
Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs

 ,
,  ,
,  , , , ,
, , , , {kind=link}
Abstract
1. Introduction
2. Epidemiology
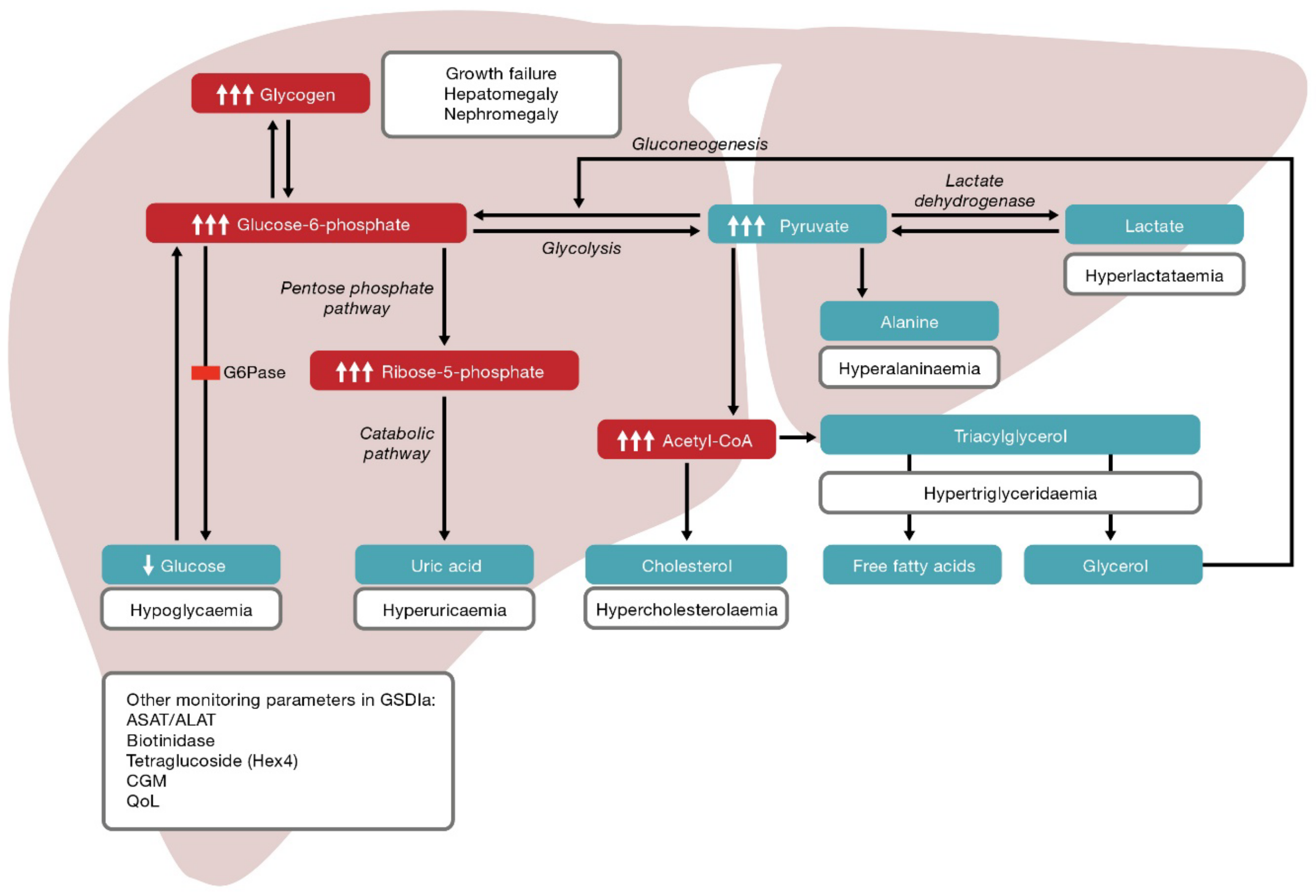
3. Pathophysiology and Clinical Manifestations
4. Current Strategies for Disease Management
- 1.
- Hypoglycaemia associated with the glycogenolysis defect.
- 2.
- Hepatomegaly, adenomas, adenocarcinomas and chronic kidney disease associated with glycogen accumulation.
- 3.
- Hypoglycaemia and hyperlactataemia associated with the gluconeogenesis defect.
- 4.
- Dyslipidaemia (hypertriglyceridaemia), hyperuricaemia and hepatic steatosis associated with glucose-6-phosphate accumulation.
- 5.
- Obesity, metabolic syndrome, anorexia and malnutrition associated with current disease management.
5. Burden of Disease, Management and Monitoring
- 1.
- 2.
- 3.
- Patients may experience hypoglycaemia despite adhering to dietary treatment [20].
- 4.
- 5.
- The therapeutic range between dietary under treatment and overtreatment is complicated by imprecision in dosing of UCCS, which can lead to fluctuating glucose levels [48].
- 6.
- 7.
- 8.
- Self-monitoring of glucose levels may not accurately capture periods of asymptomatic hypoglycaemia, particularly when lactate levels are elevated, and literature supporting the use of continuous glucose monitoring is currently lacking [61].
- 9.
- 10.
- Patients may experience reimbursement issues, e.g., for dietary products and/or blood glucose monitoring, including continuous blood glucose monitoring.
- 11.
- 12.
- Patients and parents/caregivers suffer disrupted sleep due to the need for night feeds [62].
- 13.
- Patients and parents/caregivers experience fear, anxiety and stress associated with missed doses of UCCS and whether they might prove fatal [62].
- 14.
- 15.
- Patients may have concerns or experience low self-esteem related to their physical appearance [64].
- 16.
- Patients may experience disrupted schooling due to time off/hospitalisation relating to the disease, which may impact future career choices [62].
5.1. Burden of Disease
5.2. Burden of Disease Management and Monitoring
6. Quality of Life
7. Future Directions
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Froissart, R.; Piraud, M.; Boudjemline, A.M.; Vianey-Saban, C.; Petit, F.; Hubert-Buron, A.; Eberschweiler, P.T.; Gajdos, V.; Labrune, P. Glucose-6-phosphatase deficiency. Orphanet J. Rare Dis. 2011, 6, 27. [Google Scholar] [CrossRef]
- De León, D.D.; Stanley, C.A.; Sperling, M.A. Hypoglycemia in neonates and infants. In Pediatric Endocrinology, 3rd ed.; Sperling, M.A., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2008; pp. 165–197. [Google Scholar]
- Burr, I.M.; O’Neill, J.A.; Karzon, D.T.; Howard, L.J.; Greene, H.L. Comparison of the effects of total parenteral nutrition, continuous intragastric feeding, and portacaval shunt on a patient with type I glycogen storage disease. J. Pediatr. 1974, 85, 792–795. [Google Scholar] [CrossRef]
- Greene, H.L.; Slonim, A.E.; O’Neill, J.A.; Burr, I.M. Continuous Nocturnal Intragastric Feeding for Management of Type 1 Glycogen-Storage Disease. N. Engl. J. Med. 1976, 294, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Smit, G.P.; Berger, R.; Potasnick, R.; Moses, S.W.; Fernandes, J. The Dietary Treatment of Children with Type I Glycogen Storage Disease with Slow Release Carbohydrate. Pediatr. Res. 1984, 18, 879–881. [Google Scholar] [CrossRef]
- Harari, S. Why we should care about ultra-rare disease. Eur. Respir. Rev. 2016, 25, 101–103. [Google Scholar] [CrossRef]
- Hughes, D.; Tunnage, B.; Yeo, S. Drugs for exceptionally rare diseases: Do they deserve special status for funding? Qjm: Int. J. Med. 2005, 98, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.-J.; Shelly, L.L.; Pan, C.-J.; Sidbury, J.B.; Chou, J.Y. Mutations in the Glucose-6-Phosphatase Gene that Cause Glycogen Storage Disease Type 1a. Science 1993, 262, 580–583. [Google Scholar] [CrossRef] [PubMed]
- National Organization for Rare Disorders (NORD). Glycogen Storage Disease Type I. Available online: https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/ (accessed on 13 August 2021).
- Chou, J.Y.; Jun, H.S.; Mansfield, B. Type I glycogen storage diseases: Disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes. J. Inherit. Metab. Dis. 2014, 38, 511–519. [Google Scholar] [CrossRef]
- Chen, M.A.; Weinstein, D.A. Glycogen storage diseases: Diagnosis, treatment and outcome. Transl. Sci. Rare Dis. 2016, 1, 45–72. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Steuerwald, U.; De Souza, C.F.; Derks, T. Inborn Errors of Metabolism with Hypoglycemia: Glycogen storage diseases and inherited disorders of gluconeogenesis. Pediatr. Clin. N. Am. 2018, 65, 247–265. [Google Scholar] [CrossRef]
- Bali, D.S.; Chen, Y.T.; Austin, S.; Goldstein, J.L. Glycogen Storage Disease Type I. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Scott, S.A.; Edelmann, L.; Liu, L.; Luo, M.; Desnick, R.J.; Kornreich, R. Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum. Mutat. 2010, 31, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Streeper, R.S.; Svitek, C.A.; Goldman, J.K.; O’Brien, R.M. Differential Role of Hepatocyte Nuclear Factor-1 in the Regulation of Glucose-6-phosphatase Catalytic Subunit Gene Transcription by cAMP in Liver- and Kidney-derived Cell Lines. J. Biol. Chem. 2000, 275, 12108–12118. [Google Scholar] [CrossRef]
- Parikh, N.S.; Ahlawat, R. Glycogen Storage Disease Type I. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hoogerland, J.A.; Peeks, F.; Hijmans, B.S.; Wolters, J.C.; Kooijman, S.; Bos, T.; Bleeker, A.; van Dijk, T.H.; Wolters, H.; Gerding, A.; et al. Impaired Very-Low-Density Lipoprotein catabolism links hypoglycemia to hypertriglyceridemia in Glycogen Storage Disease type Ia. J. Inherit. Metab. Dis. 2021, 44, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.A.; Ahmed, A.; Lavery, G.G.; Tomlinson, J.; Kim, S.Y.; Cooper, M.S.; Ride, J.P.; Hughes, B.A.; Shackleton, C.H.L.; McKiernan, P.; et al. 11β-Hydroxysteroid Dehydrogenase Type 1 Regulation by Intracellular Glucose 6-Phosphate Provides Evidence for a Novel Link between Glucose Metabolism and Hypothalamo-Pituitary-Adrenal Axis Function. J. Biol. Chem. 2007, 282, 27030–27036. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Simeoli, C.; Salerno, M.; Ferrigno, R.; Della Casa, R.; Colao, A.; Strisciuglio, P.; Parenti, G.; Pivonello, R.; Melis, D. Imbalanced cortisol concentrations in glycogen storage disease type I: Evidence for a possible link between endocrine regulation and metabolic derangement. Orphanet J. Rare Dis. 2020, 15, 99. [Google Scholar] [CrossRef]
- Rake, J.P.; Visser, G.; Labrune, P.; Leonard, J.V.; Ullrich, K.; Smit, A.G.P. Glycogen storage disease type I: Diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur. J. Nucl. Med. Mol. Imaging 2002, 161, S20–S34. [Google Scholar] [CrossRef]
- Özen, H. Glycogen Storage Diseases: New Perspectives. World J. Gastroenterol. 2007, 13, 2541–2553. [Google Scholar] [CrossRef] [PubMed]
- UpToDate®. Glucose-6-Phosphatase Deficiency (Glycogen Storage Disease I, von Gierke Disease). Available online: https://www.uptodate.com/contents/glucose-6-phosphatase-deficiency-glycogen-storage-disease-i-von-gierke-disease (accessed on 13 August 2021).
- Chou, J.Y.; Jun, H.S.; Mansfield, B. Glycogen storage disease type I and G6Pase-β deficiency: Etiology and therapy. Nat. Rev. Endocrinol. 2010, 6, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1. [Google Scholar] [CrossRef]
- Labrune, P.; Ullrich, K.; Smit, P.G.; Rake, J.; Visser, G.; Leonard, J.V. Guidelines for management of glycogen storage disease type I—European Study on Glycogen Storage Disease Type I (ESGSD I). Eur. J. Nucl. Med. Mol. Imaging 2002, 161, S112–S119. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Austin, S.L.; Arn, P.; Bali, D.S.; Boney, A.; Case, L.; Chung, W.K.; Desai, D.M.; El-Gharbawy, A.; Haller, R.; et al. Glycogen Storage Disease Type III diagnosis and management guidelines. Genet. Med. 2010, 12, 446–463. [Google Scholar] [CrossRef]
- Wang, D.Q.; Fiske, L.M.; Carreras, C.T.; Weinstein, D.A. Natural History of Hepatocellular Adenoma Formation in Glycogen Storage Disease Type I. J. Pediatr. 2011, 159, 442–446. [Google Scholar] [CrossRef]
- Sechi, A.; Deroma, L.; Lapolla, A.; Paci, S.; Melis, D.; Burlina, A.; Carubbi, F.; Rigoldi, M.; Di Rocco, M. Fertility and pregnancy in women affected by glycogen storage disease type I, results of a multicenter Italian study. J. Inherit. Metab. Dis. 2012, 36, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Ferrecchia, I.A.; Guenette, G.; Potocik, E.A.; Weinstein, D.A. Pregnancy in Women With Glycogen Storage Disease Ia and Ib. J. Périnat. Neonatal Nurs. 2014, 28, 26–31. [Google Scholar] [CrossRef]
- Lawrence, N.T.; Chengsupanimit, T.; Brown, L.M.; Derks, T.; Smit, G.P.A.; Weinstein, D.A. Inflammatory Bowel Disease in Glycogen Storage Disease Type Ia. J. Pediatr. Gastroenterol. Nutr. 2017, 64, e52–e54. [Google Scholar] [CrossRef]
- Wang, D.Q.; Carreras, C.T.; Fiske, L.M.; Austin, S.; Boree, D.; Kishnani, P.S.; Weinstein, D.A. Characterization and pathogenesis of anemia in glycogen storage disease type Ia and Ib. Genet. Med. 2012, 14, 795–799. [Google Scholar] [CrossRef]
- Jacoby, J.T.; dos Santos, B.B.; Nalin, T.; Colonetti, K.; Refosco, L.F.; de Souza, C.F.M.; Spritzer, P.M.; Poloni, S.; Hack-Mendes, R.; Schwartz, I.V.D. Bone Mineral Density in Patients with Hepatic Glycogen Storage Diseases. Nutrients 2021, 13, 2987. [Google Scholar] [CrossRef]
- Peeks, F.; Steunenberg, T.A.H.; de Boer, F.; Rubio-Gozalbo, M.E.; Williams, M.; Burghard, R.; Rajas, F.; Oosterveer, M.H.; Weinstein, D.A.; Derks, T.G.J. Clinical and biochemical heterogeneity between patients with glycogen storage disease type IA: The added value of CUSUM for metabolic control. J. Inherit. Metab. Dis. 2017, 40, 695–702. [Google Scholar] [CrossRef]
- Cassiman, D.; Libbrecht, L.; Verslype, C.; Meersseman, W.; Troisi, R.; Zucman-Rossi, J.; Van Vlierberghe, H. An adult male patient with multiple adenomas and a hepatocellular carcinoma: Mild Glycogen Storage Disease type Ia. J. Hepatol. 2010, 53, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Moest, W.; Van Der Deure, W.; Koster, T.; Spee-Dropková, M.; Swart-Busscher, L.; De Haas, R.J.; Derks, T.G. Glycogen storage disease type Ia: Adult presentation with microcytic anemia and liver adenomas. Hepatology 2018, 68, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; He, W.; Huang, X.; Wu, Y.; Lei, Y.; Yu, C.; Görgülü, K.; Diakopoulos, K.N.; Lu, N.; Zhu, Y. A case report of acute pancreatitis with glycogen storage disease type IA in an adult patient and review of the literature. Medicine 2020, 99, e22644. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.-J.; Terzioglu, M.; Hiraiwa, H.; Marsh, J.; Pan, C.-J.; Chen, L.-Y.; Chou, J.Y. The Molecular Basis of Glycogen Storage Disease Type 1a: Structure and function analysis of mutations in glucose-6-phosphatase. J. Biol. Chem. 2002, 277, 5047–5053. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ozawa, T.; Kawasaki, T.; Yasumi, K.; Wang, N.-Y.; Kitagawa, M.; Takehira, Y.; Tamakoshi, K.; Yamada, M.; Kida, H.; et al. Case report: Hepatocellular carcinoma in type 1a glycogen storage disease with identification of a glucose-6-phosphatase gene mutation in one family. J. Gastroenterol. Hepatol. 2002, 14, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Matern, D.; Seydewitz, H.; Bali, D.; Lang, C.; Chen, Y.-T. Glycogen storage disease type I: Diagnosis and phenotype/genotype correlation. Eur. J. Pediatr. 2002, 161 (Suppl. 1), S10–S19. [Google Scholar] [CrossRef]
- Weston, B.W.; Lin, J.-L.; Muenzer, J.; Cameron, H.S.; Arnold, R.R.; Seydewitz, H.H.; Mayatepek, E.; Van Schaftingen, E.; Veiga-Da-Cunha, M.; Matern, D.; et al. Glucose-6-Phosphatase Mutation G188R Confers an Atypical Glycogen Storage Disease Type 1b Phenotype. Pediatr. Res. 2000, 48, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Storch, E.; Keeley, M.; Merlo, L.; Jacob, M.; Correia, C.; Weinstein, D. Psychosocial Functioning in Youth with Glycogen Storage Disease Type I. J. Pediatr. Psychol. 2008, 33, 728–738. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Sun, B.; Koeberl, D.D. Gene therapy for glycogen storage diseases. Hum. Mol. Genet. 2019, 28, R31–R41. [Google Scholar] [CrossRef] [PubMed]
- Starzl, T.E.; Marchioro, T.L.; Sexton, A.W.; Illingworth, B.; Waddell, W.R.; Faris, T.D.; Herrmann, T.J. The effect of portacaval transposition on carbohydrate metabolism: Experimental and clinical observations. Surgery 1965, 57, 687–697. [Google Scholar]
- Bérat, C.-M.; Roda, C.; Brassier, A.; Bouchereau, J.; Wicker, C.; Servais, A.; Dubois, S.; Assoun, M.; Belloche, C.; Barbier, V.; et al. Enteral tube feeding in patients receiving dietary treatment for metabolic diseases: A retrospective analysis in a large French cohort. Mol. Genet. Metab. Rep. 2021, 26, 100655. [Google Scholar] [CrossRef]
- Leonard, J.V.; Dunger, D.B. Hypoglycæmia complicating feeding regimens for glycogen-storage disease. Lancet 1978, 312, 1203–1204. [Google Scholar] [CrossRef]
- Dunger, P.D.; Sutton, P.; Leonard, J.V. Hypoglycaemia complicating treatment regimens for glycogen storage disease. Arch. Dis. Child. 1995, 72, 274–275. [Google Scholar] [CrossRef][Green Version]
- Derks, T.G.; Martens, D.H.; Sentner, C.P.; Van Rijn, M.; De Boer, F.; Smit, G.P.A.; Van Spronsen, F.J. Dietary treatment of glycogen storage disease type Ia: Uncooked cornstarch and/or continuous nocturnal gastric drip-feeding? Mol. Genet. Metab. 2013, 109, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.M.; Ferrecchia, I.A.; Dahlberg, K.R.; Dambska, M.; Ryan, P.T.; Weinstein, A.D. Dietary Management of the Glycogen Storage Diseases: Evolution of Treatment and Ongoing Controversies. Adv. Nutr. 2019. [Google Scholar] [CrossRef]
- Dahlberg, K.R.; Ferrecchia, I.A.; Dambska-Williams, M.; Resler, T.E.; Ross, K.M.; Butler, G.L.; Kuo, C.; Ryan, P.T.; Weinstein, D.A. Cornstarch requirements of the adult glycogen storage disease Ia population: A retrospective review. J. Inherit. Metab. Dis. 2019, 43, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Orton, R.C.; Qi, X.; Mundy, H.; Morley, D.W.; Champion, M.P.; Eaton, S.; Tester, R.F.; Lee, P.J. A novel starch for the treatment of glycogen storage diseases. J. Inherit. Metab. Dis. 2007, 30, 350–357. [Google Scholar] [CrossRef]
- Correia, C.E.; Bhattacharya, K.; Lee, P.J.; Shuster, J.J.; Theriaque, D.W.; Shankar, M.N.; Smit, G.P.A.; Weinstein, D.A. Use of modified cornstarch therapy to extend fasting in glycogen storage disease types Ia and Ib. Am. J. Clin. Nutr. 2008, 88, 1272–1276. [Google Scholar] [PubMed]
- Ross, K.M.; Brown, L.M.; Corrado, M.M.; Chengsupanimit, T.; Curry, L.M.; Ferrecchia, I.A.; Porras, L.Y.; Mathew, J.T.; Weinstein, D.A.; Baumgartner, M.; et al. Safety and Efficacy of Chronic Extended Release Cornstarch Therapy for Glycogen Storage Disease Type I. JIMD Rep. 2015, 26, 85–90. [Google Scholar] [CrossRef]
- Nalin, T.; Venema, K.; Weinstein, D.A.; De Souza, C.F.M.; Perry, I.D.S.; Van Wandelen, M.T.R.; Van Rijn, M.; Smit, G.P.A.; Schwartz, I.V.D.; Derks, T.G.J. In vitro digestion of starches in a dynamic gastrointestinal model: An innovative study to optimize dietary management of patients with hepatic glycogen storage diseases. J. Inherit. Metab. Dis. 2014, 38, 529–536. [Google Scholar] [CrossRef]
- Monteiro, V.C.L.; de Oliveira, B.M.; dos Santos, B.B.; Sperb-Ludwig, F.; Refosco, L.F.; Nalin, T.; Derks, T.G.J.; de Souza, C.F.M.; Schwartz, I.V.D. A triple-blinded crossover study to evaluate the short-term safety of sweet manioc starch for the treatment of glycogen storage disease type Ia. Orphanet J. Rare Dis. 2021, 16. [Google Scholar] [CrossRef]
- Mathew, T.K.; Tadi, P. Blood Glucose Monitoring. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Peeks, F.; Hoogeveen, I.J.; Feldbrugge, R.L.; Burghard, R.; de Boer, F.; Fokkert-Wilts, M.J.; van der Klauw, M.M.; Oosterveer, M.H.; Derks, T.G.J. A retrospective in-depth analysis of continuous glucose monitoring datasets for patients with hepatic glycogen storage disease: Recommended outcome parameters for glucose management. J. Inherit. Metab. Dis. 2021, 44, 1136–1150. [Google Scholar] [CrossRef]
- Fernandes, J. The effect of disaccharides on the hyperlactacidaemia of glucose-6-phosphatase-deficient children. Acta Paediatr. 1974, 63, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Cederholm, T.; Jensen, G.; Correia, M.; Gonzalez, M.C.; Fukushima, R.; Higashiguchi, T.; Baptista, G.; Barazzoni, R.; Blaauw, R.; Coats, A.; et al. GLIM criteria for the diagnosis of malnutrition—A consensus report from the global clinical nutrition community. J. Cachex Sarcopenia Muscle 2019, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Colonetti, K.; Dos Santos, B.B.; Nalin, T.; De Souza, C.F.M.; Triplett, E.W.; Dobbler, P.C.T.; Schwartz, I.V.D.; Roesch, L.F.W. Hepatic glycogen storage diseases are associated to microbial dysbiosis. PLoS ONE 2019, 14, e0214582. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, N.; Seaquist, E.R. Neuroendocrine responses to hypoglycemia. Ann. N. Y. Acad. Sci. 2010, 1212, 12–28. [Google Scholar] [CrossRef]
- Herbert, M.; Pendyal, S.; Rairikar, M.; Halaby, C.; Benjamin, R.W.; Kishnani, P.S. Role of continuous glucose monitoring in the management of glycogen storage disorders. J. Inherit. Metab. Dis. 2018, 41, 917–927. [Google Scholar] [CrossRef]
- US Food & Drug Administration. Patient Listening Summaries: Glycogen Storage Disease (GSD) Type 1. Available online: https://www.fda.gov/patients/learn-about-fda-patient-engagement/patient-listening-session-summaries (accessed on 13 August 2021).
- Jelaska, B.K.; Ostojić, S.B.; Berović, N.; Kokić, V. Continuous glucose monitoring in the treatment of obesity in patients with glycogen storage disease type Ia. Endocrinol. Diabetes Metab. Case Rep. 2014, 2014, 130056. [Google Scholar] [CrossRef][Green Version]
- Flanagan, T.B.; Sutton, J.A.; Brown, L.M.; Weinstein, D.A.; Merlo, L.J.; Zschocke, J. Disordered Eating and Body Esteem Among Individuals with Glycogen Storage Disease. JIMD Rep. 2014, 19, 23–29. [Google Scholar] [CrossRef]
- Fernandes, J.; Berger, R.; Smit, G. Lactate as energy source for brain in glucose-6-phosphatase deficient child. Lancet 1982, 319, 113. [Google Scholar] [CrossRef]
- Kasapkara, S.; Demir, G.C.; Hasanoğlu, A.; Tümer, L. Continuous glucose monitoring in children with glycogen storage disease type I. Eur. J. Clin. Nutr. 2013, 68, 101–105. [Google Scholar] [CrossRef]
- Kaiser, N.; Gautschi, M.; Bosanska, L.; Meienberg, F.; Baumgartner, M.R.; Spinas, G.A.; Hochuli, M. Glycemic control and complications in glycogen storage disease type I: Results from the Swiss registry. Mol. Genet. Metab. 2019, 126, 355–361. [Google Scholar] [CrossRef]
- Maran, A.; Crepaldi, C.; Avogaro, A.; Catuogno, S.; Burlina, A.; Poscia, A.; Tiengo, A. Continuous glucose monitoring in conditions other than diabetes. Diabetes/Metab. Res. Rev. 2004, 20 (Suppl. 2), S50–S55. [Google Scholar] [CrossRef] [PubMed]
- White, F.J.; Jones, S.A. The use of continuous glucose monitoring in the practical management of glycogen storage disorders. J. Inherit. Metab. Dis. 2011, 34, 631–642. [Google Scholar] [CrossRef]
- Melis, D.; Rossi, A.; Pivonello, R.; Salerno, M.; Balivo, F.; Spadarella, S.; Muscogiuri, G.; Della Casa, R.; Formisano, P.; Andria, G.; et al. Glycogen storage disease type Ia (GSDIa) but not Glycogen storage disease type Ib (GSDIb) is associated to an increased risk of metabolic syndrome: Possible role of microsomal glucose 6-phosphate accumulation. Orphanet J. Rare Dis. 2015, 10, 1–8. [Google Scholar] [CrossRef]
- Minarich, L.A.; Kirpich, A.; Fiske, L.M.; Weinstein, D.A. Bone mineral density in glycogen storage disease type Ia and Ib. Genet. Med. 2012, 14, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Krieger, N.S.; Sessler, N.E.; Bushinsky, D.A. Acidosis inhibits osteoblastic and stimulates osteoclastic activity in vitro. Am. J. Physiol. Physiol. 1992, 262, F442–F448. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef]
- Rossi, A.; Hoogeveen, I.J.; Lubout, C.M.A.; de Boer, F.; Fokkert-Wilts, M.J.; Rodenburg, I.L.; van Dam, E.; Grünert, S.C.; Martinelli, D.; Scarpa, M.; et al. A generic emergency protocol for patients with inborn errors of metabolism causing fasting intolerance: A retrospective, single-center study and the generation of www.emergencyprotocol.net. J. Inherit. Metab. Dis. 2021, 44, 1124–1135. [Google Scholar] [CrossRef]
- Sechi, A.; Deroma, L.; Paci, S.; Lapolla, A.; Carubbi, F.; Burlina, A.; Rigoldi, M.; Di Rocco, M. Quality of Life in Adult Patients with Glycogen Storage Disease Type I: Results of a Multicenter Italian Study. JIMD Rep. 2013, 14, 47–53. [Google Scholar] [CrossRef]
- Venema, A.; Peeks, F.; der Veen, M.d.B.-v.; de Boer, F.; Fokkert-Wilts, M.J.; Lubout, C.M.A.; Huskens, B.; Dumont, E.; Mulkens, S.; Derks, T.G.J. A retrospective study of eating and psychosocial problems in patients with hepatic glycogen storage diseases and idiopathic ketotic hypoglycemia: Towards a standard set of patient-reported outcome measures. JIMD Rep. 2021, in press. [Google Scholar] [CrossRef]
- Garbade, S.F.; Ederer, V.; Burgard, P.; Wendel, U.; Spiekerkoetter, U.; Haas, D.; Grünert, S.C. Impact of glycogen storage disease type I on adult daily life: A survey. Orphanet J. Rare Dis. 2021, 16, 1–10. [Google Scholar] [CrossRef]
- Nemours. Children’s Health System. Surgeries and Procedures: Gastrostomy Tube (G-Tube). Available online: https://kidshealth.org/Nemours/en/parents/gastrostomy.html (accessed on 22 September 2021).
- Butler, J.; Dress, A.; Theodore-Oklota, C.; Egan, S.; Paulich, M.; Byrd, C.; Evans, C. The Humanistic Burden of Glycogen Storage Disease Type IA—The Impact on Symptoms, Diet and Health-Related Quality of Life. In Proceedings of the Virtual ISPOR 2020, Orlando, FL, USA, 18–20 May 2020. [Google Scholar]
- Martinez, C.C.; Tonon, T.; Nalin, T.; Refosco, L.F.; de Souza, C.F.M.; Schwartz, I.V.D. Feeding Difficulties and Orofacial Myofunctional Disorder in Patients with Hepatic Glycogen Storage Diseases. JIMD Rep. 2018, 45, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.M.S.; Silva, N.J.M.M.; Dias, P.; Porto, J.F.C.; Santos, L.C.; Costa, J.M.N. Glycogen Storage Disease type 1a—A secondary cause for hyperlipidemia: Report of five cases. J. Diabetes Metab. Disord. 2013, 12, 25. [Google Scholar] [CrossRef]
- Boers, S.J.; Visser, G.; Smit, P.G.; Fuchs, A.S. Liver transplantation in glycogen storage disease type I. Orphanet J. Rare Dis. 2014, 9, 47. [Google Scholar] [CrossRef]
- Wilke, M.V.M.B.; De Kleine, R.H.; Wietasch, J.K.G.; Van Amerongen, C.C.A.; Blokzijl, H.; Van Spronsen, F.J.; Schwartz, I.V.D.; Derks, T.G.J. Orthotopic Liver Transplantation in Glycogen Storage Disease Type 1a. J. Inborn Errors Metab. Screen. 2016, 4, 1–5. [Google Scholar] [CrossRef]
- Fisher, R.A.; Strom, S. Human Hepatocyte Transplantation: Worldwide Results. Transplantation 2006, 82, 441–449. [Google Scholar] [CrossRef]
- Muraca, M.; Burlina, A.B. Liver and liver cell transplantation for glycogen storage disease type IA. Acta Gastro-Enterol. Belg. 2006, 68, 469–472. [Google Scholar]
- Oishi, K.; Arnon, R.; Wasserstein, M.P.; Diaz, G.A. Liver transplantation for pediatric inherited metabolic disorders: Considerations for indications, complications, and perioperative management. Pediatr. Transplant. 2016, 20, 756–769. [Google Scholar] [CrossRef]
- Rodriguez-Buritica, D.F.; Ahmad, A.; Couce Pico, M.L.; Derks, T.G.; Mitchell, J.; Riba-Wolman, R.; Mou, J.; Poma, A.; Valayannopoulos, V.; Crombez, E. AAV8-Mediated Liver-Directed Gene Therapy as a Potential Therapeutic Option in Adults with Glycogen Storage Disease Type Ia (GSDIa): Updated Phase 1/2 Clinical Trial Results. In Proceedings of the American Society of Gene + Cell Therapy, Portland, OR, USA, 12–15 May 2021. [Google Scholar]
- Augustine, E.F.; Dorsey, E.R.; Saltonstall, P.L. The Care Continuum: An Evolving Model for Care and Research in Rare Diseases. Pediatrics 2017, 140, e20170108. [Google Scholar] [CrossRef]
- Peeks, F.; Boonstra, W.F.; de Baere, L.; Carøe, C.; Casswall, T.; Cohen, D.; Cowan, K.; Ferrecchia, I.; Ferriani, A.; Gimbert, C.; et al. Research priorities for liver glycogen storage disease: An international priority setting partnership with the James Lind Alliance. J. Inherit. Metab. Dis. 2019, 43, 279–289. [Google Scholar] [CrossRef]
- Nano, J.; Carinci, F.; Okunade, O.; Whittaker, S.; Walbaum, M.; Barnard-Kelly, K.; Barthelmes, D.; Benson, T.; Calderon-Margalit, R.; Dennaoui, J.; et al. A standard set of person-centred outcomes for diabetes mellitus: Results of an international and unified approach. Diabet. Med. 2020, 37, 2009–2018. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derks, T.G.J.; Rodriguez-Buritica, D.F.; Ahmad, A.; de Boer, F.; Couce, M.L.; Grünert, S.C.; Labrune, P.; López Maldonado, N.; Fischinger Moura de Souza, C.; Riba-Wolman, R.; et al. Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs. Nutrients 2021, 13, 3828. https://doi.org/10.3390/nu13113828
Derks TGJ, Rodriguez-Buritica DF, Ahmad A, de Boer F, Couce ML, Grünert SC, Labrune P, López Maldonado N, Fischinger Moura de Souza C, Riba-Wolman R, et al. Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs. Nutrients. 2021; 13(11):3828. https://doi.org/10.3390/nu13113828
Chicago/Turabian StyleDerks, Terry G. J., David F. Rodriguez-Buritica, Ayesha Ahmad, Foekje de Boer, María L. Couce, Sarah C. Grünert, Philippe Labrune, Nerea López Maldonado, Carolina Fischinger Moura de Souza, Rebecca Riba-Wolman, and et al. 2021. "Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs" Nutrients 13, no. 11: 3828. https://doi.org/10.3390/nu13113828
APA StyleDerks, T. G. J., Rodriguez-Buritica, D. F., Ahmad, A., de Boer, F., Couce, M. L., Grünert, S. C., Labrune, P., López Maldonado, N., Fischinger Moura de Souza, C., Riba-Wolman, R., Rossi, A., Saavedra, H., Gupta, R. N., Valayannopoulos, V., & Mitchell, J. (2021). Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs. Nutrients, 13(11), 3828. https://doi.org/10.3390/nu13113828