
Impact of Newborn Screening and Early Dietary Management on Clinical Outcome of Patients with Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency and Medium Chain Acyl-CoA Dehydrogenase Deficiency—A Retrospective Nationwide Study

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Methods
2.2.1. LCHADD/MTPD Acute Events
2.2.2. LCHADD/MTPD Chronic Complications
2.2.3. MCADD
2.2.4. Statistical Analysis
3. Results
3.1. LCHADD/MTPD and MCADD Demographic Data
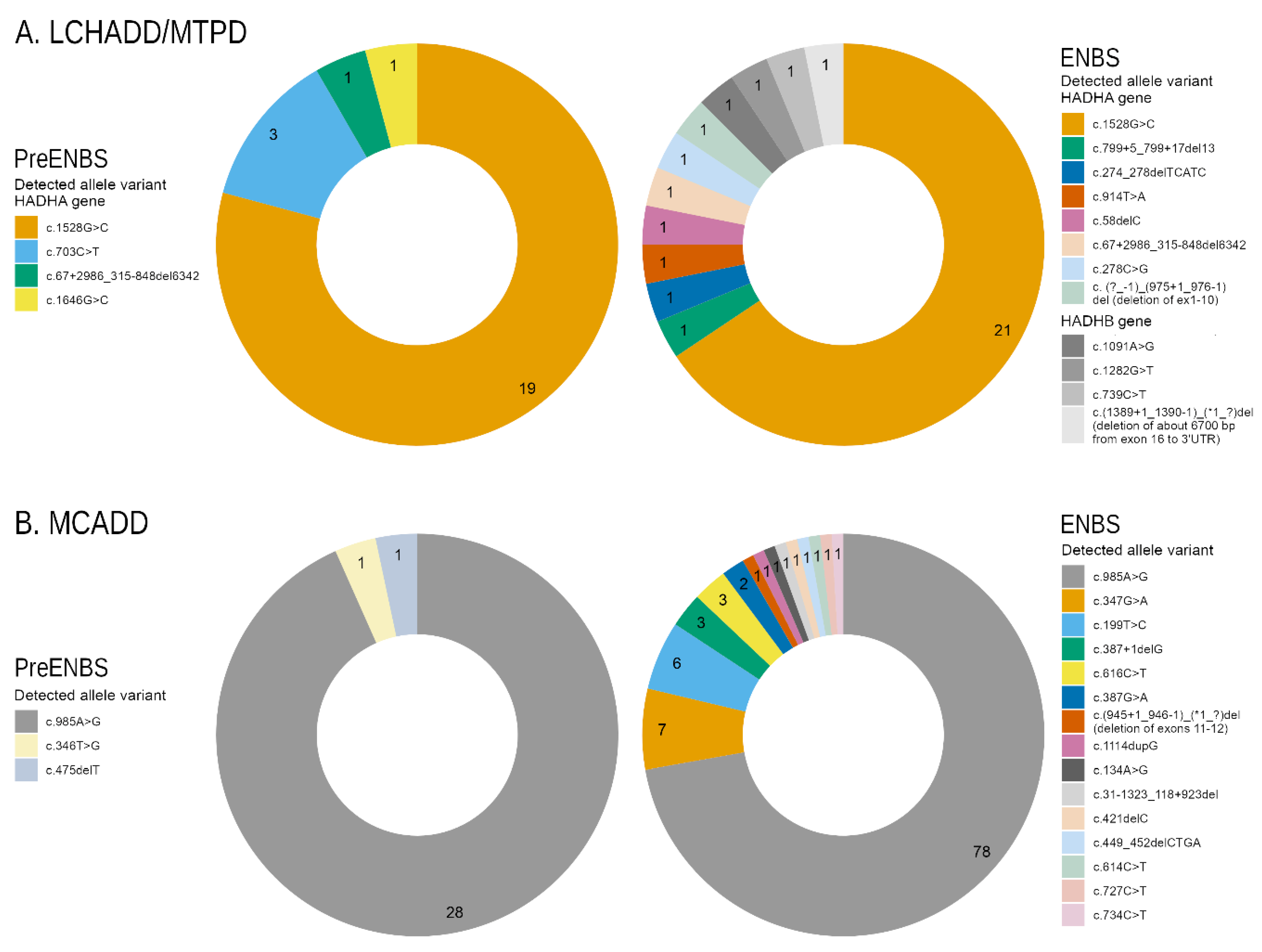
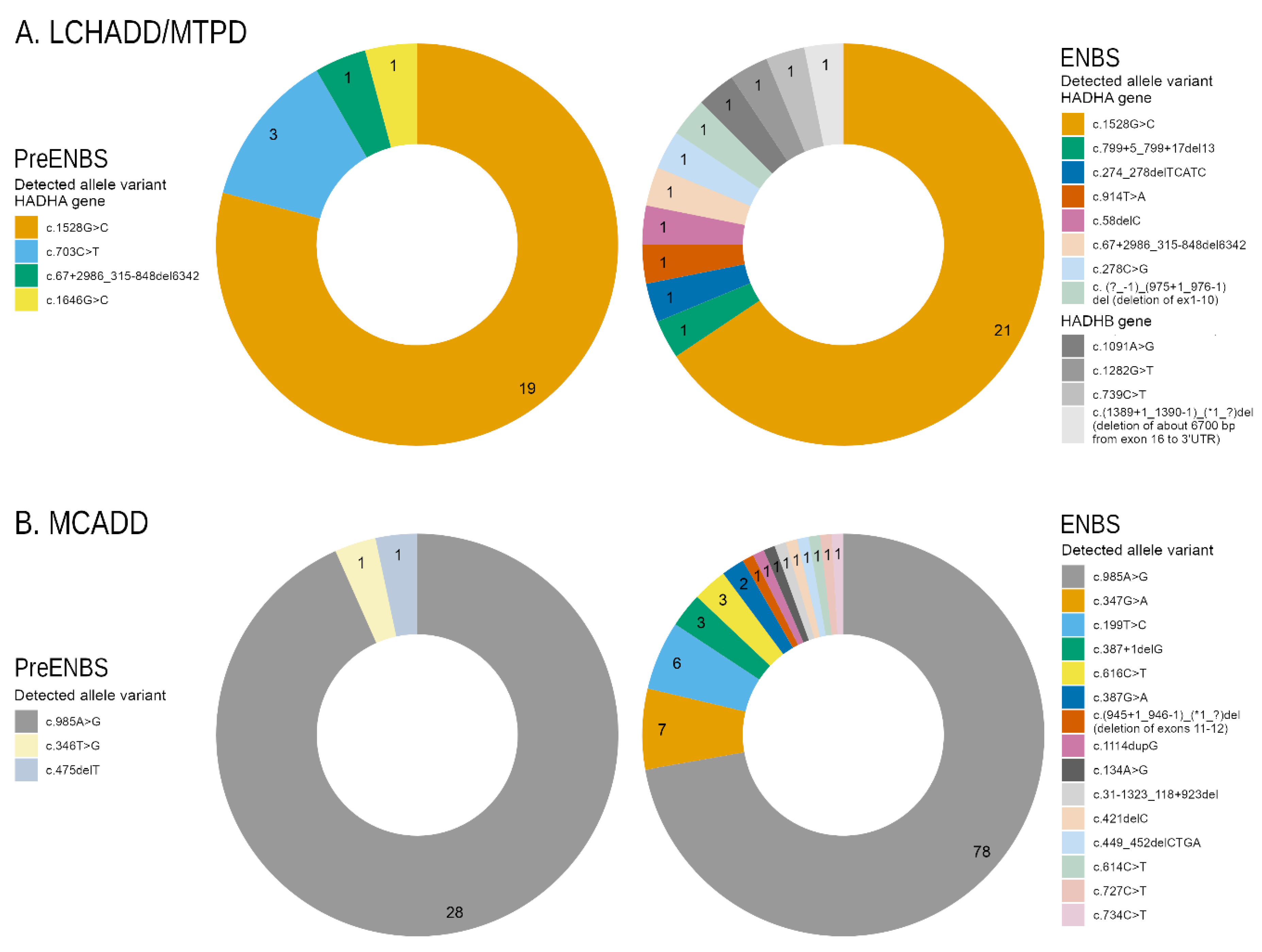
3.2. LCHADD/MTPD and MCADD Genotypes
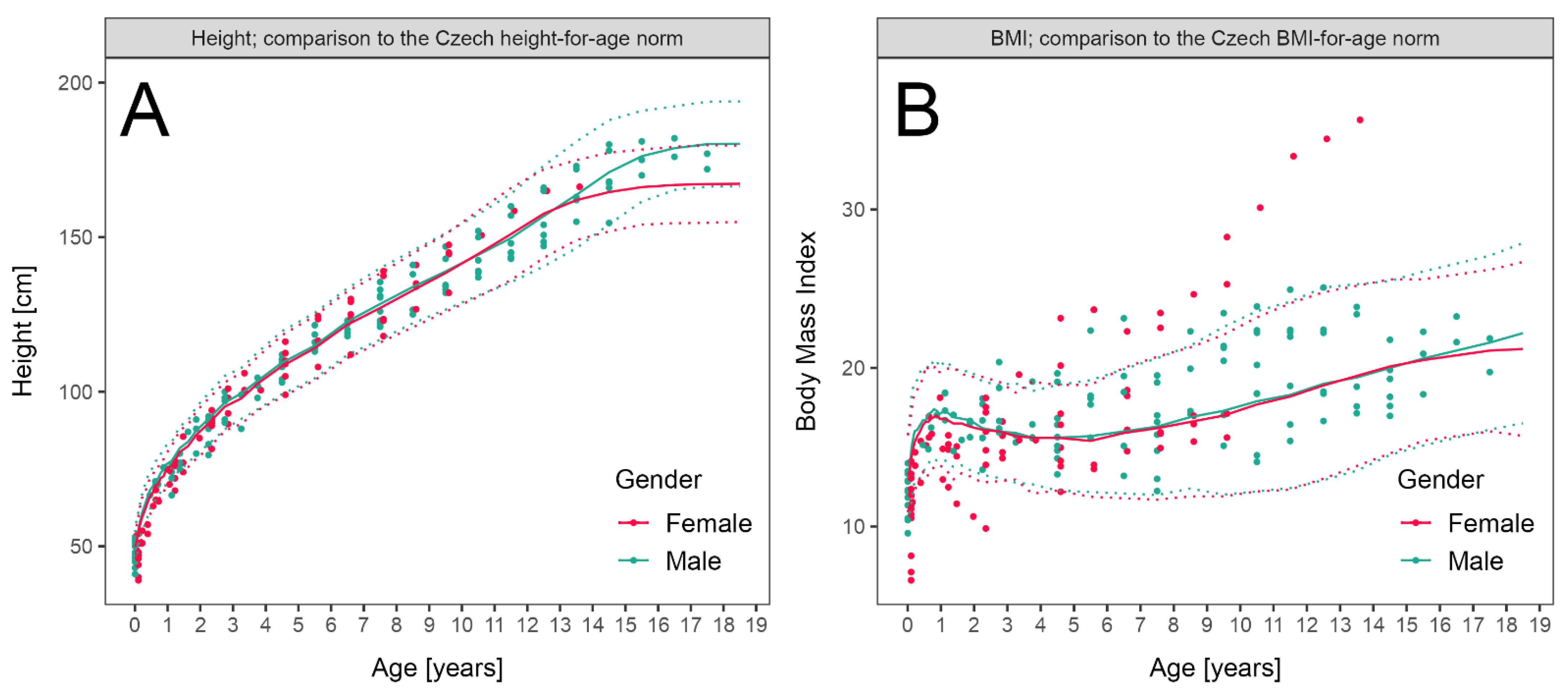
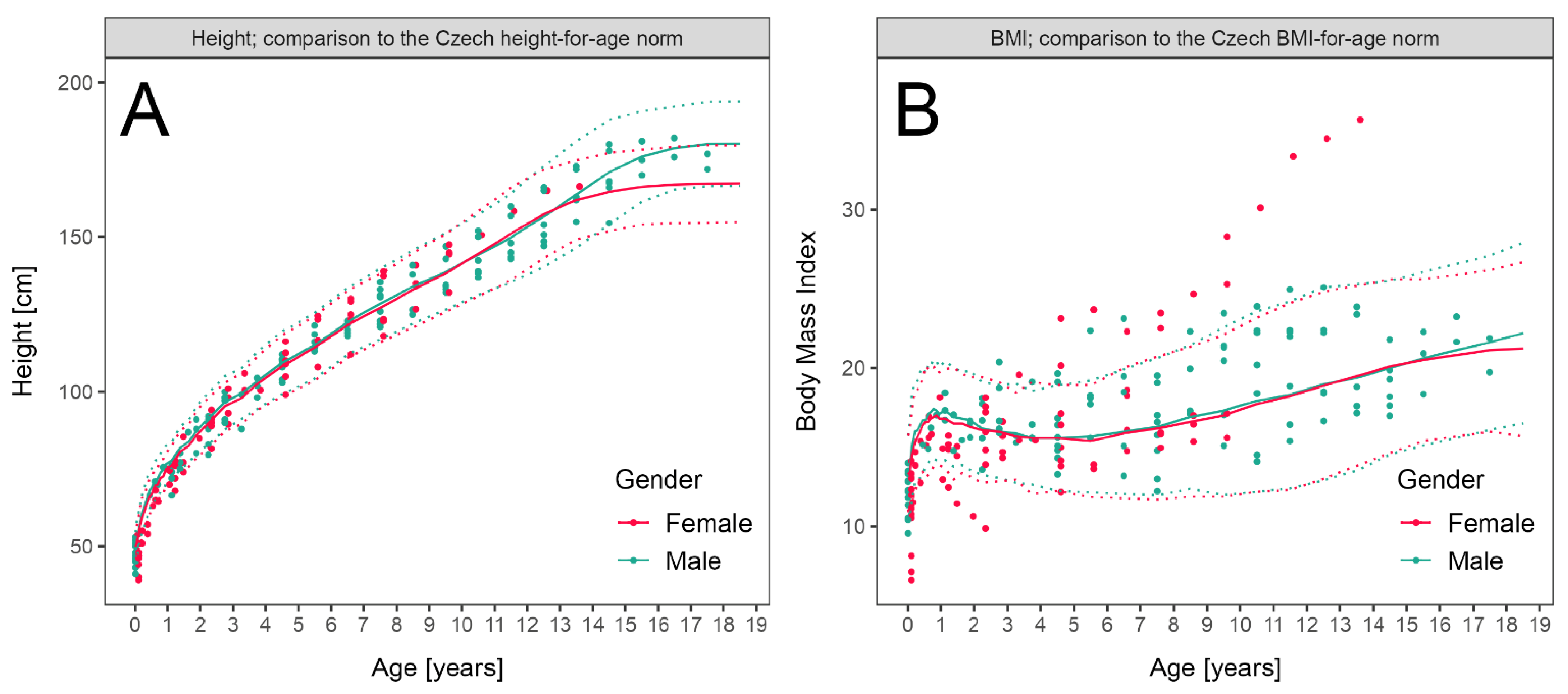
3.3. LCHADD/MTPD Anthropometric Parameters
3.4. LCHADD/MTPD and MCADD Clinical Outcome
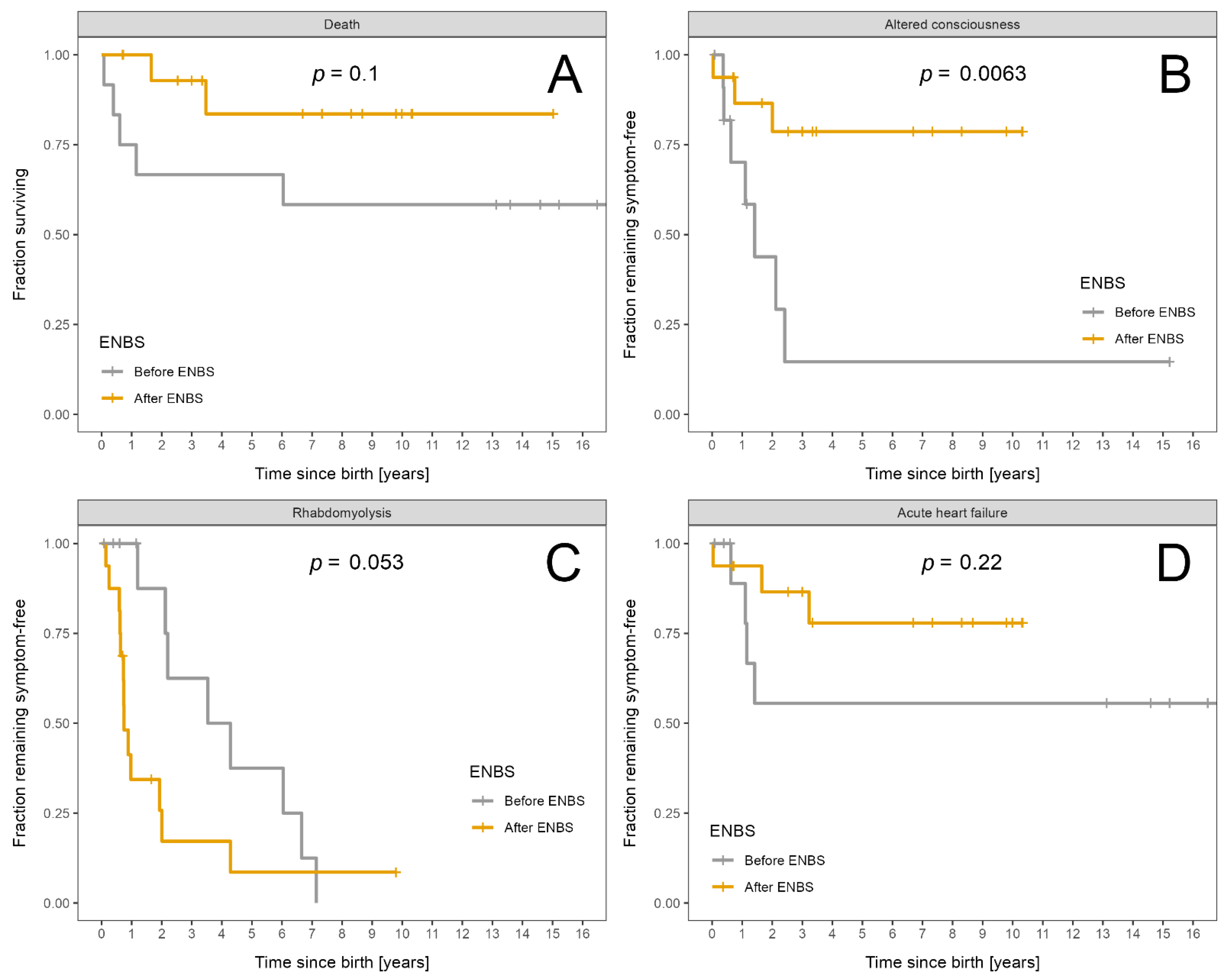
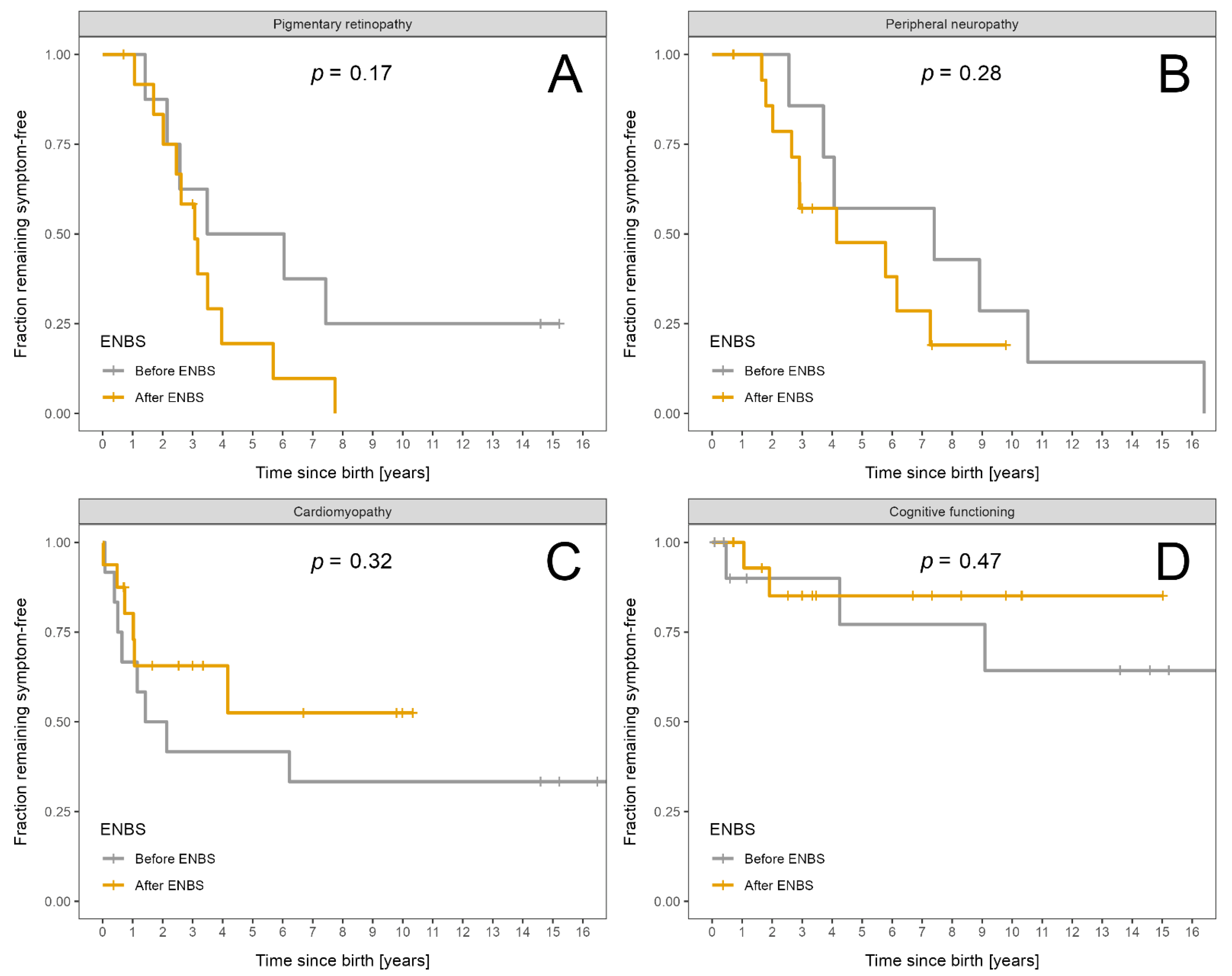
3.4.1. LCHADD/MTPD
3.4.2. MCADD
3.5. Relationship between Acute Events and Chronic Complications
3.6. Genotype-Phenotype Correlation
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merritt, J.L.; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef]
- Wajner, M.; Amaral, A.U. Mitochondrial dysfunction in fatty acid oxidation disorders: Insights from human and animal studies. Biosci. Rep. 2015, 36, e00281. [Google Scholar] [CrossRef] [Green Version]
- Hickmann, F.H.; Cecatto, C.; Kleemann, D.; Monteiro, W.O.; Castilho, R.F.; Amaral, A.U.; Wajner, M. Uncoupling, metabolic inhibition and induction of mitochondrial permeability transition in rat liver mitochondria caused by the major long-chain hydroxyl monocarboxylic fatty acids accumulating in LCHAD deficiency. Biochim. Biophys. Acta 2015, 1847, 620–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derks, T.G.; Reijngoud, D.J.; Waterham, H.R.; Gerver, W.J.; van den Berg, M.P.; Sauer, P.J.; Smit, G.P. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: Clinical presentation and outcome. J. Pediatr. 2006, 148, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Sperk, A.; Mueller, M.; Spiekerkoetter, U. Outcome in six patients with mitochondrial trifunctional protein disorders identified by newborn screening. Mol. Genet. Metab. 2010, 101, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U. Mitochondrial fatty acid oxidation disorders: Clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J. Inherit. Metab. Dis. 2010, 33, 527–532. [Google Scholar] [CrossRef]
- Tyni, T.; Kivelä, T.; Lappi, M.; Summanen, P.; Nikoskelainen, E.; Pihko, H. Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: A new type of hereditary metabolic chorioretinopathy. Ophthalmology 1998, 105, 810–824. [Google Scholar] [CrossRef]
- Immonen, T.; Turanlahti, M.; Paganus, A.; Keskinen, P.; Tyni, T.; Lapatto, R. Earlier diagnosis and strict diets improve the survival rate and clinical course of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 2016, 105, 549–554. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Lindner, M.; Santer, R.; Grotzke, M.; Baumgartner, M.R.; Boehles, H.; Das, A.; Haase, C.; Hennermann, J.B.; Karall, D.; et al. Treatment recommendations in long-chain fatty acid oxidation defects: Consensus from a workshop. J. Inherit. Metab. Dis. 2009, 32, 498–505. [Google Scholar] [CrossRef]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Chaplin, M.; Black, C.; Fletcher, J.; McGill, J.; Boneh, A. Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: A cohort study. Lancet 2007, 369, 37–42. [Google Scholar] [CrossRef]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Bowling, F.; Carpenter, K.; Christodoulou, J.; Cowley, D.; Ellaway, C.; Fletcher, J.; et al. Expanded newborn screening: Outcome in screened and unscreened patients at age 6 years. Pediatrics 2009, 124, e241–e248. [Google Scholar] [CrossRef]
- Janeiro, P.; Jotta, R.; Ramos, R.; Florindo, C.; Ventura, F.V.; Vilarinho, L.; Tavares de Almeida, I.; Gaspar, A. Follow-up of fatty acid β-oxidation disorders in expanded newborn screening era. Eur. J. Pediatr. 2019, 178, 387–394. [Google Scholar] [CrossRef]
- Landau, Y.E.; Waisbren, S.E.; Chan, L.M.; Levy, H.L. Long-term outcome of expanded newborn screening at Boston children’s hospital: Benefits and challenges in defining true disease. J. Inherit. Metab. Dis. 2017, 40, 209–218. [Google Scholar] [CrossRef]
- Fraser, H.; Geppert, J.; Johnson, R.; Johnson, S.; Connock, M.; Clarke, A.; Taylor-Phillips, S.; Stinton, C. Evaluation of earlier versus later dietary management in long-chain 3-hydroxyacyl-CoA dehydrogenase or mitochondrial trifunctional protein deficiency: A systematic review. Orphanet. J. Rare Dis. 2019, 14, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Burton, B.; Berry, G.T.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; Bowden, A.; et al. Results from a 78-week, single-arm, open-label phase 2 study to evaluate UX007 in pediatric and adult patients with severe long-chain fatty acid oxidation disorders (LC-FAOD). J. Inherit. Metab. Dis. 2019, 42, 169–177. [Google Scholar] [CrossRef]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.A.; McCracken, E.; Conway, R.; Enns, G.M.; Starr, J.; Wang, R.; et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef]
- Vockley, J.; Marsden, D.; McCracken, E.; DeWard, S.; Barone, A.; Hsu, K.; Kakkis, E. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment--A retrospective chart review. Mol. Genet. Metab. 2015, 116, 53–60. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Heitner, S.B.; Martin, J.; Rose, S.; Goldstein, A.; El-Gharbawy, A.H.; Deward, S.; Lasarev, M.R.; Pollaro, J.; DeLany, J.P.; et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial. J. Inherit. Metab. Dis. 2017, 40, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Kobzova, J.; Vignerova, J.; Blaha, P.; Krejcovsky, L.; Riedlova, J. The 6th nationwide anthropological survey of children and adolescents in the Czech Republic in 2001. Cent. Eur. J. Public Health 2004, 12, 126–130. [Google Scholar] [PubMed]
- Stahl, K.; Rastelli, E.; Schoser, B. A systematic review on the definition of rhabdomyolysis. J. Neurol. 2020, 267, 877–882. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Cantinotti, M.; Scalese, M.; Murzi, B.; Assanta, N.; Spadoni, I.; Festa, P.; De Lucia, V.; Crocetti, M.; Marotta, M.; Molinaro, S.; et al. Echocardiographic nomograms for ventricular, valvular and arterial dimensions in caucasian children with a special focus on neonates, infants and toddlers. J. Am. Soc. Echocardiogr. 2014, 27, 179–191.e2. [Google Scholar] [CrossRef]
- Michot, C.; Hubert, L.; Romero, N.B.; Gouda, A.; Mamoune, A.; Mathew, S.; Kirk, E.; Viollet, L.; Rahman, S.; Bekri, S.; et al. Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise-induced myalgia. J. Inherit. Metab. Dis. 2012, 35, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Janković, S.R.; Stosić, J.J.; Vucinić, S.; Vukcević, N.P.; Ercegović, G.V. Causes of rhabdomyolysis in acute poisonings. Vojnosanit. Pregl. 2013, 70, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G.M.; Zeng, X.; Waikar, S.S. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern. Med. 2013, 173, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Haglind, C.B.; Stenlid, M.H.; Ask, S.; Alm, J.; Nemeth, A.; Döbeln, U.; Nordenström, A. Growth in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. JIMD Rep. 2013, 8, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguolo, A.; Rodella, G.; Dianin, A.; Nurti, R.; Monge, I.; Rigotti, E.; Cantalupo, G.; Salviati, L.; Tucci, S.; Pellegrini, F.; et al. Diagnosis, genetic characterization and clinical follow up of mitochondrial fatty acid oxidation disorders in the new era of expanded newborn screening: A single centre experience. Mol. Genet. Metab. Rep. 2020, 24, 100632. [Google Scholar] [CrossRef]
- Sturm, M.; Herebian, D.; Mueller, M.; Laryea, M.D.; Spiekerkoetter, U. Functional Effects of Different Medium-Chain Acyl-CoA Dehydrogenase Genotypes and Identification of Asymptomatic Variants. PLoS ONE 2012, 7, e45110. [Google Scholar] [CrossRef]
- Gramer, G.; Haege, G.; Fang-Hoffmann, J. Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Evaluation of Genotype-Phenotype Correlation in Patients Detected by Newborn Screening. JIMD Rep. 2015, 23, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Sykut-Cegielska, J.; Gradowska, W.; Piekutowska-Abramczuk, D.; Andresen, B.S.; Olsen, R.K.; Ołtarzewski, M.; Pronicki, M.; Pajdowska, M.; Bogdańska, A.; Jabłońska, E.; et al. Urgent metabolic service improves survival in long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency detected by symptomatic identification and pilot newborn screening. J. Inherit. Metab. Dis. 2011, 34, 185–195. [Google Scholar] [CrossRef]
- Kang, E.; Kim, Y.M.; Kang, M.; Heo, S.H.; Kim, G.H.; Choi, I.H.; Choi, J.H.; Yoo, H.W.; Lee, B.H. Clinical and genetic characteristics of patients with fatty acid oxidation disorders identified by newborn screening. BMC Pediatr. 2018, 18, 103. [Google Scholar] [CrossRef] [Green Version]
- Fahnehjelm, K.T.; Liu, Y.; Olsson, D.; Amrén, U.; Haglind, C.B.; Holmström, G.; Halldin, M.; Andreasson, S.; Nordenström, A. Most patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency develop pathological or subnormal retinal function. Acta Paediatr. 2016, 105, 1451–1460. [Google Scholar] [CrossRef]
- Baruteau, J.; Sachs, P.; Broué, P.; Brivet, M.; Abdoul, H.; Vianey-Saban, C.; de Baulny, O.H. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: A French pediatric study from 187 patients. Complementary data. J. Inherit. Metab. Dis. 2014, 37, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Boese, E.A.; Jain, N.; Jia, Y.; Schlechter, C.L.; Harding, C.O.; Gao, S.S.; Patel, R.C.; Huang, D.; Weleber, R.G.; Gillingham, M.B.; et al. Characterization of Chorioretinopathy Associated with Mitochondrial Trifunctional Protein Disorders: Long-Term Follow-up of 21 Cases. Ophthalmology 2016, 123, 2183–2195. [Google Scholar] [CrossRef] [Green Version]
- Karall, D.; Brunner-Krainz, M.; Kogelnig, K.; Konstantopoulou, V.; Maier, E.M.; Möslinger, D.; Plecko, B.; Sperl, W.; Volkmar, B.; Scholl-Bürgi, S. Clinical outcome, biochemical and therapeutic follow-up in 14 Austrian patients with Long-Chain 3-Hydroxy Acyl CoA Dehydrogenase Deficiency (LCHADD). Orphanet. J. Rare Dis. 2015, 10, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biase, I.; Viau, K.S.; Liu, A.; Yuzyuk, T.; Botto, L.D.; Pasquali, M.; Longo, N. Diagnosis, Treatment, and Clinical Outcome of Patients with Mitochondrial Trifunctional Protein/Long-Chain 3-Hydroxy Acyl-CoA Dehydrogenase Deficiency. JIMD Rep. 2017, 31, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulz, S.; Atiskova, Y.; Engel, P.; Wildner, J.; Tsiakas, K.; Santer, R. Retained visual function in a subset of patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). Ophthalmic Genet. 2021, 42, 23–27. [Google Scholar] [CrossRef]
- Immonen, T.; Ahola, E.; Toppila, J.; Lapatto, R.; Tyni, T.; Lauronen, L. Peripheral neuropathy in patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency—A follow-up EMG study of 12 patients. Eur. J. Paediatr. Neurol. 2016, 20, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal. Screen 2021, 7, 15. [Google Scholar] [CrossRef]
- Guffon, N.; Mochel, F.; Schiff, M.; De Lonlay, P.; Douillard, C.; Vianey-Saban, C. Clinical outcomes in a series of 18 patients with long chain fatty acids oxidation disorders treated with triheptanoin for a median duration of 22 months. Mol. Genet. Metab. 2021, 132, 227–233. [Google Scholar] [CrossRef]
- Nennstiel-Ratzel, U.; Arenz, S.; Maier, E.M.; Knerr, I.; Baumkötter, J.; Röschinger, W.; Liebl, B.; Hadorn, H.B.; Roscher, A.A.; von Kries, R. Reduced incidence of severe metabolic crisis or death in children with medium chain acyl-CoA dehydrogenase deficiency homozygous for c.985A > G identified by neonatal screening. Mol. Genet. Metab. 2005, 85, 157–159. [Google Scholar] [CrossRef]
- Iafolla, A.K.; Thompson, R.J., Jr.; Roe, C.R. Medium-chain acyl-coenzyme A dehydrogenase deficiency: Clinical course in 120 affected children. J. Pediatr. 1994, 124, 409–415. [Google Scholar] [CrossRef]
- Schatz, U.A.; Ensenauer, R. The clinical manifestation of MCAD deficiency: Challenges towards adulthood in the screened population. J. Inherit. Metab. Dis. 2010, 33, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Pennesi, M.E.; Harding, C.O.; Weleber, R.G.; Gillingham, M.B. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Mol. Genet. Metab. 2012, 106, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentler, K.; Zhai, S.; Elsbecker, S.A.; Arnold, G.L.; Burton, B.K.; Vockley, J.; Cameron, C.A.; Hiner, S.J.; Edick, M.J.; Berry, S.A. Inborn Errors of Metabolism Collaborative. 221 newborn-screened neonates with medium-chain acyl-coenzyme A dehydrogenase deficiency: Findings from the Inborn Errors of Metabolism Collaborative. Mol. Genet. Metab. 2016, 119, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couce, M.L.; Sánchez-Pintos, P.; Diogo, L.; Leão-Teles, E.; Martins, E.; Santos, H.; Bueno, M.A.; Delgado-Pecellín, C.; Castiñeiras, D.E.; Cocho, J.A.; et al. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: Regional experience and high incidence of carnitine deficiency. Orphanet. J. Rare Dis. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LCHADD/MTPD | MCADD | |||||
|---|---|---|---|---|---|---|
| Total | Pre-ENBS | ENBS | Total | Pre-ENBS | ENBS | |
| N (%) | N (%) | |||||
| All patients | 28 (100) | 12 (100) | 16 (100) | 69 (100) | 15 (100) | 54 |
| Male | 15 (53.6) | 9 (75) | 6 (37.5) | 34 (49.3) | 9 (60) | 25 (46.3) |
| Female | 13 (46.4) | 3 (25) | 10 (62.5) | 35 (50.7) | 6 (40) | 29 (53.7) |
| Homozygotes * | 15 (53.6) | 8 (66.7) | 7 (43.8) | 43 (62.3) | 13 (86.7) | 30 (55.6) |
| Compound heterozygotes * | 10 (35.7) | 3 (25) | 7 (43.8) | 20 (29) | 2 (13.3) | 18 (33.3) |
| Other genotypes | 3 (10.7) | 1 (8.3) | 2 (12.5) (both MTPD) | 6 (8.7) | 0 (0) | 6 (11.1) |
| Median (range) | Median (range) | |||||
| Age at diagnosis (months) | 10 (1.2–91) | <0.33 | 1.8 (0.1–17) | <0.33 | ||
| Present age (years) | 13.4 (0.1–27.8) | 7 (0.7–15) | 19.5 (1.2–36.6) | 4.9 (0–12.1) | ||
| Follow-up (years) | 9.3 (0–27.4) | 7.01 (0.7–15.02) | 14.57 (0.25–25.88) | 4.86 (0.23–11.65) | ||
| Follow-up (patient-years) | 228.4 | 126.6 | 101.8 | 565.1 | 275.6 | 289.5 |
| Total | Pre-ENBS | ENBS | p Value | |
|---|---|---|---|---|
| Acute events | N; log-rank test (time to event) | |||
| Patients | 28 | 12 | 16 | |
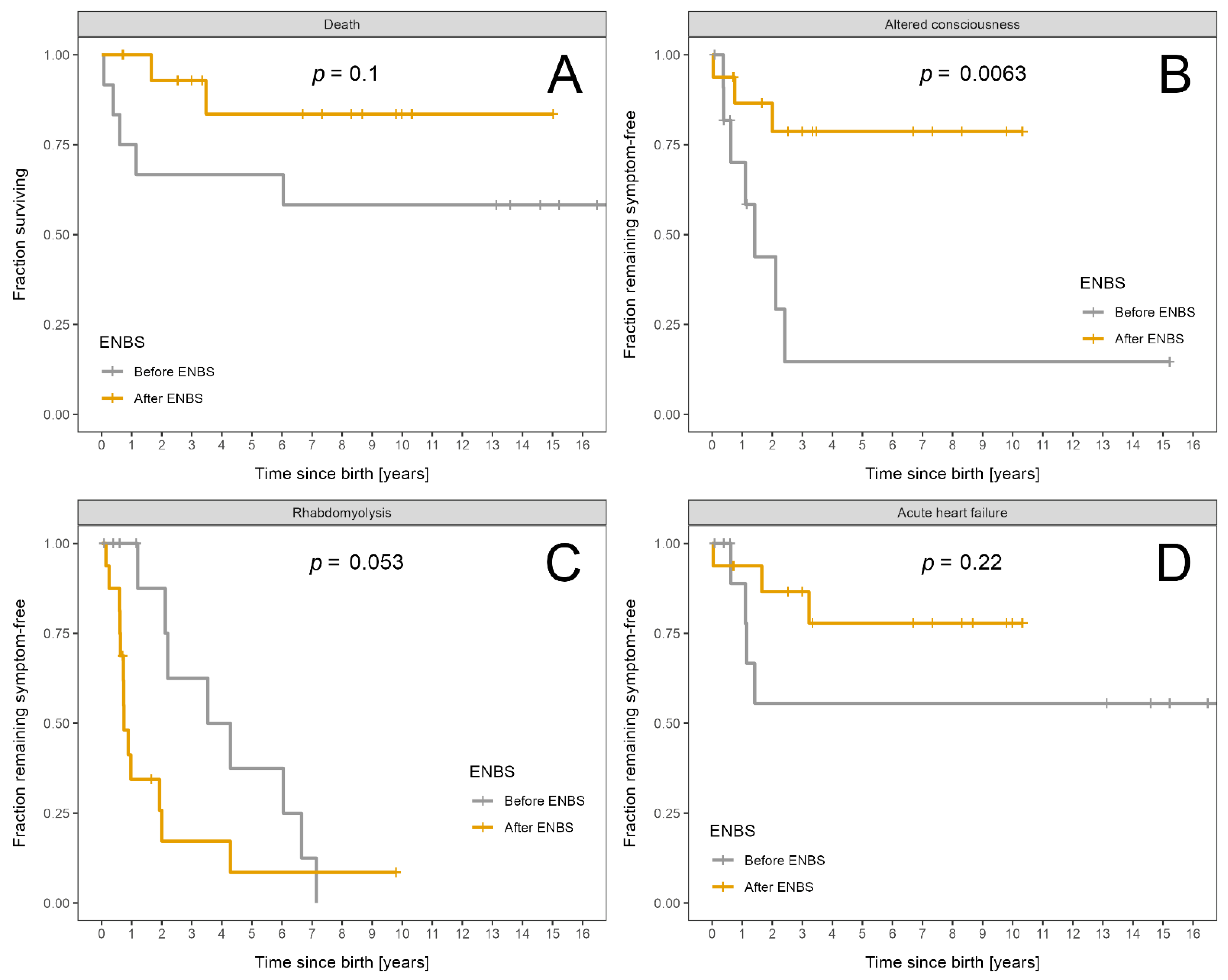
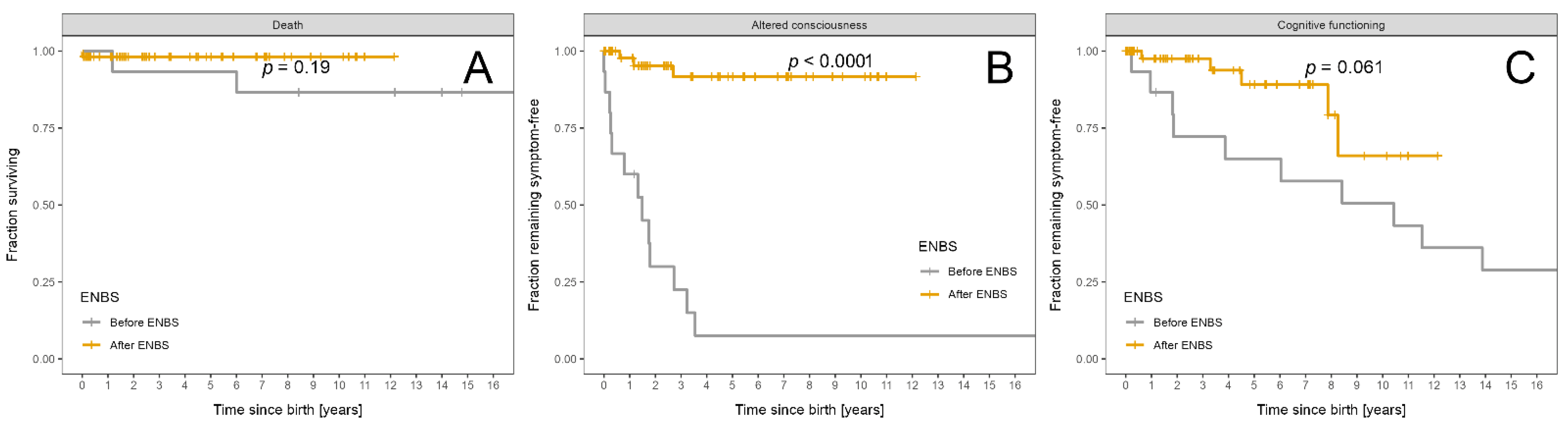
| Death | 7 | 5 | 2 | 0.10 |
| Altered consciousness | 10 | 7 | 3 | 0.006 |
| Rhabdomyolysis | 21 | 8 | 13 | 0.053 |
| Acute heart failure | 7 | 4 | 3 | 0.22 |
| Incidence of acute events | N of events/patient-years; χ2-test | |||
| Death | 7/229 | 5/127 | 2/102 | 0.40 |
| Altered consciousness | 24/229 | 15/127 | 9/102 | 0.51 |
| Rhabdomyolysis | 292/229 | 157/127 | 135/102 | 0.70 |
| Severe and critical rhabdomyolysis only | 37/229 | 13/127 | 24/102 | 0.02 |
| Acute heart failure | 10/229 | 5/127 | 5/102 | 0.73 |
| Severity of acute events | Severity score/patient-years; χ2-test | |||
| Altered consciousness | 46/229 | 29/127 | 17/102 | 0.34 |
| Rhabdomyolysis | 733/229 | 364/127 | 369/102 | 0.13 |
| Severe and critical rhabdomyolysis only | 148/229 | 52/127 | 96/102 | <0.0001 |
| Chronic complications | N | |||
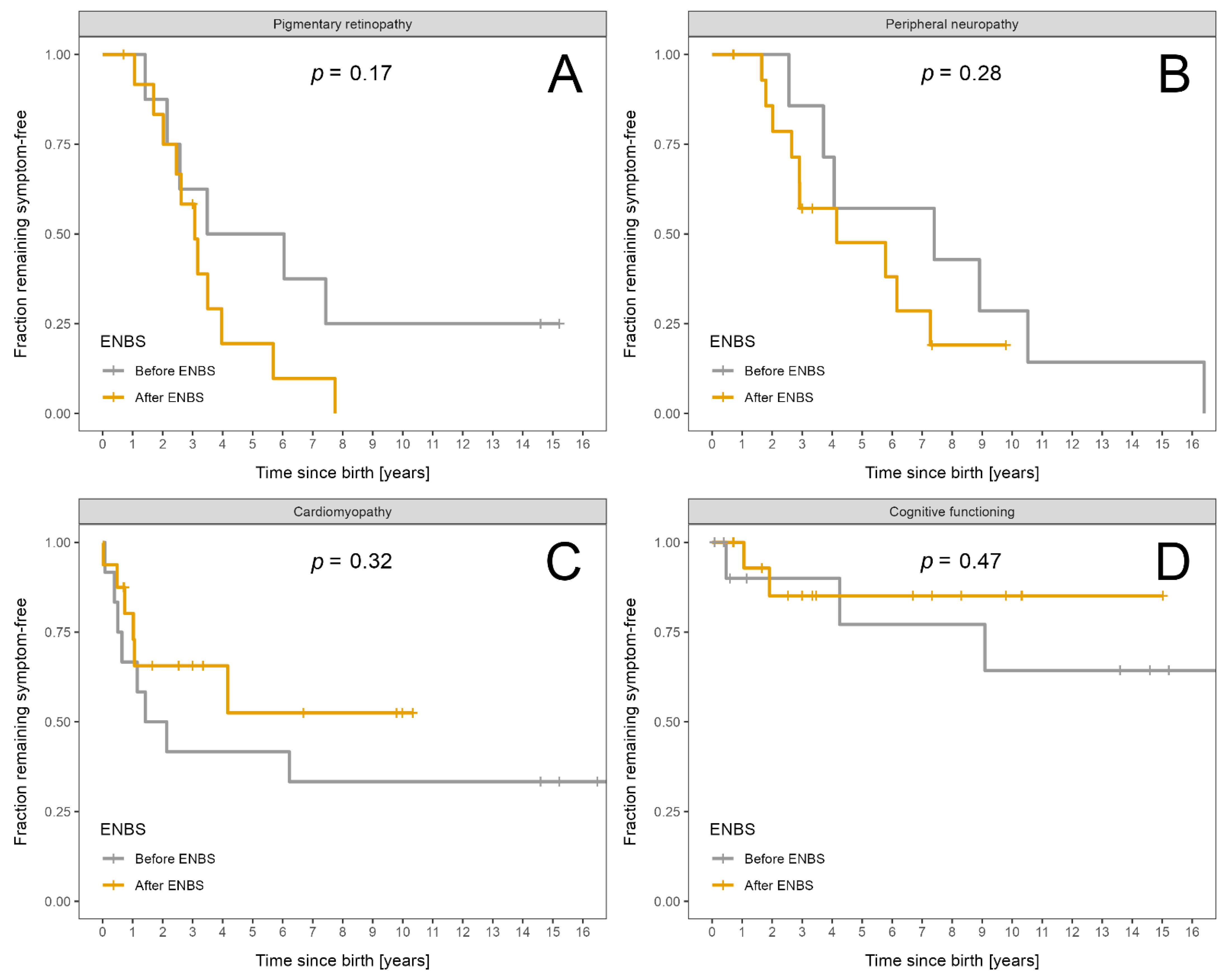
| Retinopathy (patients examined) | 21 | 8 | 13 | |
| Retinopathy | 17 | 6 | 11 | |
| Neuropathy (patients examined) | 23 | 7 | 16 | |
| Neuropathy | 17 | 7 | 10 | |
| Cardiomyopathy (patients examined) | 28 | 12 | 16 | |
| Cardiomyopathy | 14 | 8 | 6 | |
| Intellectual disability (patients examined) | 28 | 12 | 16 | |
| Intellectual impairment | 5 | 3 | 2 | |
| Severity of chronic complications | AUC/patient-year- mean (SD); two-sample t-test | |||
| Retinopathy | 0.53 (0.39) | 0.46 (0.36) | 0.57 (0.42) | 0.53 |
| Neuropathy | 0.53 (0.54) | 0.82 (0.66) | 0.39 (0.42) | 0.07 |
| Cardiomyopathy | 0.22 (0.28) | 0.33 (0.26) | 0.15 (0.27) | 0.09 |
| Intellectual disability | 0.05 (0.14) | 0.05 (0.11) | 0.06 (0.15) | 0.97 |
| Total | Pre-ENBS | Post-ENBS | p Value | |
|---|---|---|---|---|
| Acute events | N; log-rank test (time to event) | |||
| Patients | 69 | 15 | 54 | |
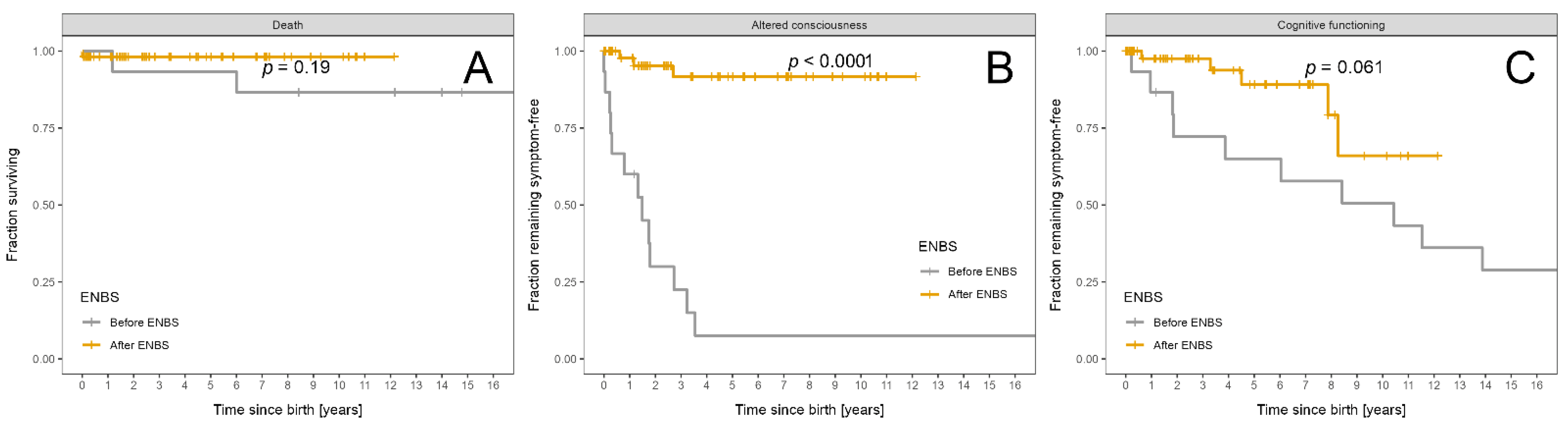
| Death | 3 | 2 | 1 | 0.19 |
| Altered consciousness | 16 | 13 | 3 | <0.0001 |
| Incidence of acute events | N of events/patient-years; χ2-test | |||
| Altered consciousness | 33/565 | 29/276 | 4/289 | <0.0001 |
| Severity of acute events | Severity score/patient-years; χ2-test | |||
| Altered consciousness | 67/565 | 61/276 | 6/289 | <0.0001 |
| Chronic complications | N | |||
| Intellectual disability | 16 | 10 | 6 * | |
| Severity of chronic complications | AUC/patient-year- mean (SD); two-sample t-test | |||
| Intellectual disability | 0.1 (0.3) | 0.4 (0.5) | 0.0 (0.1) | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rücklová, K.; Hrubá, E.; Pavlíková, M.; Hanák, P.; Farolfi, M.; Chrastina, P.; Vlášková, H.; Kousal, B.; Smolka, V.; Foltenová, H.; et al. Impact of Newborn Screening and Early Dietary Management on Clinical Outcome of Patients with Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency and Medium Chain Acyl-CoA Dehydrogenase Deficiency—A Retrospective Nationwide Study. Nutrients 2021, 13, 2925. https://doi.org/10.3390/nu13092925
Rücklová K, Hrubá E, Pavlíková M, Hanák P, Farolfi M, Chrastina P, Vlášková H, Kousal B, Smolka V, Foltenová H, et al. Impact of Newborn Screening and Early Dietary Management on Clinical Outcome of Patients with Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency and Medium Chain Acyl-CoA Dehydrogenase Deficiency—A Retrospective Nationwide Study. Nutrients. 2021; 13(9):2925. https://doi.org/10.3390/nu13092925
Chicago/Turabian StyleRücklová, Kristina, Eva Hrubá, Markéta Pavlíková, Petr Hanák, Martina Farolfi, Petr Chrastina, Hana Vlášková, Bohdan Kousal, Vratislav Smolka, Hana Foltenová, and et al. 2021. "Impact of Newborn Screening and Early Dietary Management on Clinical Outcome of Patients with Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency and Medium Chain Acyl-CoA Dehydrogenase Deficiency—A Retrospective Nationwide Study" Nutrients 13, no. 9: 2925. https://doi.org/10.3390/nu13092925
APA StyleRücklová, K., Hrubá, E., Pavlíková, M., Hanák, P., Farolfi, M., Chrastina, P., Vlášková, H., Kousal, B., Smolka, V., Foltenová, H., Adam, T., Friedecký, D., Ješina, P., Zeman, J., Kožich, V., & Honzík, T. (2021). Impact of Newborn Screening and Early Dietary Management on Clinical Outcome of Patients with Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency and Medium Chain Acyl-CoA Dehydrogenase Deficiency—A Retrospective Nationwide Study. Nutrients, 13(9), 2925. https://doi.org/10.3390/nu13092925