef]
- Franko, K.L.; Giussani, D.A.; Forhead, A.J.; Fowden, A.L. Effects of dexamethasone on the glucogenic capacity of fetal, pregnant and non-pregnant adult sheep. J. Endocrinol. 2007, 191, 67–73. [Google Scholar] [CrossRef]
- Langdown, M.L.; Sugden, M.C. Enhanced placental GLUT1 and GLUT3 expression in dexamethasone-induced fetal growth retardation. Mol. Cell Endocrinol. 2001, 185, 109–117. [Google Scholar] [CrossRef]
- Jellyman, J.K.; Martin-Gronet, M.S.; Cripps, R.L.; Giussani, D.A.; Ozanne, S.E.; Shen, Q.W.; Du, M.; Fowden, A.L.; Forhead, A.J. Effects of cortisol and dexamethasone on insulin signaling pathways in skeletal muscle of the ovine fetus during late gestation. PLoS ONE 2012, 7, e52363. [Google Scholar] [CrossRef]
- Franko, K.L.; Forhead, A.J.; Fowden, A.L. Differential effects of prenatal stress and glucocorticoid administration on postnatal growth and glucose metabolism in rats. J. Endocrinol. 2010, 204, 319–329. [Google Scholar] [CrossRef]
- Gray, S.; Stonestreet, B.S.; Thamotharan, S.; Sadowska, G.B.; Daood, M.; Watchko, J.; Devaskar, S.U. Skeletal muscle glucose transporter protein responses to antenatal glucocorticoids in the ovine fetus. J. Endocrinol. 2006, 189, 219–229. [Google Scholar] [CrossRef]
- Gnanalingham, M.G.; Hyatt, M.A.; Bispham, J.; Mostyn, A.; Clarke, L.; Budge, H.; Symonds, M.E.; Stephenson, T. Maternal dexamethasone administration and the maturation of perinatal adipose tissue of the neonatal sheep. Organogenesis 2008, 4, 188–194. [Google Scholar] [CrossRef]
- Sloboda, D.M.; Newnham, J.P.; Challis, J.R.G. Repeated maternal glucocorticoid administration and the developing liver in fetal sheep. J. Endocrinol. 2002, 175, 535–543. [Google Scholar] [CrossRef]
- Hahn, T.; Barth, S.; Graf, R.; Engelmann, M.; Beslagic, D.; Reul, J.M.H.M.; Holsboer, F.; Dohr, G.; Desoye, G. Placental glucose transporter expression is regulated by glucocorticoids. J. Clin. Endocrinol. Metab. 1999, 84, 1445–1452. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Fisher, H.M.; Dionelis, K.N.; Jeffreys, E.L.C.; Higgins, J.S.; Musial, B.; Sferruzzi-Perri, A.N.; Fowden, A.L. Corticosterone alters materno-fetal glucose partitioning and insulin signalling in pregnant mice. J. Physiol. 2015, 593, 1307–1321. [Google Scholar] [CrossRef]
- Bartho, L.A.; Holland, O.J.; Moritz, K.M.; Perkins, A.V.; Cuffe, J.S.M. Maternal corticosterone in the mouse alters oxidative stress markers, antioxidant function and mitochondrial content in placentas of female fetuses. J. Physiol. 2019, 597, 3053–3067. [Google Scholar] [CrossRef]
- Jensen, E.C.; Gallaher, B.W.; Breir, B.H.; Harding, J.E. The effect of a chronic maternal cortisol infusion on the late-gestation fetal sheep. J. Endocrinol. 2002, 174, 27–36. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Davies, K.L.; Ward, J.W.; de Blasio, M.J.; Fowden, A.L. A physiological increase in maternal cortisol alters uteroplacental metabolism in pregnant ewes. J. Physiol. 2016, 594, 6407–6418. [Google Scholar] [CrossRef]
- Richards, E.M.; Wood, C.E.; Rabaglino, M.B.; Antolic, A.; Keller-Wood, M. Mechanism for the adverse effects of later gestational increases in maternal cortisol on the heart revealed by transcriptomic analyses of the fetal septum. Physiol. Genom. 2014, 46, 547–559. [Google Scholar] [CrossRef]
- Joseph, S.; Walejko, J.M.; Zhang, S.; Edison, A.S.; Keller-Wood, M. Maternal hypercortisolemia alters placental metabolism: A multiomics view. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E950–E960. [Google Scholar] [CrossRef]
- Joseph, S.; Alava, B.; Antolic, A.; Richards, E.M.; Wood, C.E.; Keller-Wood, M. Fetal ovine skeletal and cardiac muscle transcriptomics are differentially altered by increased maternal cortisol during gestation. Physiol. Genom. 2020, 52, 178–190. [Google Scholar] [CrossRef]
- Timmerman, M.; Teng, C.; Wilkening, R.B.; Fennessey, P.; Battaglia, F.C.; Meschia, G. Effects of dexamethasone on fetal hepatic glutamine-glutamate exchange. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E829–E845. [Google Scholar] [CrossRef]
- Barbera, A.; Wilkening, R.B.; Teng, C.; Battaglia, F.C.; Meschia, M. Metabolic alterations in the fetal hepatic and umbilical circulations during glucocorticoid-induced parturition in sheep. Pediatr. Res. 1997, 41, 242–248. [Google Scholar] [CrossRef]
- Milley, J.R. Effects of increased cortisol concentration on ovine fetal leucine kinetics and protein metabolism. Am. J. Physiol. Endocrinol. Metab. 1995, 268, E1114–E1122. [Google Scholar] [CrossRef]
- Milley, J.R. Fetal substrate uptake during increased ovine fetal cortisol concentration. Am. J. Physiol. Endocrinol. Metab. 1996, 271, E186–E191. [Google Scholar] [CrossRef]
- Fowden, A.L.; Mijovic, J.; Silver, M. The effects of cortisol on hepatic and renal gluconeogenic enzyme activities in the sheep fetus during late gestation. J. Endocrinol. 1993, 137, 213–222. [Google Scholar] [CrossRef]
- Ward, J.W.; Wooding, F.P.B.; Fowden, A.L. Ovine feto-placental metabolism. J. Physiol. 2004, 554, 529–541. [Google Scholar] [CrossRef]
- Vaughan, O.R.; De Blasio, M.J.; Fowden, A.L. Ovine uteroplacental and fetal metabolism during and after cortisol overexposure in late gestation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R791–R801. [Google Scholar] [CrossRef] [PubMed]
- Mostyn, A.; Pearce, S.; Budge, H.; Elmes, M.; Forhead, A.J.; Fowden, A.L.; Stephenson, T.; Symonds, M.E. Influence of cortisol on adipose tissue development in the fetal sheep during late gestation. J. Endocrinol. 2003, 176, 23–30. [Google Scholar] [CrossRef]
- Davies, K.L.; Camm, E.J.; Smith, D.J.; Vaughan, O.R.; Forhead, A.J.; Murray, A.J.; Fowden, A.L. Glucocorticoid maturation of mitochondrial respiratory capacity in skeletal muscle before birth. J. Endocrinol. 2021, 251, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Lumbers, E.R.; Boyce, A.C.; Joulianos, G.; Kumarasamy, V.; Barner, E.; Segar, J.L.; Burrell, J.H. Effects of cortisol on cardiac myocytes and on expression of cardiac genes in fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R567–R574. [Google Scholar] [CrossRef] [PubMed]
- Townsend, S.F.; Rudolph, C.D.; Rudolph, A.M. Cortisol induces perinatal hepatic gluconeogenesis in the lamb. J. Dev. Physiol. 1991, 16, 71–79. [Google Scholar] [PubMed]
- Fowden, A.L.; Mundy, L.; Silver, M. Developmental regulation of glucogenesis in the sheep fetus during late gestation. J. Physiol. 1998, 508, 937–947. [Google Scholar] [CrossRef]
- Wyrwoll, C.S.; Seckl, J.R.; Homes, M.C. Altered placental function of 11β-hydroxysteroid dehydrogenase 2 knockout mice. Endocrinology 2009, 150, 1287–1293. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Phillips, H.M.; Everden, A.J.; Sferruzzi-Perri, A.N.; Fowden, A.L. Dexamethasone treatment of pregnant F0 mice leads to parent of origin-specific changes in placental function of the F2 generation. Reprod. Fert. Develop. 2015, 27, 704–711. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Fowden, A.L. Placental metabolism: Substrate requirements and the response to stress. Reprod. Dom. Anim. 2016, 51 (Suppl. 2), 25–35. [Google Scholar] [CrossRef]
- Nyirenda, M.J.; Welberg, L.A.; Seckl, J.R. Programming hyperglycaemia in the rat through prenatal exposure to glucocorticoids -fetal effects or maternal influence. J. Endocrinol. 2001, 170, 653–660. [Google Scholar] [CrossRef]
- Monica Shih, M.C.; Huang, C.C.J.; Chu, H.P.; Hsu, N.C.; Chung, B.C. Embryonic steroids control developmental programming of energy balance. Endocrinology 2021, 162, bqab196. [Google Scholar] [CrossRef]
- Fowden, A.L.; Forhead, A.J. Adrenal glands are essential for activation of glucogenesis during undernutrition in fetal sheep near term. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E94–E102. [Google Scholar] [CrossRef]
- Houin, S.S.; Rozance, P.J.; Brown, L.D.; Hay, W.W.; Wilkening, R.B.; Thorn, S.R. Coordinated changes in hepatic amino acid metabolism and endocrine signals support hepatic glucose production during fetal hypoglycemia. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E306–E314. [Google Scholar] [CrossRef]
- Rudolph, C.D.; Roman, C.; Rudolph, A.M. Effect of cortisol on hepatic gluconeogenesis in the fetal sheep. J. Dev. Physiol. 1989, 11, 219–223. [Google Scholar]
- Sparks, J.W.; Hay, W.W.; Bonds, D.; Meschia, G.; Battaglia, F.C. Simultaneous measurements of lactate turnover rate and umbilical lactate uptake in the fetal lamb. J. Clin. Investig. 1982, 70, 179–192. [Google Scholar] [CrossRef]
- Meznarich, M.K.; Hay, W.W.; Sparks, J.W.; Sparks, G.; Battaglia, F.C. Fructose disposal and oxidation rates in the ovine fetus. Quart. J. Exp. Physiol. 1987, 72, 617–625. [Google Scholar] [CrossRef]
- McGowan, J.E.; Aldoretta, P.W.; Hay, W.W. Contribution of fructose and lactate produced in the placenta to calculation of fetal glucose oxidation rate. Am. J. Physiol. Endocrinol. Metab. 1995, 269, E834–E839. [Google Scholar] [CrossRef]
- Jensen, E.; Wood, C.E.; Keller-Wood, M. Chronic alterations in ovine uteroplacental corticosteroid levels influence uterine blood flow and placental and fetal growth. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R54–R61. [Google Scholar] [CrossRef]
- Fowden, A.L. The role of insulin in fetal growth. Early Hum. Dev. 1992, 29, 177–181. [Google Scholar] [CrossRef]
- Shen, C.-N.; Seckl, J.R.; Slack, J.M.W.; Tosh, D. Glucocorticoids suppress beta-cell development and induces metaplasia in embryonic pancreas. Biochem. J. 2003, 375, 41–50. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Huan, Y.-H.; Sheen, J.-M.; Tain, Y.-L.; Yu, H.-R.; Chen, C.-C.; Tiao, M.M.; Kuo, H.-C.; Huang, L.-T. Prenatal dexamethasone exposure programs the development of the pancreas and the secretion of insulin in rats. Pediatr. Neonatol. 2017, 58, 135–144. [Google Scholar] [CrossRef]
- Cleal, J.K.; Lofthouse, E.M.; Sengers, B.G.; Lewis, R.M. A systems perspective on placental amino acid transport. J. Physiol. 2018, 596, 5511–5522. [Google Scholar] [CrossRef] [PubMed]
- Walejko, J.M.; Antolic, A.; Koelmel, J.P.; Garrett, T.J.; Edison, A.S.; Keller-Wood, M. Chronic maternal cortisol excess during late gestation leads to metabolic alterations in the newborn heart. Am. J. Physiol. Endocrinol. Metab. 2019, 136, E546–E556. [Google Scholar] [CrossRef] [PubMed]
- Franko, K.L.; Forhead, A.J.; Fowden, A.L. Effects of stress during pregnancy on hepatic glucogenic capacity of rat dams and their fetuses. Physiol. Rep. 2017, 5, e13293. [Google Scholar] [CrossRef] [PubMed]
- Audette, M.C.; Challis, J.R.G.; Jones, R.L.; Sibley, C.P.; Matthews, S.G. Synthetic glucocorticoid reduces human placental systems transport in women treated with antenatal therapy. J. Clin. Endocrinol. Metab. 2014, 99, E2226–E2233. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.; Pintado, E.; Ferre, P. Fuel metabolism in the mammalian fetus. Ann. Biol. Anim. Bioch. Biophys. 1979, 19, 181–197. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Lapp, H.E.; Barlett, A.A.; Hunter, R.G. Stress and glucocorticoid receptor regulation of mitochondrial gene expression. J. Mol. Endocrinol. 2019, 62, R121–R128. [Google Scholar] [CrossRef]
- Rog-Zielinkska, E.A.; Graig, M.A.; Manning, J.R.; Richardson, R.V.; Gowans, G.J.; Dunbar, D.R.; Gharbi, K.; Kenyon, C.J.; Holmes, M.C.; Hardie, D.G.; et al. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: A role of PGC-1α. Cell Death Diff. 2015, 22, 1106–1116. [Google Scholar] [CrossRef]
- Grilo, L.F.; Tocantin, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafrome, P.; Pereira, S.P. Metabolic disease programming: From mitochondria to epigenetics, glucocorticoid signalling and beyond. Eur. J. Clin. Investig. 2021, 51, e13625. [Google Scholar] [CrossRef]
- Prieur, B.; Bismuth, J.; Delaval, E. Effects of adrenal steroid hormones on mitochondrial maturation during the late fetal period. Eur. J. Biochem. 1998, 252, 194–199. [Google Scholar] [CrossRef]
- Synder, J.E.; Rodgers, H.F.; O’Brien, J.A.; Mahil, N.; Magliato, S.A.; Gurham, P.L. Glucocorticoid effects on rabbit lung maturation in vivo: An ultrastructural morphometric study. Anat. Rec. 1992, 232, 133–140. [Google Scholar] [CrossRef]
- Lane, R.H.; Chandorkar, A.K.; Flozak, A.S.; Simmons, R.A. Intrauterine growth retardation alters mitochondrial gene expression and function in fetal and juvenile rat skeletal muscle. Pediatr. Res. 1998, 43, 563–570. [Google Scholar] [CrossRef]
- Lane, R.H.; Flozak, A.S.; Ogata, E.S.; Bell, G.I.; Simmons, R.A. Altered hepatic gene expression of enzymes involved in energy metabolism in the growth retarded fetal rat. Pediatr. Res. 1996, 39, 390–394. [Google Scholar] [CrossRef]
- Pendleton, A.L.; Antolic, A.T.; Kelly, A.C.; Davis, M.A.; Camacho, L.E.; Doubleday, K.; Anderson, M.J.; Langlais, P.R.; Lynch, R.M.; LImesand, S.W. Lower oxygen consumption and Complex I activity in mitochondria isolated from skeletal muscle of fetal sheep with intrauterine growth restriction. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E67–E80. [Google Scholar] [CrossRef]
- Busada, J.T.; Cidlowski, J.A. Mechanisms of glucocorticoid action during development. Curr. Top. Dev. Biol. 2007, 125, 147–170. [Google Scholar]
- Moisiadis, V.G.; Matthews, S.G. Glucocorticoids and fetal programming part 2: Mechanisms. Nat. Rev. Endocrinol. 2014, 10, 403–4011. [Google Scholar] [CrossRef]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef]
- Weaver, I.C.G. Shaping adult phenotypes through early life environments. Birth Defects Res. C Embryo Today 2009, 87, 314–326. [Google Scholar] [CrossRef]
- Drake, A.J.; Liu, L.; Kerrigan, D.; Meehan, R.R.; Seckl, J.R. Multigenerational programming in the glucocorticoid programmed rat is associated with generation-specific and parent of origin effects. Epigenetics 2011, 6, 1334–1343. [Google Scholar] [CrossRef]
- Matthews, S.G.; McGowan, P.O. Developmental programming of the HPA axis and related behaviours: Epigenetic mechanisms. J. Endocrinol. 2019, 242, T69–T79. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Cuffe, J.S.M.; Moritz, K.M. Short- and long-term effects of exposure to natural and synthetic glucocorticoids during development. Clin. Exp. Exp. Pharm. Physiol. 2012, 39, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Forhead, A.J. Endocrine regulation of fetal metabolism towards term. Dom. Anim. Endocrinol. 2022, 78, 106657. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Aye, I.L.M.H.; Goberdhan, D.C.I. The emerging role of mTORC1 signalling in placental nutrient-sensing. Placenta 2012, 33 (Suppl. 2), e23–e29. [Google Scholar] [CrossRef]
- Roos, R.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T.J. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am. J. Physiol. Cell Physiol. 2009, 296, C142–C150. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Powell, T.L.; Jansson, T. Glucocorticoid regulation of amino acid transport in primary human trophoblast cells. J. Mol. Endocrinol. 2019, 63, 239–248. [Google Scholar] [CrossRef]
- Blanco, C.L.; Moreira, A.G.; McGill-Vargas, L.L.; Anzueto, D.G.; Nathanielsz, P.; Musi, N. Antenatal corticosteroids alter insulin signaling pathways in fetal baboon skeletal muscle. J. Endocrinol. 2014, 221, 253–260. [Google Scholar] [CrossRef][Green Version]
- Ain, R.; Canham, L.N.; Soares, M.J. Dexamethasone-induced intrauterine growth restriction impacts the placental prolactin family, insulin-like growth factor-II and the Akt signalling pathway. J. Endocrinol. 2005, 185, 253–263. [Google Scholar] [CrossRef]
- Rog-Zielinkska, E.A.; Thomson, A.; Kenyon, C.J.; Brownstein, D.G.; Moran, C.M.; Szumska, D.; Michailidou, Z.; Richardson, J.; Owen, E.; Watt, A.; et al. Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 2013, 22, 3239–3282. [Google Scholar]
- Opherk, C.; Tronche, F.; Kellendork, C.; Kohmuller, D.; Schulze, A.; Schmid, W.; Schutz, G. Inactivation of the glucocorticoid receptor hepatocytes leads to fasting hypoglycaemia and ameliorates hyperglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol. Endocrinol. 2004, 18, 1346–1353. [Google Scholar] [CrossRef]
- Keller-Wood, M.; Feng, X.; Wood, C.E.; Richards, E.; Anthony, R.V.; Dahl, G.E.; Tao, S. Elevated maternal cortisol leads to relative maternal hyperglycemia and increased stillbirth in ovine pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R405–R413. [Google Scholar] [CrossRef]
- Fowden, A.L.; Forhead, A.J. Endocrine interactions in the control of fetal growth. Nestlé Nutr. Inst. Workshop Ser. 2013, 74, 91–102. [Google Scholar]
- Ward, J.W.; Forhead, A.J.; Wooding, F.B.P.; Fowden, A.L. Functional significance and cortisol dependence of the gross morphology of ovine placentomes during late gestation. Biol. Reprod. 2006, 74, 137–145. [Google Scholar] [CrossRef]
- Wyrwoll, C.S.; Kerrigan, D.; Holmes, M.C.; Seckl, J.R.; Drake, A.J. Altered placental methyl donor transport in the dexamethasone programmed rat. Placenta 2012, 33, 220–223. [Google Scholar] [CrossRef][Green Version]
- Cuffe, J.S.M.; O’Sullivan, L.; Simmons, D.G.; Anderson, S.T.; Moritz, K.M. Maternal corticosterone exposure in the mouse has sex-specific effects on placental growth and mRNA expression. Endocrinology 2012, 153, 5500–5511. [Google Scholar] [CrossRef]
- Braun, T.; Li, S.; Moss, T.J.M.; Newnham, J.P.; Challis, J.R.G.; Gluckman, P.D.; Sloboda, D.M. Maternal betamethasone administration reduces binucleate cell number and placental lactogen in sheep. J. Endocrinol. 2007, 194, 337–347. [Google Scholar] [CrossRef]
- Shang, H.; Meng, W.; Sloboda, D.M.; Li, S.; Ehrlich, L.; Plagemann, A.; Dudenhausen, J.W.; Henrich, W.; Newnham, J.P.; Challis, J.R.G.; et al. Effects of maternal dexamethasone treatment early in pregnancy on glucocorticoid receptors in the ovine placenta. Reprod. Sci. 2015, 22, 534–544. [Google Scholar] [CrossRef]
- Fletcher, A.J.W.; McGarrigle, H.; Edwards, M.; Fowden, A.L.; Giussani, D.A. Effects of low dose dexamethasone treatment on basal cardiovascular and endocrine function in fetal sheep during late gestation. J. Physiol. 2002, 545, 649–660. [Google Scholar] [CrossRef]
- Forhead, A.J.; Curtis, K.; Kapstein, E.; Visser, T.J.; Fowden, A.L. Developmental control of iodothyronine deiodinases by cortisol in the ovine fetus and placenta near term. Endocrinology 2006, 147, 5988–5994. [Google Scholar] [CrossRef]
- Forhead, A.J.; Jellyman, J.K.; Gardner, D.S.; Giussani, D.A.; Kaptein, E.; Visser, T.J.; Fowden, A.L. Differential effects of maternal dexamethasone treatment on circulating thyroid hormone concentrations and tissue deiodinase activity in the pregnant ewe and fetus. Endocrinology 2007, 148, 800–805. [Google Scholar] [CrossRef]
- Forhead, A.J.; Thomas, L.; Crabtree, J.; Hoggard, N.; Gardner, D.S.; Giussani, D.A.; Fowden, A.L. Plasma leptin concentration in fetal sheep during late gestation: Ontogeny and effect of glucocorticoids. Endocrinology 2002, 143, 1166–1173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Braun, T.; Li, S.; Sloboda, D.M.; Li, W.; Audette, M.C.; Moss, T.J.M.; Matthews, S.G.; Polglase, G.; Nitsos, I.; Newnham, J.P.; et al. Effects of maternal dexamethasone treatment in early pregnancy on pituitary-adrenal axis in fetal sheep. Endocrinology 2009, 150, 5466–5477. [Google Scholar] [CrossRef] [PubMed]
- Jellyman, J.K.; Gardner, D.S.; Edwards, C.M.B.; Fowden, A.L.; Giussani, D.A. Fetal cardiovascular, endocrine and metabolic responses to acute hypoxaemia during and following maternal treatment with dexamethasone in sheep. J. Physiol. 2005, 567, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Sugden, M.C.; Langdown, M.L.; Munns, M.J.; Holness, M.J. Maternal glucocorticoid treatment modulates placental leptin and leptin receptor expression and maternal-fetal leptin physiology during late gestation, and elicits hypertension associated with hyperleptinaemia in the early-growth-retarded adult offspring. Eur. J. Endocrinol. 2001, 145, 529–539. [Google Scholar] [CrossRef]
- Forhead, A.J.; Cutts, S.; Matthews, P.; Fowden, A.L. Role of thyroid hormones in the developmental control of tissue glycogen in fetal sheep near term. Exp. Physiol. 2009, 94, 1079–1087. [Google Scholar] [CrossRef]
- Forhead, A.J.; Poore, K.R.; Mapstone, J.; Fowden, A.L. Developmental regulation of hepatic and renal gluconeogenic enzymes by thyroid hormones in fetal sheep during late gestation. J. Physiol. 2003, 548, 941–947. [Google Scholar] [CrossRef]
- Davies, K.L.; Camm, E.J.; Atkinson, E.V.; Lopez, T.; Forhead, A.J.; Murray, A.J.; Fowden, A.L. Developmental and thyroid hormone dependence of skeletal muscle mitochondrial function towards birth. J. Physiol. 2020, 598, 2453–2468. [Google Scholar] [CrossRef]
- Fowden, A.L.; Szemere, J.; Hughes, P.; Gilmour, R.S.; Forhead, A.J. The effects of cortisol on the growth rate of the sheep fetus during late gestation. J. Endocrinol. 1996, 151, 97–105. [Google Scholar] [CrossRef]
- Reynolds, R.M. Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis—2012 Curt Riches Award winner. Psychoneuroendocrinology 2013, 38, 1–11. [Google Scholar] [CrossRef]
- Valsamakis, G.; Papatheodorou, D.; Chalarakis, N.; Manolikaki, M.; Margeli, A.; Papassotiriou, I.; Barber, T.M.; Kumar, S.; Kalamtaridou, S.; Mastorakos, G. Maternal chronic stress correlates with serum levels of cortisol, glucose and C-peptide in the fetus, and maternal non chronic stress with fetal growth. Psychoneuroendocrinology 2020, 114, 104591. [Google Scholar] [CrossRef]
- Jellyman, J.K.; Gardner, D.S.; McGarrigle, H.H.; Fowden, A.L.; Giussani, D.A. Antenatal glucocorticoid therapy increases glucose delivery to cerebral circulations during acute hypoxaemia in fetal sheep during late gestation. Am. J. Obstet. Gynec. 2009, 201, e1–e8. [Google Scholar] [CrossRef]
- Rakers, F.; Frauendorf, V.; Rupprecht, S.; Bischoff, S.J.; Kiehntopf, M.; Reinhold, P.; Witte, O.W.; Schubert, H.; Schwarb, M. Effects of early- and late-gestational maternal stress and synthetic glucocorticoid on development of the fetal hypothalamus-pituitary-adrenal axis in sheep. Stress 2013, 16, 122–129. [Google Scholar] [CrossRef]
- Li, S.; Moss, T.J.M.; Nitsos, I.; Matthews, S.G.; Challis, J.R.G.; Newnham, J.P.; Sloboda, D.M. The impact of maternal synthetic glucocorticoid administration in late pregnancy on fetal and early neonatal hypothalamic-pituitary-adrenal axes regulatory genes is dependent upon the dose and gestational age at exposure. J. Dev. Orig. Health Dis. 2013, 4, 77–89. [Google Scholar] [CrossRef]
- Cuffe, J.S.M.; Salf, S.; Perkins, A.V.; Moritz, K.M.; Clifton, V.L. Dexamethasone and sex regulate placental glucocorticoid receptor isoforms in mice. J. Endocrinol. 2017, 234, 89–100. [Google Scholar] [CrossRef]
- Clarke, K.; Ward, J.W.; Forhead, A.J.; Giussani, D.A.; Fowden, A.L. Regulation of 11β-hydoxysteroid dehydrogenase type 2 (11β-HSD2) activity in ovine placenta by fetal cortisol. J. Endocrinol. 2002, 172, 527–534. [Google Scholar] [CrossRef][Green Version]
- Drake, A.J.; Walker, B.R.; Seckl, J.R. Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am. J. Regul. Integr. Comp. Physiol. 2005, 288, R34–R38. [Google Scholar] [CrossRef]
- McGowan, P.O.; Matthews, S.G. Prenatal stress, glucocorticoids and developmental programming of the stress response. Endocrinology 2018, 159, 69–82. [Google Scholar] [CrossRef]
- Hamada, H.; Matthews, S.G. Prenatal programming of stress responsiveness and behaviours: Progress and perspectives. J. Neuroendocrinol. 2018, 13, e12674. [Google Scholar] [CrossRef]
- De Blasio, M.J.; Dodic, M.; Jefferies, A.J.; Moritz, K.M.; Wintour, E.M.; Owens, J.A. Maternal exposure to dexamethasone or cortisol in early pregnancy differentially alters insulin secretion and glucose homeostasis in adult male sheep offspring. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E75–E82. [Google Scholar] [CrossRef]
- Cleasby, M.E.; Kelly, P.A.; Walker, B.R.; Seckl, J.R. Programming of rat muscle and fat metabolism by in utero exposure to glucocorticoids. Endocrinology 2003, 144, 999–1007. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Hu, Y.; Yang, Q.-Y.; Son, J.S.; Liu, X.-D.; de Avila, J.M.; Zhu, M.-J.; Du, M. Excessive glucocorticoids during pregnancy impair fetal brown fat development and predispose offspring to metabolic dysfunction. Diabetes 2020, 69, 1662–1673. [Google Scholar] [CrossRef]
- Long, N.M.; Shasa, D.R.; Ford, S.P.; Nathanielsz, P.W. Growth and insulin dynamics in two generations of female offspring of mothers receiving a single course of synthetic glucocorticoids. Am. J. Obstet. Gynecol. 2012, 207, e1–e8. [Google Scholar] [CrossRef]
- O’Regan, D.; Kenyon, C.J.; Seckl, J.R.; Holmes, M.C. Glucocorticoid exposure in late gestation in the rat permanently programs gender-specific differences in adult cardiovascular and metabolic physiology. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E853–E870. [Google Scholar] [CrossRef]
- Kuo, A.H.; Li, J.; Li, C.; Huber, H.F.; Schwarb, M.; Nathanielsz, P.W.; Clarke, G.D. Prenatal steroid administration leads to adult pericardial and hepatic steatosis in male baboons. Int. J. Obes. 2017, 41, 1299–1302. [Google Scholar] [CrossRef]
- Sloboda, D.M.; Moss, T.J.M.; Li, S.; Doherty, D.; Nitsos, I.; Challis, J.R.G.; Newnham, J.P. Hepatic glucose regulation and metabolism in adult sheep: Effects of prenatal betamethasone. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E721–E728. [Google Scholar] [CrossRef]
- Sloboda, D.M.; Moss, T.J.; Gurrin, L.C.; Newnham, J.P.; Challis, J.R.G. The effect of prenatal betamethasone administration on postnatal ovine hypothalamic-pituitary-adrenal function. J. Endocrinol. 2002, 172, 71–81. [Google Scholar] [CrossRef]
- Sloboda, D.M.; Moss, T.J.M.; Li, S.; Doherty, D.; Nitsos, I.; Challis, J.R.G.; Newnham, J.P. Prenatal betamethasone exposure results in pituitary-adrenal hyporesponsiveness in adult sheep. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E61–E70. [Google Scholar] [CrossRef]
- Santos-Silva, J.C.; da Silva, P.M.R.; de Souza, D.N.; Teixeira, C.J.; Bordin, S.; Anhe, G.F. In utero exposure to dexamethasone programs the development of the pancreatic α- and β-cells during early postnatal life. Life Sci. 2020, 255, 117810. [Google Scholar] [CrossRef]
- Riveline, J.-P.; Baz, B.; Nguewa, J.-L.; Vidal-Trecan, T.; Ibrahim, F.; Boudou, P.; Vicaut, E.; de la Perriere, A.B.; Fetita, S.; Breant, B.; et al. Exposure to glucocorticoids in the first part of fetal life with insulin secretory defect in adult humans. J. Clin. Endocrinol. Metab. 2020, 105, dgz145. [Google Scholar] [CrossRef]
- de Vires, A.; Holmes, M.C.; Heijins, A.; Seier, J.V.; Heerden, J.; Louw, J.; Wolfe-Coote, S.; Meaney, M.J.; Levitt, N.S.; Seckl, J.R. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J. Clin. Investig 2007, 117, 1058–1067. [Google Scholar] [CrossRef]
- Cuffe, J.S.M.; Turton, E.L.; Akison, L.K.; Bielefeldt-Ohmann, H.; Moritz, K.M. Prenatal corticosterone exposure programs sex-specific adrenal adaptations in mouse offspring. J. Endocrinol. 2017, 232, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Langdown, M.L.; Smith, N.D.; Sugden, M.C.; Holness, M.J. Excessive glucocorticoid exposure during late intrauterine development modulates the expression of cardiac uncoupling proteins in adult hypertensive male offspring. Pflugers Arch. 2001, 442, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Huber, H.F.; Kuo, A.H.; Li, C.; Jenkins, S.L.; Gerow, K.G.; Clarke, G.D.; Nathanielsz, P.W. Antenatal synthetic glucocorticoid exposure at human therapeutic equivalent doses predisposes middle-age male offspring baboons to an obese phenotype that emerges with aging. Reprod. Sci. 2019, 26, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.-M.; Hsieh, C.-S.; Tain, Y.-L.; Li, S.-W.; Yu, H.-R.; Chen, C.-C.; Tiao, M.-M.; Chen, Y.-C.; Huang, L.-T. Programming effects of prenatal glucocorticoid exposure with a postnatal high-fat diet in diabetes mellitus. Int. J. Mol. Sci. 2016, 17, 533. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.J.; Raubenheimer, P.J.; Kerrigan, D.; McInnes, K.J.; SEckl, J.R.; Walker, B.R. Prenatal dexamethasone programs expression of genes in liver and adipose tissue and increased hepatic lipid accumulation but not obesity on a high-fat diet. Endocrinology 2010, 151, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Long, N.M.; Ford, S.P.; Nathanielsz, P.W. Multigenerational effects of fetal dexamethasone exposure on the hypothalamic-pituitary-adrenal axis of first- and second-generation female offspring. Am. J. Obstet. Gynecol. 2013, 208, 217.e1–217.e8. [Google Scholar] [CrossRef]
- Iqbal, M.; Moisiadis, V.G.; Kostaki, A.; Matthews, S.G. Transgenerational effects of prenatal synthetic glucocorticoids on hypothalamic-pituitary-adrenal function. Endocrinology 2012, 153, 3295–3307. [Google Scholar] [CrossRef][Green Version]
- Ilg, L.; Kirschbaum, C.; Le, S.-C.; Rosenlocher, F.; Miller, R.; Alexander, N. Persistent effects of antenatal synthetic glucocorticoids on endocrine stress reactivity from childhood to adolescence. J. Clin. Endocrinol. Metab. 2019, 104, 827–834. [Google Scholar] [CrossRef]
- Moss, T.M.J.; Sloboda, D.M.; Gurrin, L.C.; Harding, R.; Challis, J.R.G.; Newnham, J.P. Programming effects in sheep of prenatal growth restriction and glucocorticoid exposure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R960–R970. [Google Scholar] [CrossRef]
- Irwin, J.L.; Meyering, A.L.; Peterson, G.; Glynn, L.M.; Sandman, C.A.; Hicks, L.M.; Davis, E.P. Maternal prenatal cortisol programs the infant hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology 2021, 125, 105106. [Google Scholar] [CrossRef]
- Davis, E.P.; Glynn, L.M.; Waffarn, F.; Sandman, C.A. Prenatal maternal stress programs infant stress regulation. J. Child Psychol. Psychiatry 2011, 52, 119–129. [Google Scholar] [CrossRef]
- Krontira, A.C.; Cruceanu, C.; Binder, E.B. Glucocorticoids as mediators of adverse outcomes of prenatal stress. Trends Neurosci. 2020, 43, 394–405. [Google Scholar] [CrossRef]
- Davis, E.P.; Waffarn, F.; Sandman, C.A. Prenatal treatment with glucocorticoids sensitizes the hpa axis response to stress among full-term infants. Dev. Psychobiol. 2011, 53, 175–183. [Google Scholar] [CrossRef]
- Long, N.M.; Smith, D.T.; Ford, S.P.; Nathanielsz, P.W. Elevated glucocorticoids during ovine pregnancy increase appetite and produce glucose dysregulation and adiposity in their granddaughters in response to ad libitum feeding at one year of age. Am. J. Obstet. Gynecol. 2013, 209, e1–e9. [Google Scholar] [CrossRef]
- Liu, L.; Matthews, S.G. Maternal glucocorticoid treatment programs HPA regulation in adult offspring: Sex-specific effects. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E729–E739. [Google Scholar] [CrossRef]
- Moisiadis, V.G.; Mouratidis, A.; Kostaki, A.; Matthews, S.G. A single course of synthetic glucocorticoids in pregnant guinea pigs programs behaviour and stress response in two generations of offspring. Endocrinology 2018, 159, 4065–4076. [Google Scholar] [CrossRef]
- Buchwald, U.; Teupser, D.; Kuehnel, F.; Grohmann, J.; Schmieder, N.; Beindirff, N.; Schlumbohm, C.; Fuhrmann, H.; Einspanier, A. Prenatal stress programs lipid metabolism in the female F1, F2, and F3 generation in the primate model common marmoset (Callithrix jacchus). J. Med. Primatol. 2012, 41, 231–240. [Google Scholar] [CrossRef]
- Moisiadis, V.G.; Constantinof, A.; Kostaki, A.; Szyf, M.; Matthews, S.G. Prenatal glucocorticoid exposure modifies endocrine function and behaviour for 3 generations following maternal or paternal transmission. Sci. Rep. 2017, 7, 11814. [Google Scholar] [CrossRef]
- Sasaki, A.; Eng, M.E.; Lee, A.H.; Kastaki, A.; Matthews, S.G. DNA methylome signatures of prenatal exposure to synthetic glucocorticoids in hippocampus and peripheral whole blood of female guinea pigs in early life. Transl. Psychiatry 2021, 11, 63. [Google Scholar] [CrossRef]
- Constantinof, A.; Boureau, L.; Moisiadis, V.G.; Kostaki, A.; Szyf, M.; Matthews, S.G. Prenatal glucocorticoid exposure results in changes in gene transcription and DNA methylation in the female juvenile guinea pig hippocampus across three generations. Sci. Rep. 2019, 9, 18211. [Google Scholar] [CrossRef]
- Baxter, F.A.; Drake, A.J. Non-genomic inheritance via the male germline in mammals. Philo. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180118. [Google Scholar] [CrossRef] [PubMed]
- Szyl, M. Perinatal stress and epigenetics. Handb. Clin. Neurol. 2021, 180, 125–148. [Google Scholar]
- Jobe, A.H. Antenatal corticosteroids—A concern for lifelong outcomes. J. Pediatr. 2020, 217, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Dagklis, T.; Sen, C.; Tsakiridis, I.; Villalain, C.; Allegaert, K.; Wellman, S.; Kusada, S.; Serra, B.; Luna, M.S.; Huertas, E.; et al. The use of antenatalk corticosteroid for fetal maturation: Clinical practice guidelines by the WAPM—World Association of Perinatal Medicine- and the PMF- Perinatal Medical Foundation. J. Pernatal Med. 2022, 50, 375–385. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
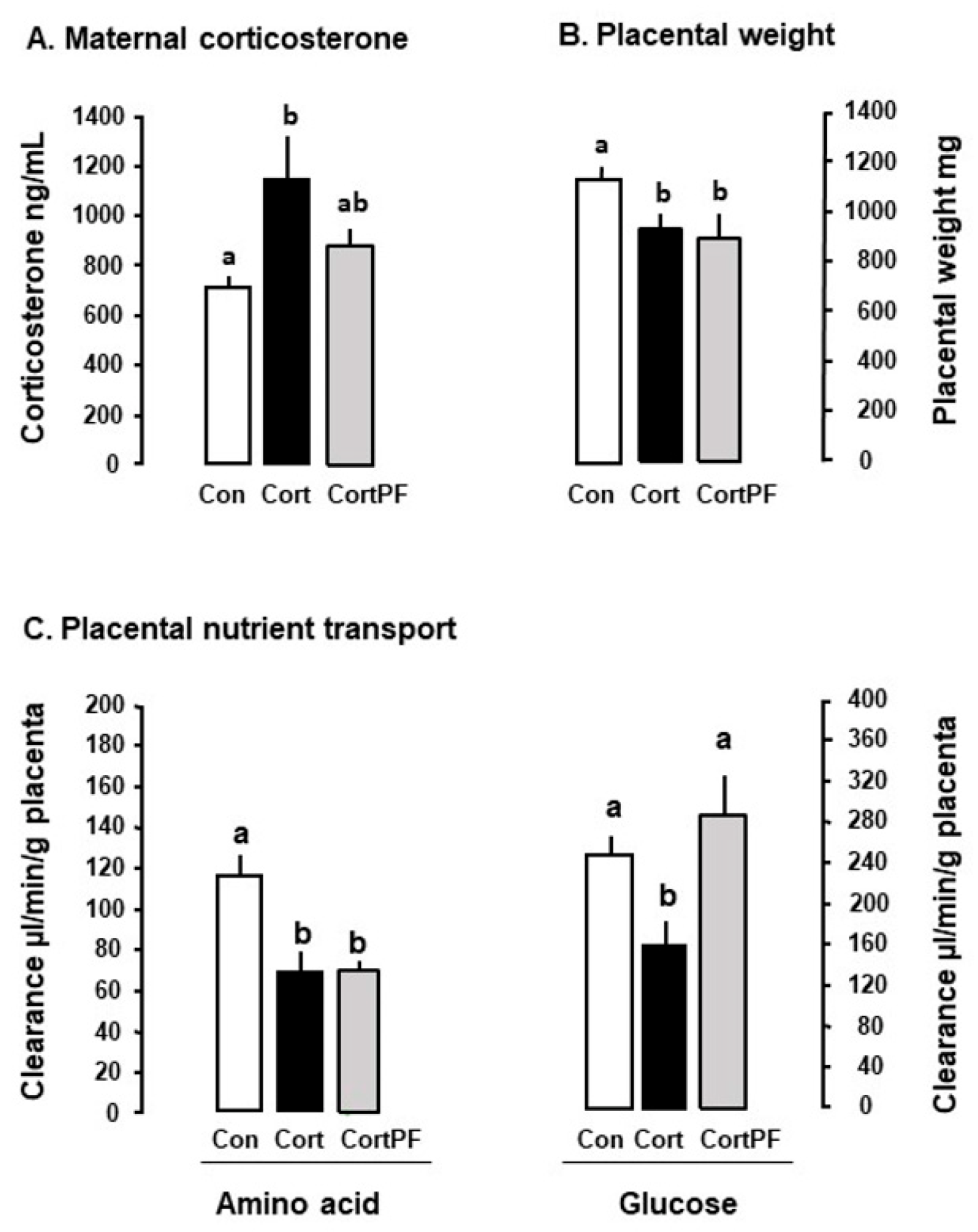{kind=link}
| Treatment | Species | Period of Treatment | Gestational Age at Study | Metabolic Effects | Reference |
|---|---|---|---|---|---|
| Maternal Treatments | |||||
| Synthetic Glucocorticoids | |||||
| Dexamethasone | Mouse | 14 and 15 day sc | 19 day | ↓ Placental MeAIB transport No Δ placental Slc38 a1, Slc38 a2 or Slc38 a4 | [38] |
| 11–16 days po | 19 day | ↑ Placental MeAIB clearance ↓ Slc38 a2 | [39] | ||
| 11–16 days sc | 19 day | No Δ placental MeAIB clearance ↓ Slc38 a2 | |||
| 14–19 days po | 19 day | No Δ placental MeAIB clearance Maternal hyperglycemia | |||
| 14–19 days sc | 19 day | No Δ placental MeAIB clearance Maternal hyperglycemia | |||
| 16 day sc | 17 day | ↓ Placental glycogen ↑ Fetal hepatic glycogen content | [40] | ||
| 17 day ip | 18 day | Fetal heart: ↓ fatty acid translocase ↓ fatty acid oxidation genes ↓ PGC1α (regulator of mitochondrial biogenesis) No Δ mitochondrial morphology | [41] | ||
| Spiny mouse | 21 day sc | 23 day 37 day | ↓ placental GLUT1 expression Sex-linked ↓ placental glycogen Females: ↑ GLUT1 Males: ↓ GLUT1 | [42,43] | |
| Rat | 13–21 days po | 22 day | Fetal skeletal muscle: ↓ protein content ↓ protein synthetic rate, Fetal Liver: ↑ tyrosine aminotransferase expression | [44] | |
| 14 and 15 day im | 16 day | Fetal Brain: ↑ ADP stimulated mitochondrial respiration | [45] | ||
| 16, 19 and 21 day sc | 22 day | ↑ Fetal hepatic and cardiac glycogen | [46] | ||
| 2 × daily 18–19 days sc | 21 day | Fetal heart: ↑ ATP synthase activity, ↑ ATP content, ↑ glycolysis enzymes, ↑ pyruvate content | [47] | ||
| 2 × daily 18–20 days sc | 20 day | No Δ placental transport No Δ fetal uptake of glucose | [48] | ||
| 9–20 days sc | 20 day | Males: ↓ cholesterol, amino acid and TG transporters Females: ↑ cholesterol, TG, amino acid and glucose transporters | [49] | ||
| 9–20 days sc | 20 day | ↓ fetal insulin concentration, ↓ pancreatic β cells ↓ β cell sensitivity to glucose | [50] | ||
| 15–20 days sc | 20 day | ↑ Fetal hepatic glycogen, G6 Pase and PEPCK | [51] | ||
| 15–21 days sc Infusion | 21 day | ↑ Placental GLUT1 and GLUT3 expression | [52] | ||
| Sheep | 125 and 126 days im | 127 day | Fetal skeletal muscle ↑ GLUT4. Δ No muscle glycogen content | [53] | |
| 125 and 126 days im | 127 day | ↑ Fetal and maternal hyperglycemia ↑ hepatic glycogen and G6 Pase, No Δ hepatic PEPCK | [54] | ||
| 106–107 days im 4 doses | 107 day | Maternal and fetal hyperinsulinemia, Fetal hyperglycemia. ↑ Fetal muscle specific GLUT1 and GLUT4 | [55] | ||
| 138 day im | 140 day | Fetal perirenal adipose tissue: ↑ UCP1 and 2 mRNA, VDAC and cytochrome c proteins | [56] | ||
| Betamethasone | Sheep | 104, 111 and 118 days im | 125 day | Decreased fetal insulin concentration with fetal normoglycemia | [57] |
| 121 day im | 122 day | Fetal heart: ↓ PGC1α mRNA (regulator of mitochondrial biogenesis) | [41] | ||
| Triamcinolone-acetonide | Rat | 16 day ip | 21 day | Initial maternal hyperglycemia then hypoglycemia ↓ placental GLUT1 and GLUT3 mRNA and protein | [58] |
| Natural Glucocorticoids | |||||
| Corticosterone | Mouse | 11–16 days po | 16 day | ↑ Placental MeAIB accumulation, ↑ Slc38 a1 and Slc38 a2 mRNA ↑ GLUT1 and GLUT3 mRNA | [14] |
| 11–16 days po | 19 day | ↑ Placental MeAIB clearance, ↑ Slc38 a1 | |||
| 14–19 days po | 19 day | ↓ Placental MeAIB clearance | |||
| Maternal hyperinsulinemia ↓ Placental glucose clearance. No Δ GLUT transporters | [59] | ||||
| 13–15 days minipump | 15 day | ↓ Placental mitochondrial DNA and Complex III sex-linked | [60] | ||
| Cortisol | Sheep | 115–130 days iv infusion | 130 day | ↑ Maternal and fetal lactate concentrations ↑ fetal urea concentrations No Δ α-amino acid concentrations | [61] |
| 125–130 days iv Infusion | 130 day | Maternal hyperglycemia Maternal hyperinsulinemia ↑ Maternal lactate concentration ↓ umbilical glucose uptake ↑ uteroplacental lactate production ↑ increased feto-placental glucose clearance ↑ Placental GLUT8 mRNA ↑ Fetal hepatic lactate dehydrogenase activity No Δ uteroplacental or fetal oxygen consumption | [62] | ||
| 115–140 days iv Infusion | 140 day | Fetal cardiac muscle: ↓ mitochondrial DNA | [63] | ||
| Altered placental metabolism of glutamate, branched chain amino acids and glycerophospholipids | [64] | ||||
| Fetal muscle specific ↓ mitochondrial DNA and metabolism, ↓ cytochrome c ↓ GLUT4 protein, ↓ insulin sensitivity | [65] | ||||
| Fetal treatments | |||||
| Synthetic Glucocorticoids | |||||
| Dexamethasone | Sheep | 126 day iv infusion | 127 day | ↑ fetal gluconeogenic amino acid levels ↓ umbilical alanine uptake ↑ umbilical lactate uptake No Δ umbilical glucose uptake No Δ umbilical oxygen uptake ↓ Placental glutamate uptake from fetal circulation Fetal liver: ↓ Glutamine and gluconeogenic amino acid uptake, ↓ Glutamate output | [66] |
| 130 day iv infusion | 131 day | Fetal and maternal hyperglycemia ↑ fetal gluconeogenic amino acid levels ↓ Placental glutamate uptake from fetal circulation ↓ Umbilical glucose uptake ↓ Glucose/oxygen quotient | [67] | ||
| Natural Glucocorticoids | |||||
| Cortisol | Sheep | 121 day iv | 121 day +6 h | ↑ Fetal proteolysis, ↓ protein accretion, ↑ Fetal leucine oxidation No Δ umbilical glucose or lactate uptake ↓ Umbilical α-amino nitrogen uptake | [68,69] |
| 122–125 days iv infusion | 125 day | ↑ Fetal hepatic glycogen content | [22] | ||
| 125–130 days iv infusion | 130 day | Fetal hepatic G6 Pase, PEPCK, FDP and aminotransferases | [70] | ||
| ↓ Umbilical glucose uptake No Δ uteroplacental or fetal oxygen consumption No Δ uteroplacental production of lactate ↓ Umbilical uptake of lactate | [71] | ||||
| ↓ Umbilical glucose uptake ↑ Uteroplacental glucose utilization No Δ umbilical α-amino nitrogen uptake ↑ Hepatic glycogen deposition ↑ Hepatic mitochondrial pyruvate carboxylase mRNA | [72] | ||||
| ↑ UCP1 and UCP2 mRNA in fetal adipose tissue | [73] | ||||
| Selective fetal skeletal muscles: ↑ mitochondrial density and OXPHOS capacity ↑ Complex I protein ↑ Adenine nucleotide transporter | [74] | ||||
| 128–130 days iv infusion | 130 day | Fetal cardiac muscle: ↓ GLUT1 | [75] | ||
| 138 d iv infusion | 138 day +6 h | ↑ Hepatic gluconeogenesis from lactate | [76] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fowden, A.L.; Vaughan, O.R.; Murray, A.J.; Forhead, A.J. Metabolic Consequences of Glucocorticoid Exposure before Birth. Nutrients 2022, 14, 2304. https://doi.org/10.3390/nu14112304
Fowden AL, Vaughan OR, Murray AJ, Forhead AJ. Metabolic Consequences of Glucocorticoid Exposure before Birth. Nutrients. 2022; 14(11):2304. https://doi.org/10.3390/nu14112304
Chicago/Turabian StyleFowden, Abigail L., Owen R. Vaughan, Andrew J. Murray, and Alison J. Forhead. 2022. "Metabolic Consequences of Glucocorticoid Exposure before Birth" Nutrients 14, no. 11: 2304. https://doi.org/10.3390/nu14112304
APA StyleFowden, A. L., Vaughan, O. R., Murray, A. J., & Forhead, A. J. (2022). Metabolic Consequences of Glucocorticoid Exposure before Birth. Nutrients, 14(11), 2304. https://doi.org/10.3390/nu14112304